 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of energy transfer nanostructures using gelation-driven dynamic combinatorial libraries†
Siva Krishna Mohan
Nalluri
* and
Rein V.
Ulijn
*
WestCHEM, Department of Pure & Applied Chemistry, Thomas Graham Building, University of Strathclyde, 295 Cathedral Street, G1 1XL Glasgow, UK. E-mail: siva.nalluri@strath.ac.uk; rein.ulijn@strath.ac.uk; Fax: +44 141 548 4822; Tel: +44 141 548 2110
First published on 2nd July 2013
Abstract
Peptide self-assembly provides a useful approach to control the organization of functional molecular components, as relevant to future opto-electronic or photonic nanostructures. In this article, we report on the discovery of efficient energy transfer nanostructures using a dynamic combinatorial library (DCL) approach driven by molecular self-assembly, demonstrating an enhanced self-selection and amplification of effective energy transfer nanostructures from complex mixtures of dipeptide derivatives. By taking advantage of an enzyme-catalysed fully reversible amide formation reaction, we show how gelation shifts the equilibrium in favour of the formation of short aromatic dipeptide derivatives in the DCL system, as confirmed by reversed-phase high pressure liquid chromatography (HPLC), fluorescence emission spectroscopy, atomic force microscopy (AFM), transmission force microscopy (TEM) and circular dichroism (CD) spectroscopy. This approach enabled us to identify a two-component donor–acceptor hydrogel, which forms within minutes and exhibits efficient energy transfer.
Introduction
The fabrication of functional nanomaterials by molecular self-assembly is attracting increasing attention.1,2 The use of short peptide derivatives as versatile building blocks offers a suitable platform due to the chemical versatility of amino acid building blocks combined with simplicity and chemical accessibility of peptides.3–8 There is an on-going effort to develop molecular design rules for the basic building blocks. Many new self-assembling structures are discovered by serendipity, rather than by rational design. One approach that may accelerate discovery is combining molecular self-assembly with dynamic combinatorial chemistry (DCC).9,10 This technique allows for the continuous interconversion of building blocks that leads to the formation of several dynamic combinatorial library (DCL) members under thermodynamic control through a reversible, yet covalent, chemical reaction. Recently, DCLs involving peptide derivatives have been developed by using disulphide,11–13 metal binding14,15 and amide16 exchange reactions. We have focused on a fully reversible enzymatic amide exchange that provides access to peptide sequences in gelation-driven17 DCLs. These systems have the ability to self-select the most stable self-assembling peptide nanomaterials from a library of several amino acid components.4,18 (Opto-)electronic nanostructures rely on the effective stacking of electron-rich and electron-deficient components. Peptides are potentially suited to precisely positioning these components in a configuration that optimizes their function. Hence the use of a peptide DCL approach for the identification of nanomaterials with improved function19–23 (in addition to stability) from complex mixtures provides a potentially useful tool for the discovery of functional materials and this has not been explored yet in the literature.One area where designed nanomaterials have received a great attention is in mimics of the light-harvesting portion of natural photosynthesis.24 The light-harvesting and energy transfer between donor and acceptor molecules have been reported in various nanoscale architectures such as micelles,25 vesicles,26,27 dendrimers,28,29 conjugated polymers,30 peptide fibres,31 organogels32–35 and hydrogels,36–39 with a first short peptide-based system described by Adams.40 In these supramolecular systems, the efficient energy transfer occurs in a state where the self-organization of aromatic groups (donors and acceptors) should represent a local thermodynamic minimum of a free energy landscape and also the defects arising during the self-assembly process are expected to be minimized (self-correction due to reversibility)41 to increase the overall energy transfer efficiency.
