 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel nanographene/porphyrin hybrids – preparation, characterization, and application in solar energy conversion schemes†
Daniel Kiessling‡a, Rubén D. Costa‡a, Georgios Katsukis‡a, Jenny Malig‡a, Fabian Lodermeyera, Sebastian Feihla, Alexandra Rotha, Leonie Wibmera, Matthias Kehrera, Michel Vollanda, Pawel Wagnerb, Gordon G. Wallaceb, David L. Officerb and Dirk M. Guldi*a
aDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany. E-mail: guldi@fau.de; Web: http://www.chemie.uni-erlangen.de/dcp/assets/pdf/DCP-Profile2010.pdf Fax: +49 (0)9131 8528307; Tel: +49 (0)9131 8527340
bARC Centre of Excellence for Electromaterials Science and the Intelligent Polymer Research Institute, University of Wollongong, NSW 2522, Australia. E-mail: davido@uow.edu.au; Fax: +61 (2)42213114; Tel: +61 (2)42213127
First published on 28th May 2013
Abstract
Four novel nanographene/porphyrin hybrids were prepared, characterized, and probed in solar energy conversion schemes. Exfoliation of graphite by means of immobilizing four different porphyrins onto the basal plane of graphene is accompanied by distinct electronic interactions in both the ground and the excited states. In the ground state, a strong loss in oscillator strength goes hand-in-hand with a notable broadening of the porphyrin transitions and, as such, attests to the shift of electron density from the electron donating porphyrins to nanographene. In the excited state, a nearly quantitative quenching of the porphyrin fluorescence is indicative of full charge transfer. The latter is corroborated by femtosecond transient absorption measurements, which reveal the generation of the one-electron oxidized radical cation of the porphyrins with absorption maxima at 490 and 625 nm in the visible region and conduction band electrons in nanographene with features at 890 and 1025 nm in the near infrared region. We have demonstrated the applicability of the new nanographene/porphyrin hybrids in, for example, solar cells. In this regard, the presence of flakes is crucial in terms of influencing the injection processes, preventing aggregation, and reducing recombination losses, which are commonly encountered in porphyrin-based DSSCs.
Introduction
Nature chose carbon to provide the basis for life on earth. As a matter of fact, carbon is the key to many technological applications, which over the years have become indispensable in our daily life and have influenced the world's civilization for centuries. Notably, the structural diversity of organic building blocks results in sheer endless chemical and physical properties. Altering the periodic binding motifs in sp3-, sp2-, and sp-hybridized networks represents the toolbox for constructing a wide palette of carbon allotropes. To this end, the past two decades have served as a test-bed for probing the physico-chemical properties of nanocarbons in reduced dimensions starting with the advent of 0D fullerenes, followed by 1D carbon nanotubes (CNTs) and by 2D graphene.1–5Turning to 2D graphene, single layers of graphene were first prepared successfully in 2004 by mechanical exfoliation of graphite using Scotch Tape.6 Alternative fabrication strategies – epitaxial growth7 and solubilization from bulk graphite8 – have recently been demonstrated and they are, indeed, paving the way to systematic experiments and technological applications.9 At room temperature, charge transport measurements show that a flat single layer graphene is a zero-gap semiconductor and exhibits a remarkably high electron mobility with values exceeding values from 2000 (CVD10) to 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (suspended11) cm2 V−1 s−1. The symmetry of the experimentally measured conductance indicates high mobility not only for electrons but also for holes. In addition, an ideal monolayer has an optical transmittance of 97.7%.12 The aforementioned calls for its implementation into transparent conducting electrodes as a viable alternative to indium tin oxide.13
000 (suspended11) cm2 V−1 s−1. The symmetry of the experimentally measured conductance indicates high mobility not only for electrons but also for holes. In addition, an ideal monolayer has an optical transmittance of 97.7%.12 The aforementioned calls for its implementation into transparent conducting electrodes as a viable alternative to indium tin oxide.13
Graphite is the most desired starting material en route towards fabricating graphene. Going from graphite to graphene requires, however, an activation step owing to the thermodynamic stabilization of the former. The activation might come in the form of oxidizing graphite to graphitic acid, which evolved providing valuable insights into the structure and constitution of graphite.14 At basic pH, exfoliation results in monolayers of graphene oxide (GO).14 Nevertheless, the final step, namely reduction of GO monolayers, fails to completely remove the introduced oxygen atoms and reduction leads irreversibly to partially amorphous carbon.15 Not surprisingly, the correspondingly reduced GO (rGO) features properties different than those predicted for real graphene. Another form of activation involves the intercalation of graphite with alkali metals.16 Dissolving the resulting negatively charged graphene sheets in solution, enables solutions of bi- and trilayer graphene.17 To avoid any of the aforementioned pre-treatments, but still to activate graphite requires ultrasound as a chemical free treatment in liquids.8,18 A common denominator is a suitable amphiphilic intercalator and/or solvent that guarantees the efficient exfoliation.19 As such, the versatility lies in the use of water and/or organic solvents to exfoliate graphite and/or to directly functionalize nanographene sheets by a variety of non-covalent means.4
Nevertheless, recent results have documented that charge transfer emerges as a necessity to harvest the full potential of single layer graphene by tailoring the electronic properties for new complex electronics.20,21 To this day, only a few scattered reports are known that come, however, short to provide a coherent picture.20,22–24 Particularly relevant are advances on self-ordering/self-assembling,25–28 on one hand, and π-stacking29,30 as well as charge transfer interactions,30,31 on the other hand, to yield high-quality single layer flakes of nanographene that are stable and dispersible.