Herein, we report a gelation-driven peptide-based DCL approach for the unprecedented discovery of stable functional nanomaterials which exhibit efficient energy transfer. Our design strategy is focused on the molecular self-assembly of various aromatic dipeptide amphiphiles (naphthoxy-substituted dipeptide amide derivatives) which are formed by direct condensation of amino acid derivatives (Scheme 1). We demonstrate that the gelation-driven DCL approach has the potential ability to self-select and amplify the most stable self-assembling nanostructures (Fig. 1). We further demonstrate that these nanostructures are formed rapidly (within minutes) that remarkably show superior energy transfer behaviour (more rapid and efficient) compared to literature reports.40 In particular, we emphasize that this system operates under physiological conditions and no organic solvents are necessary to dissolve the donor–acceptor building blocks.
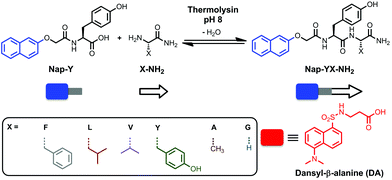 | ||
| Scheme 1 Formation of naphthoxy-substituted dipeptide amphiphiles by a fully reversible thermolysin-catalysed condensation reaction and the molecular structure of dansyl-β-alanine (DA) used in this study. | ||
Results and discussion
The first objective was to develop a DCL based on the self-assembly of short aromatic dipeptide amphiphiles, to identify the most stable structures, in the absence of the acceptor. The self-assembly of naphthoxy-substituted dipeptides driven by an enzyme-catalysed condensation reaction was selectively chosen for this purpose. As shown in Fig. 1, the precursor solution consisted of naphthoxy-substituted tyrosine derivative (Nap-Y; 20 mM) with a 4-fold excess of an amino acid amide nucleophile (X-NH22222222222222222, where “X” denotes phenylalanine, F; leucine, L; valine, V; tyrosine, Y; alanine, A and/or glycine, G) in 100 mM phosphate buffer (pH 8). A non-specific endoprotease, thermolysin from Bacillus thermoproteolyticus rokko, was used (1 mg ml−1)4,18,42 to catalyse the condensation reaction of the amino acid amide derivatives. This ultimately converts the non-assembling precursors (Nap-Y and X-NH22222222222222222) into self-assembling building blocks (Nap-YX-NH222222222), provided that the reaction is thermodynamically driven by the free energy contribution of the self-assembly process.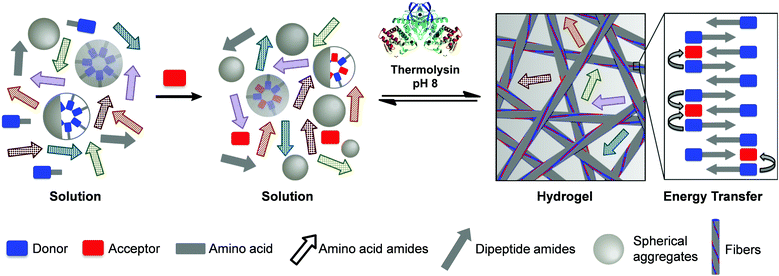 | ||
| Fig. 1 Schematic representation of thermolysin-triggered development of a bi-component hydrogel composed of naphthalene donors and dansyl acceptors, showing efficient energy transfer from a library of eight different amino acid non-assembling precursor components. | ||
At first, the enzymatic formation of various Nap-YX-NH222222222 derivatives (also simply represented as “YX”) from each combination of Nap-Y and X-NH22222222222222222 was investigated in isolation. The molecular self-assembly of the building blocks upon the addition of thermolysin was directly identified by the naked eye observation of a rapid transformation from a clear and transparent solution into a self-supporting hydrogel (as recognized by vial inversion). The percentage conversion was investigated by reversed-phase high-pressure liquid chromatography (HPLC). As shown in Fig. 2A, some of the dipeptide derivatives, YF (89%), YL (72%) and YV (81%) were formed in very good yields, while the other derivatives, YY (5%), YA (0.9%) and YG (0.7%) were formed in very low yields after 48 h. The high yields obtained for YF, YL and YV indicate that gelation-driven self-assembly of these dipeptide derivatives is thermodynamically favourable, as a result of the presence of strong π–π stacking between naphthalene chromophores and the hydrogen bonding between peptide motifs. Notably, only YF, YL and YV derivatives were self-assembled to form self-supporting hydrogels (Fig. S1 in ESI†).43 Interestingly, these hydrogels were formed rapidly (within minutes after the addition of thermolysin).44 The percentage formation of these dipeptide derivatives was further monitored over time and in each case the maximum conversions were reached after 24 h (Fig. S2 in ESI†). The low yields obtained for the other dipeptide derivatives (YY, YA and YG) are likely due to their less favourable self-assembly; these systems lack the thermodynamic driving force to overcome the bias for amide hydrolysis in aqueous systems.4 It should be noted that thermolysin has a kinetic preference45 for more hydrophobic amino acids (X = phenylalanine in the case of YX) on the amine side of the peptide bond, whereas it is non-specific for carboxylic acid residue in condensation reactions. However, an equilibrium distribution of the products should eventually be reached.