With the help of recent studies a number of challenges in studying nanographene have been unveiled. Clearly, the lack of spectroscopic signatures, which render spectroscopic assignments quite ambiguous – similar to early contributions to the field of CNTs – stands out among these challenges. In preliminary investigations, we have realized high-quality flakes of nanographene, which were, however, few layer graphene rather than single layer, by means of solution processing graphite with tightly interacting porphycenes, phthalocyanines and porphyrins.29,32–34 In terms of spectroscopy, we were able to establish spectroscopic as well as kinetic evidence not only for ground-state interactions but also for interactions in the excited state. In the excited state, radical ion pair states are formed including the radical cation of the electron donating phthalocyanines and/or porphyrins and electrons that are delocalized within the basal plane of graphene. As a matter of fact, planar, aromatic macrocycles are highly versatile to exfoliate graphite and to afford functional nanographene based charge-transfer hybrids.4,29,35
In the current work, we report on the formation and characterization of novel nanographene/porphyrin hybrids and their implementation into solar cells. Particular emphasis was placed on a top-down preparation of stable dispersions – starting from natural graphite rather than graphene oxide – whilst preserving the intrinsic properties of graphene. To the best of our knowledge, the benefits of introducing such hybrid materials in DSSCs have not been established to date.
Experimental
Materials
Natural graphite was purchased from Graphit Kropfmühl AG. Spectroscopic quality solvents tetrahydrofuran (THF) and toluene were ordered from Sigma-Aldrich and were used without further purification. The 5,10,15,20-tetrabutylporphyrin and its copper complex (CuTBP) were synthesized according to literature procedures.36 The other reagents were obtained commercially.Synthesis
Preparation of nanographene hybrids
Natural graphite (1 mg) was added to a 2 × 10−5 M stock solution (3 mL) of 6a/b and 7a/b in THF or THF–water mixtures (1:1, 1:10 or 1:100 v/v) and subsequently ultrasonicated for 10 min with a Bransonic 52 from BRANSON at room temperature with 37 kHz and an effective power of 330 W. The considered solvents were THF and THF–water mixtures. Next, graphite (1 mg) was added again and the dispersion was kept under ultrasonic treatment for additional 10 min. Afterwards, the heterogeneous dispersion was centrifuged for 15 min at 500 rpm with a Fresco 21 centrifuge from Thermo Scientific at room temperature. The steps were repeated two times with the resulting supernatant. The final step included centrifugation at 5000 rpm for 15 min to remove any larger particles of graphite.Physico-chemical characterization
Steady-state absorption spectra were recorded with a Perkin-Elmer Lambda 35. Steady-state emission spectra were recorded with a Fluoromax-P-spectrometer from HORIBA Jobin Yvon. Time-correlated single photon counting (TCSPC) spectra were taken with a Fluorolog system (HORIBA Jobin Yvon). Signal acquisition was gathered by a Hamamatsu MCP photomultiplier (type R3809U-50). The time profiles were recorded at the emission maxima. All samples were measured in a quartz glass cuvette with a width of 10 mm. Femtosecond transient absorption spectra were obtained with a Ti:sapphire laser system CPA-2101 (Clark-MXR Inc.) in combination with a Helios TAPPS detection unit from Ultrafast Inc. The initial laser excitation wavelength was 775 nm with a pulse width of 150 fs. The excitation wavelength employed was 387 nm, which was generated with a SHG crystal. For the generation of the white light, a sapphire crystal of adequate thickness was used. The chirp-effect between 420 and 770 nm was approximately 350 fs. The detection was carried out with two CCD cameras, each for a specific measuring range. The spectral windows were therefore 415 to 770 nm and 770 to 1600 nm. The delay line allowed spectral acquisition up to time delays of 8000 ps. All samples were measured in a fused quartz glass cuvette with a width of 2 mm. Data acquisition was done with the software HELIOS visible/nIR (Newport/Ultrafast Systems).Raman measurements were carried out with a LabRAM ARAMIS Raman-spectrometer from HORIBA Jobin Yvon that was equipped with a confocal microscope and an automated XYZ-table, using laser excitations of 532 and 633 nm. The sample preparation involved drop casting the hybrids on a Si substrate with a 300 nm oxide layer. In transmission electron microscopy (TEM), sample preparation was performed by drop casting and drying the hybrids on holey carbon-coated copper grids. TEM images were recorded with an 80 kV EM 900 TEM from Carl Zeiss AG.