![The enzymatic conversion of Nap-Y and X-NH22222222222222222 into Nap-YX-NH222222222 amphiphiles was shown by (A) part of the reversed-phase HPLC traces in isolation (all except top two traces) as well as in DCL system (top two traces) and (B) time course of the percentage conversion in DCL system by HPLC as measured in the absence (solid traces) and presence (dashed traces) of DA. Conditions: [Nap-Y] = 20 mM, [each X-NH22222222222222222] = 80 mM, [DA] = 20 mM and [thermolysin] = 1 mg ml−1.](/image/article/2013/SC/c3sc51036k/c3sc51036k-f2.gif) | ||
| Fig. 2 The enzymatic conversion of Nap-Y and X-NH22222222222222222 into Nap-YX-NH222222222 amphiphiles was shown by (A) part of the reversed-phase HPLC traces in isolation (all except top two traces) as well as in DCL system (top two traces) and (B) time course of the percentage conversion in DCL system by HPLC as measured in the absence (solid traces) and presence (dashed traces) of DA. Conditions: [Nap-Y] = 20 mM, [each X-NH22222222222222222] = 80 mM, [DA] = 20 mM and [thermolysin] = 1 mg ml−1. | ||
For the development of DCL, Nap-Y (20 mM) and all six amino acid amide derivatives (80 mM for each X-NH22222222222222222 where X = F, L, V, Y, A and G) were mixed together in one-pot and a thermolysin-catalysed condensation reaction was carried out in competition. As shown in Fig. 2A, after 48 h, it was found out that YF was preferentially produced in 52% conversion, corresponding to 77% of the total peptide yield obtained, indicating that this dipeptide derivative represents the lowest free energy well of the free energy landscape. Conversely, YL was formed in 23% yield (of total peptide formed), while all the other derivatives, YV (2%), YY (0.3%), YA (0.1%) and YG (0.04%) were formed in negligible amounts. These results remarkably show that YV was exclusively screened out from the library, despite the fact that it was formed in very good yield (81%) when carried out in isolation. Furthermore, in order to determine whether the enzyme has any kinetic preference for a specific sequence of the dipeptide, we also investigated the evolution of all dipeptide derivatives over time (solid traces, Fig. 2B). It was also found out that only YF was produced predominantly right from the beginning stages of the experiment, while YL was produced in low yield and YV, YY, YA and YG were remained relatively constant just above the baseline. These results demonstrate that this peptide-based DCL approach has the potential ability for the selection of the most stable self-assembling nanostructures from a library of several amino acid precursor components.