Square wave voltammetry measurements were performed using a three-electrode setup consisting of a Teflon covered (d = 10 mm) glassy carbon (d = 2 mm) working electrode, a Pt-wire as counter electrode, and an Ag-wire as pseudo-reference electrode. Ferrocene served as internal standard and was measured in separate measurements under ambient conditions. 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) was applied as electrolyte in dichloromethane. All solutions were deoxygenated under an argon stream before measuring. The scan rate was set to 0.125 V s−1 and the amplitude to 0.2 V. A μAutolab Methrom PG3stat with an impedance unit was used for all experiments. The applied software was NOVA 1.6.
Photocurrent measurements were carried out under AM 1.5 conditions using a custom-made solar simulator, including a 350–1000 watt adjustable Xe lamp source (LOT) combined with appropriate filters. Current–voltage measurements were made by using a Keithley Sourcemeter 2400 in the range of −0.7 to 0.3 V with a rate of 0.037 V s−1. The cells were measured without a black mask with an error of around 10% for the current density values.
Device preparation
FTO TEC 8 plates (Xop Glass Company) were extensively cleaned by means of ultrasonication in subsequent baths of detergent, acetone and isopropanol for 15 min each, just before the deposition of the TiO2 electrodes. TiO2-T and TiO2-S electrodes were prepared from Ti-nanoxide T/SP paste (Solaronix) and from “DSL 18NR-AO” paste (Dyesol), respectively. Both pastes were doctor-bladed using a circular template with a diameter of 6 mm and a thickness of 50 μm onto an FTO slide, and subsequently baked from room temperature to 450 °C with a ramp of 3.5 °C min−1. The samples were baked at 450 °C for 30 min. Film thickness of 7–8 mm was measured for both electrodes. These electrodes were immersed into different suspensions – i.e., THF and THF–water (1:1 v/v) prepared for the porphyrins and the nanographene/porphyrin hybrids at room temperature. For counter electrode fabrication, FTO plates with holes of 1 mm2 at the edge of the active area were used. Prior to the fabrication of the counter-electrodes, the FTO slides were cleaned using the following procedure. A thin film of chloroplatinic acid, namely 0.5 mmol H2PtCl6 in ethanol, prepared from chloroplatinic acid hydrate ∼38% Pt basis (Sigma-Aldrich), was spread over the FTO plates and dried in air prior to baking at 390 °C for 30 min. The thickness of the platinum counter electrode was of around 20–40 nm. Subsequently both electrodes were sealed together with a transparent film of Surlyn 1472 (DuPont Ltd., UK) cut as a frame around the nanocrystalline film. A solution of 0.6 M 1-butyl-3-methylimidazolium iodide 99% (Sigma Aldrich), 0.03 M iodine double sublimed (Merck), 0.1 M guanidine thiocyanate ≥99.0% (Fluka) and 0.5 M 4-tert-butylpyridine 96% (Sigma Aldrich) in 85% acetonitrile (Sigma) and 15% valeronitrile (Alfa Aesar) was employed as electrolyte. The latter was introduced through the aforementioned holes in the counter electrode without vacuum system and immediately sealed.Results and discussion
Scheme 1 displays the structures of porphyrins 6a/b and 7a/b, which were used in this work to realize the multifunctional nanographene hybrids. Porphyrins 6a/b and 7a/b have several key features. Firstly, the hydrophobic nature of the porphyrin core is of utmost importance for the successful exfoliation of graphite to yield stable suspensions of nanographene hybrids. Secondly, the conjugated β-pyrrolic side chain bears one or two carboxylic groups that facilitate grafting of TiO2 nanoparticles and the integration of the hybrids into the TiO2 electrodes. Additionally, the cyano group or corresponding carboxylic acid group is electron withdrawing and assists in electron transfer to the TiO2. | ||
| Scheme 1 Synthesis of porphyrin acids. Reagents and conditions: (i) N-methylformanilide, POCl3, chloroform, 60 °C; (ii) POCl3, water, 0 °C, (iii) (1,3-dioxolan-2-ylmethyl)triphenylphosphonium bromide, K2CO3, 18-crown-6, benzene, reflux; (iv) Zn(OAc)2, DCM, methanol, rt; (v) malonic or cyanoacetic acid, NH4OAc, AcOH, THF, 65 °C. | ||
The general features of the syntheses of porphyrins 6a/b and 7a/b have been previously described for the synthesis of 7a.38 As indicated in Scheme 1, the porphyrins were synthesized in an analogous fashion to what we have previously used for tetraarylporphyrin dyes.37,39 The Vilsmeier formylation of tetraalkylporphyrins has been reported previously albeit in very low yields.40,41 We also found that using the DMF–POCl3 formylation complex at 80–90 °C produces mostly decomposition products. Shortening the reaction time to 50 min gave 2 in low yield (<20%) with recovery of some unreacted 1, whilst lowering the reaction temperature below 70 °C did not give any appreciable product. However, replacing DMF with the more active N-methylformanilide allowed us to decrease the reaction temperature to 60 °C yet still provide 2 in satisfactory yield (67%) with no starting material remaining. It is notable that a much larger excess (×100) of the Vilsmeier reagent is required than normally used for porphyrin formylation.