The second objective was to exploit this peptide-based DCL approach for the discovery of energy transfer nanomaterials from component mixtures, by introducing water-soluble dansyl-β-alanine (DA) as an acceptor to utilize the well-known donor–acceptor interactions between naphthalene chromophores and dansyl chromophores.37,40,46 We first investigated the effect of the presence of DA on the enzyme-triggered evolution of Nap-YX-NH222222222 amphiphiles in DCL by reversed-phase HPLC. For this, Nap-Y and all six amino acid amide derivatives (X-NH22222222222222222) and DA were mixed together in one-pot. The addition of thermolysin triggered the rapid transformation of a free-flowing transparent solution into a self-supporting hydrogel. As shown in Fig. 2, the percentage evolution of all dipeptide derivatives in DCL was investigated at a 1:1 donor-acceptor ratio. Remarkably, only a single component (YF) was produced in 82% (corresponding to 95% of the total peptide yield obtained), while YL (8%) was formed in low yield and all the other derivatives, YV (4%), YY (0.5%), YA (0.1%) and YG (0.07%) were formed in negligible amounts (Fig. 2A). Most interestingly, the comparison of yields obtained in the absence (solid traces) and presence (dashed traces) of DA remarkably suggest that the formation of YF (82% instead of 52%) was significantly amplified and YL (8% instead of 23%) was reduced (Fig. 2B). These observations clearly indicate that the presence of both donors and acceptors in DCL provides the additional donor–acceptor interactions that makes this enzyme-assisted self-assembly more favourable and eventually allows the system to reach a lowest energy minimum state for a particular dipeptide sequence in DCL. This hypothesis was reinforced by the fact that the percentage evolution of YF in DCL is strictly dependent on the total donor to acceptor ratio. The relative percentage evolutions of YF in DCL are 61% at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 65% at 7:1 and 70% at 2:1 donor–acceptor ratios (Fig. S3 in ESI†). This further confirms that the product distribution in the library is dependent on the type and the extent of the interactions present and is entirely thermodynamically driven by the more favourable self-assembly of mixed donors and acceptors in the library. The availability of both donors and acceptors in DCL makes this approach more versatile and greatly enhances its potential ability not just for self-selection but also for the amplification of most stable energy transfer nanostructures at the expense of less stable self-assembling nanostructures.
:1, 65% at 7:1 and 70% at 2:1 donor–acceptor ratios (Fig. S3 in ESI†). This further confirms that the product distribution in the library is dependent on the type and the extent of the interactions present and is entirely thermodynamically driven by the more favourable self-assembly of mixed donors and acceptors in the library. The availability of both donors and acceptors in DCL makes this approach more versatile and greatly enhances its potential ability not just for self-selection but also for the amplification of most stable energy transfer nanostructures at the expense of less stable self-assembling nanostructures.
Next, we moved on to determine the efficiency of energy transfer within the newly discovered hydrogels. The supramolecular self-assembly of Nap-YX-NH222222222 amphiphiles in the DCL system was characterized by fluorescence emission spectroscopy both in the absence and presence of DA. As shown in Fig. 3A and B, in the absence of DA, the fluorescence emission spectrum of the starting mixture (Nap-Y + X-NH22222222222222222) exhibited two distinct characteristic peaks. One is a sharp naphthalene monomer emission peak at 350 nm and the other one is a broad shoulder peak at around 420 nm which corresponds to the excimer emission,47 emerging from the intermolecular π–π stacking between naphthalene chromophores (solid black trace).
![Fluorescence emission spectroscopy showing (A) time-dependent changes observed for DCL system upon the addition of thermolysin, and efficient energy transfer observed for both (B) DCL system before (solid traces) and at 15 min after (dashed traces) the addition of thermolysin at variable concentrations of DA as well as for (C) YF system in isolation before (solid traces) and at 2 min after (dashed traces) the addition of thermolysin at variable concentrations of DA (λex = 280 nm). Conditions: [Nap-Y] = 20 mM, [each X-NH22222222222222222] = 80 mM, [DA] = 0.6 mM to 10 mM and [thermolysin] = 1 mg ml−1.](/image/article/2013/SC/c3sc51036k/c3sc51036k-f3.gif) | ||
| Fig. 3 Fluorescence emission spectroscopy showing (A) time-dependent changes observed for DCL system upon the addition of thermolysin, and efficient energy transfer observed for both (B) DCL system before (solid traces) and at 15 min after (dashed traces) the addition of thermolysin at variable concentrations of DA as well as for (C) YF system in isolation before (solid traces) and at 2 min after (dashed traces) the addition of thermolysin at variable concentrations of DA (λex = 280 nm). Conditions: [Nap-Y] = 20 mM, [each X-NH22222222222222222] = 80 mM, [DA] = 0.6 mM to 10 mM and [thermolysin] = 1 mg ml−1. | ||
The addition of thermolysin to the above starting mixture induced a slight red-shift in the peak at 350 nm accompanied by a massive increase in the relative emission intensity (red trace, Fig. 3A). These observations indicate that excimer naphthalene chromophores were formed which eventually emit at higher wavelength when compared to monomer naphthalene chromophores. Even after 24 h, the peak at 350 nm was further red-shifted accompanied by a further increase in the relative emission intensity (green trace, Fig. 3A). Such an increase in the relative emission intensity over time indeed indicates that the self-assembly of Nap-YX-NH222222222 amphiphiles in DCL system shows an aggregation-induced emission (AIE) behaviour.48
The fluorescence emission of Nap-YX-NH222222222 amphiphiles in the presence of acceptor DA was then studied. The fluorescence emission of DA molecules (in solution state, λem = 560 nm and λex = 330 nm) is strongly dependent on the surrounding environment such as solvent, morphology, etc. and usually exhibits a blue-shift in the emission peak when migrates from aqueous to non-aqueous environment (Fig. S4 in ESI†).49 As shown in Fig. 3B, the addition of DA to the starting mixture (Nap-Y + X-NH22222222222222222) at variable concentrations induced substantial changes in the fluorescence emission spectra. For instance, when the concentration of DA was 2 mM (i.e., 10:1 donor–acceptor ratio), the monomer emission peak at 350 nm was quenched completely, while the excimer emission peak at 420 nm was enhanced exclusively and interestingly, a new broad emission peak was appeared at 535 nm (solid red trace). When the concentration of DA was increased to 10 mM (i.e., 2:1 donor–acceptor ratio), the emission peak at 420 nm was also quenched completely, while the broad peak at 535 nm was even broadened (solid blue trace). The new emission peak at 535 nm (rather than the expected peak at 560 nm for DA in solution) may suggest that the surrounding environment of DA molecules is changing from aqueous to non-aqueous environment (corresponding to blue-shifted emission). At this stage, the direct visualization of the morphology by atomic force microscopy (AFM) and transmission electron microscopy (TEM) confirmed the presence of spherical aggregates with an average diameter of 100–400 nm (Fig. 4A and C and S5 in ESI†). These results suggest that the DA molecules were incorporated in these spherical aggregates formed by Nap-Y and further indicate that there is a partial energy transfer between the naphthalene chromophores of Nap-Y and the dansyl chromophores of DA. In the solution state, efficient energy transfer occurs above 10 mM concentration of DA and precursor molecules.