Porphyrin demetallation reactions usually require strong acids such as conc. sulfuric acid.37 Usage of such harsh conditions decreases the yield of the free-base tetraalkylporphyrin.41 As a result, we have utilized here the milder procedure described by Ponomarev and Maravin.42 A cooled solution of porphyrin 2 dissolved in a small amount of POCl3 is treated with water (10% by volume). After a few minutes a vigorous reaction takes place and a large quantity of hydrogen chloride is evolved. A high yield of the resulting free-base porphyrin 3 is thus obtained. The reaction is so vigorous that using more than 300 mg of the starting materials is not advised. In addition, using larger amounts of porphyrin only results in partial demetallation.
The allylaldehyde derivative 4 was synthesized by a modified procedure of Ishkov et al.43 Porphyrin aldehyde was reacted with the ylide generated in situ from (1,3-dioxolan-2-ylmethyl)triphenylphosphonium bromide, using crown ether as a phase-transfer catalyst and potassium carbonate as a base. Since we found that the ylide was not stable under the reaction conditions, the addition of an excess of the phosphonium salt and the base had to be carried out every 2 h for 6 h. Interestingly, the attempts to replace benzene with the less toxic toluene did not give satisfactory results. The reaction in boiling toluene gave lower yields due to the apparent decomposition of the starting materials. Lowering the temperature of the reaction in toluene to 90 °C did result in some product formation but the reaction could not be driven to completion.
The product of this condensation is a mixture of E and Z isomers of the protected aldehyde 4. It was previously reported that attempts to purify a similar mixture on silica gel resulted in partial deprotection of the porphyrins.43 As a result we carried out deprotection and isomerisation on the crude Wittig reaction mixture, which gave, after purification, the porphyrin 4 in high yield (80%).
The zinc insertion to give 5 was achieved by a standard procedure with almost quantitative yield.37 The resulting zinc complex 5 and free-base 4 were then subjected to Knoevenagel condensation with malonic and cyanoacetic acids to give the corresponding porphyrin acids 6a/b and 7a/b in excellent yields. We found that the reactivity of the tetraalkyl derivatives after formylation did not differ significantly from their tetraaryl equivalents.37
To determine the interactions of the porphyrins with graphene, we decided to firstly elucidate the ground and excited state features of 6a/b and 7a/b based on steady-state absorption and fluorescence spectroscopy as well as their electrochemical characteristics. The absorption spectra of 7a and 7b in THF show asymmetric and broadened Soret bands at 419 and 417 nm, respectively. The Q-bands of free-base cyanoacetic acid 7a are found at 523, 556, 611 and 669 nm, while for malonic acid 7b they emerge at 524, 553, 612 and 669 nm – Fig. 1. The overall shape of the Soret band is a result of β-pyrrolic substitution by the conjugated linker that causes a loss in symmetry as well as some aggregation effects. Zinc porphyrins 6a and 6b show typical absorption features of metalloporphyrins in THF with Soret bands at 433 and 429 nm and Q-bands at 573/618 and 571/618 nm, respectively. Additionally, experiments were performed in THF–water mixture (1:1 v/v) – Fig. S1 in the ESI.† Comparing the absorption features of 6a/b and 7a/b in THF–water (1:1 v/v) with those in pure THF, the biscarboxyporphyrins 6b and 7b show strongly increased molar extinction coefficients by a factor of up to 2 for the Soret band, which hints at a less aggregated structure as a result of better solubility promoted by the two carboxylic acids with water.