![(A and B) AFM images of DCL + 2 mM DA system on mica surface and (C–F) TEM images of DCL and individual systems in the absence and presence of DA on carbon-coated copper grid. (A and C) Before and after the addition of thermolysin in the (D) absence and (B and E) presence of DA molecules in DCL system. (F) Isolated YF + 2 mM DA system. Inset: Photographs of the corresponding materials before (transparent and clear solution) and after (self-supporting hydrogel) the addition of thermolysin. Conditions: [Nap-Y] = 20 mM, [each X-NH22222222222222222] = 80 mM, [DA] = 2 mM and [thermolysin] = 1 mg ml−1.](/image/article/2013/SC/c3sc51036k/c3sc51036k-f4.gif) | ||
| Fig. 4 (A and B) AFM images of DCL + 2 mM DA system on mica surface and (C–F) TEM images of DCL and individual systems in the absence and presence of DA on carbon-coated copper grid. (A and C) Before and after the addition of thermolysin in the (D) absence and (B and E) presence of DA molecules in DCL system. (F) Isolated YF + 2 mM DA system. Inset: Photographs of the corresponding materials before (transparent and clear solution) and after (self-supporting hydrogel) the addition of thermolysin. Conditions: [Nap-Y] = 20 mM, [each X-NH22222222222222222] = 80 mM, [DA] = 2 mM and [thermolysin] = 1 mg ml−1. | ||
The subsequent addition of thermolysin to the starting mixture solution of DA (2 mM and 10 mM) induced the formation of a self-supporting hydrogel. As shown in Fig. 3B, both peaks at 350 nm and 420 nm were quenched completely, while the broad emission peak at 535 nm was largely blue-shifted and appeared at 510 nm (dashed traces). Interestingly, energy transfer induced a dramatic enhancement in fluorescence emission38 (more than a 150-fold increase in the relative emission intensity above 500 nm) over time (Fig. S6 and S7 in ESI†). AFM and TEM images showed that the addition of thermolysin triggered the rapid morphological transition of spherical aggregates into entangled nanofibres of up to several micrometres in length (Fig. 4B, D and E). Furthermore, similar experiments were also repeated for the most stable YF system in isolation (since it was discovered as a major component from the direct competition in DCL).
Remarkably, it was found that the energy transfer in the gel state is efficient even at 0.6 mM concentration of DA (i.e., 33:1 donor–acceptor ratio) and also in this case, such an energy transfer induced an enhanced fluorescence emission over time (Fig. 3C and also Fig. S8–S10 in ESI†). TEM images also showed the presence of entangled nanofibres of up to several micrometers in length (Fig. 4F). The striking differences observed in the emission spectra before (solid traces) and after (dashed traces) the addition of thermolysin would indeed suggest that the energy transfer occurs within the gel preparation and measurement time (<2 min), as well as being efficient in the gel state (fiber networks) when compared to the solution state (spherical aggregates).
We further investigated the successful incorporation of DA molecules into the entangled fibre networks of naphthoxy-substituted dipeptides (for instance, YF) by circular dichroism (CD) spectroscopy. The interactions of DA molecules with Nap-YF-NH2 nanostructures were characterized by monitoring the signals which appear due to the supramolecular chiral environment of naphthalene chromophores in the gel phase over time. As shown in Fig. 5, the dansyl chromophore of DA (2 mM) by itself is achiral and showed no CD signals (black trace). Similarly, no CD signals were observed even when DA was added to the starting mixture solution (Nap-Y + F-NH2) as naphthalene chromophores are also achiral (red trace). However, upon the addition of thermolysin, the corresponding CD signals were instantaneously starting to appear within the measurement time (<2 min, blue trace) which indicates that the chromophores were self-assembling into a supramolecular chiral structure.
![Time-dependent circular dichroism spectra of naphthoxy-substituted dipeptide derivative (Nap-YF-NH2) upon the addition of thermolysin measured in the absence and presence of DA (i.e., 10:1 donor-acceptor ratio). Conditions: [Nap-Y] = 20 mM, [F-NH2] = 80 mM [DA] = 2 mM, and [thermolysin] = 1 mg ml−1.](/image/article/2013/SC/c3sc51036k/c3sc51036k-f5.gif) | ||
| Fig. 5 Time-dependent circular dichroism spectra of naphthoxy-substituted dipeptide derivative (Nap-YF-NH2) upon the addition of thermolysin measured in the absence and presence of DA (i.e., 10:1 donor-acceptor ratio). Conditions: [Nap-Y] = 20 mM, [F-NH2] = 80 mM [DA] = 2 mM, and [thermolysin] = 1 mg ml−1. | ||
The strength and intensity of CD signals at 288 nm and 328 nm, originating due to the π–π* and n–π* transitions, respectively, of naphthalene chromophores were increased over time. In this case, the CD activity of dansyl chromophores of DA should be an induced CD (ICD) which would appear50 at 330 nm and it is more likely to interfere with the CD signals originating due to the n–π* transition peaks of the naphthalene chromophores. Therefore, we monitored the changes observed in the CD signals of the naphthalene chromophores both in the absence and presence of DA. Notably, the comparison of CD signals in the absence (orange trace) and presence (purple trace) of DA suggested the following changes in CD signals after 24 h: the intensity of the CD signals was increased by more than 2 times, the signal at 278 nm was red-shifted by about 10 nm and the ratio of both these π–π* and n–π* transitions peaks changed from 3 to 1.3 (purple and orange traces). These findings indicate that the key presence of DA molecules would certainly provide the crucial strong donor–acceptor interactions in addition to the fundamental non-covalent interactions that are already present in the entangled nanofibres and hence facilitates the formation of the most stable nanostructures with enhanced chirality. All these results consistently indicate that DA molecules have been successfully incorporated in the entangled fibre networks of Nap-YF-NH2 which ultimately leads to the formation of two-component hydrogels.51
Conclusions
In summary, we have demonstrated that a two-component hydrogel composed of Nap-YF-NH2 and DA emerged as an effective energy transfer gel from a library of eight competing amino acid non-assembling precursor components (Nap-Y; F-, L-, V-, Y-, A-, G-NH2 amino acid amide derivatives and DA molecules) through a fully reversible thermolysin-triggered condensation reaction. Furthermore, we also showed that the discovery of a functional (rather than just structural) system was achieved through a self-selection and amplification mechanism from component mixtures. This approach opens up the new possibility of discovering electronically conductive nanomaterials with enhanced charge transfer properties and fewer defects.Acknowledgements
Financial support was provided by US Air Force (AFOSR, grant 12448RK7359).Notes and references
- J.-M. Lehn, Science, 1993, 260, 1762–1763 CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2008, 4, 19–24 CrossRef.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- B. Bilgiçer, X. Xing and K. Kumar, J. Am. Chem. Soc., 2001, 123, 11815–11816 CrossRef.
- Y. Krishnan-Ghosh and S. Balasubramanian, Angew. Chem., Int. Ed., 2003, 42, 2171–2173 CrossRef CAS.
- E. H. C. Bromley, R. B. Sessions, A. R. Thomson and D. N. Woolfson, J. Am. Chem. Soc., 2009, 131, 928–930 CrossRef CAS.
- M. A. Case and G. L. McLendon, J. Am. Chem. Soc., 2000, 122, 8089–8090 CrossRef CAS.
- H. J. Cooper, M. A. Case, G. L. McLendon and A. G. Marshall, J. Am. Chem. Soc., 2003, 125, 5331–5339 CrossRef CAS.
- P. G. Swann, R. A. Casanova, A. Desai, M. M. Frauenhoff, M. Urbancic, U. Slomczynska, A. J. Hopfinger, G. C. Le Breton and D. L. Venton, Biopolymers, 1996, 40, 617–625 CrossRef CAS.
- N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS.
- A. K. Das, A. R. Hirst and R. V. Ulijn, Faraday Discuss., 2009, 143, 293–303 RSC.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–490 CrossRef CAS.
- A. S. Tayi, A. K. Shveyd, A. C. H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS.
- E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC.
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2010, 49, 5331–5334 CrossRef CAS.
- S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2009, 48, 7635–7638 CrossRef CAS.
- J. Barber and B. Andersson, Nature, 1994, 370, 31–34 CrossRef CAS.
- M. H. Gehlen and F. C. De Schryver, Chem. Rev., 1993, 93, 199–221 CrossRef CAS.
- X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623–629 CrossRef CAS.
- F. J. M. Hoeben, I. O. Shklyarevskiy, M. J. Pouderoijen, H. Engelkamp, A. P. H. J. Schenning, P. C. M. Christianen, J. C. Maan and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 1232–1236 CrossRef CAS.
- A. P. H. J. Schenning, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 4489–4495 CrossRef CAS.
- D.-L. Jiang and T. Aida, Nature, 1997, 388, 454–456 CrossRef CAS.
- T.-Q. Nguyen, J. Wu, V. Doan, B. J. Schwartz and S. H. Tolbert, Science, 2000, 288, 652–656 CrossRef CAS.
- K. J. Channon, G. L. Devlin and C. E. MacPhee, J. Am. Chem. Soc., 2009, 131, 12520–12521 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- A. Ajayaghosh, S. J. George and V. K. Praveen, Angew. Chem., Int. Ed., 2003, 42, 332–335 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260–6265 CrossRef CAS.
- C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef CAS.
- T. Nakashima and N. Kimizuka, Adv. Mater., 2002, 14, 1113–1116 CrossRef CAS.
- M. Montalti, L. S. Dolci, L. Prodi, N. Zaccheroni, M. C. A. Stuart, K. J. C. van Bommel and A. Friggeri, Langmuir, 2006, 22, 2299–2303 CrossRef CAS.
- K. V. Rao, K. K. Datta, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2011, 50, 1179–1184 CrossRef CAS.
- P. Bairi, B. Roy and A. K. Nandi, Chem. Commun., 2012, 48, 10850–10852 RSC.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC (this paper describes the first reported energy transfer in a peptide-based hydrogel system that occurs only at relatively low pH values (pH < 4); organic solvents are used to dissolve propyl dansylamide acceptor used in this system, however, the energy transfer is apparently rather slow and low efficient).
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS.
- R. V. Ulijn, B. Baragaña, P. J. Halling and S. L. Flitsch, J. Am. Chem. Soc., 2002, 124, 10988–10989 CrossRef CAS.
- However, the chemically synthesized (instead of enzymatic) YF and YL dipeptide derivatives formed amorphous precipitates upon cooling a heated solution in buffer which highlights the significant role of the enzyme needed for the formation of self-supporting hydrogels in this case. The enzymatic reaction starts from freely soluble building blocks rather than an insoluble precursor, which facilitates overcoming the free energy barrier to enable access to the self-assembled state.
- The presence of free terminal amide groups in dipeptide amphiphiles may accelerate the self-assembly process that eventually triggers rapid hydrogelation.
- R. H. P. Doezé, B. A. Maltman, C. L. Egan, R. V. Ulijn and S. L. Flitsch, Angew. Chem., Int. Ed., 2004, 43, 3138–3141 CrossRef.
- W.-S. Li, M.-J. Teng, X.-R. Jia and Y. Wei, Tetrahedron Lett., 2010, 51, 5336–5340 CrossRef CAS.
- H. Ikeda, Y. Iidaka and A. Ueno, Org. Lett., 2003, 5, 1625–1627 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- K. P. Ghiggino, A. G. Lee, S. R. Meech, D. V. O'Connor and D. Phillips, Biochemistry, 1981, 20, 5381–5389 CrossRef CAS.
- K. Hamasaki, H. Ikeda, A. Nakamura, A. Ueno, F. Toda, I. Suzuki and T. Osa, J. Am. Chem. Soc., 1993, 115, 5035–5040 CrossRef CAS.
- We cannot be conclusive at this point to exclude the possibility of co-assembly with the other dipeptide derivatives (for instance, YF and YL). The investigation of this would require labeling experiments which are currently ongoing.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data for new compounds, additional reversed-phase high-pressure liquid chromatography, atomic force microscopy and UV-vis as well as fluorescence emission spectroscopy results. See DOI: 10.1039/c3sc51036k |
| This journal is © The Royal Society of Chemistry 2013 |