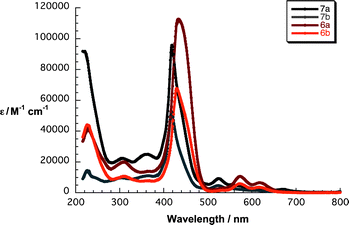 | ||
| Fig. 1 Absorption spectra of 6a/b and 7a/b in THF. | ||
The electrochemical features of 6a/b and 7a/b were measured in dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as electrolyte. A silver wire, which was calibrated vs. the Fc/Fc+ reference redox couple, was used as a quasi-reference electrode. As typically observed in porphyrins, two reductions – the first reversible, the second quasi-reversible – were noted for 7a and 7b at −1.6 and −1.8 V followed by a quasi-reversible oxidation at +0.4 V. In contrast, 6a and 6b reveal two oxidations – the first reversible, the second quasi-reversible – at +0.2 and +0.6 V. The quasi-reversible reduction was detected for 6b at −1.5 V, whereas 6a did not show any appreciable reduction within the electrochemical window of up to −2.0 V. In general, when comparing zinc porphyrins 6a and 6b with free-base porphyrins 7a and 7b, the former feature lower oxidation potentials, that is, +0.2 vs. +0.4 V, while the reduction potentials are −1.5 and −1.6 V.
The excited state characteristics of 6a/b and 7a/b were determined by fluorescence spectroscopy in THF. As Fig. 2 illustrates, upon exciting 7a and 7b at 410 nm broad fluorescence bands centered at 662/720 nm and 660/718 nm evolved, respectively. Notably, the fluorescence quantum yields are 0.035 for 7a and 0.061 for 7b. These facts are in line with the absorption measurements confirming the effect of β-pyrrolic substitution and support the notion of aggregation that even prevails at concentrations below 10−7 M. The excited state features of 6a and 6b include fluorescence maxima at 614/660 nm and 614/658 nm and fluorescence quantum yields of 0.015 and 0.023, respectively.44 Corroborating results came from time-correlated single photon-counting (TCSPC) experiments. In particular, exciting 7a and 7b at 403 nm results in fluorescence decays, which were best fitted by a two-exponential fitting function – Fig. S2 (ESI†) – revealing lifetimes of 3.9 and 9.1 ns with a relative distribution of 17 and 83% for 7a. For 7b, the lifetimes are 4.3 and 9.3 ns with a relative weight of 20 and 80%, respectively. The lifetimes of 6a and 6b, as is typical for zinc porphyrins, decreased to 1.8/3.1 ns and 1.8/4.7, respectively, with relative distributions for both of 83/17%. We rationalize the shorter lifetime by an excited state deactivation of the aggregates.45–47
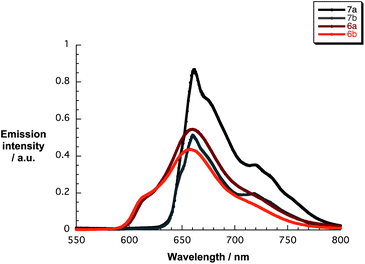 | ||
| Fig. 2 Fluorescence spectra of 6a/b and 7a/b (10−6 M) in THF upon excitation at 420 (6a/b) and 410 (7a/b) nm. | ||
Having established the key features of 6a/b and 7a/b, we turned to the preparation of the respective nanographene/porphyrin hybrids G6a/b and G7a/b following a previously reported procedure that was, however, slightly modified.38 The procedure consists of several consecutive steps, namely: (i) ultrasonication of solutions of the porphyrins with natural graphite, (ii) centrifugation of the suspension to eliminate the remaining graphite particles, (iii) ultrasonication of the supernatant with newly added natural graphite, and (iv) repetition of the steps – see Experimental section for more details. Such a treatment resulted in the direct exfoliation of graphite and the concomitant immobilization of the porphyrins onto the basal plane of graphene. For 7a and 7b, steps (i), (ii) and (iv) were sufficient to yield stable nanographene/porphyrin hybrid suspensions. However, in the case of 6a and 6b, despite the optimization of different parameters such as force, time and temperature during the ultrasonication, only the addition of step (iii) resulted in acceptable results. An overall enrichment of exfoliated graphite required the addition of new natural graphite to decrease the amount of free porphyrins. Interestingly, the stability of the nanographene/porphyrin dispersions varies as a function of relative THF–water ratios – see Fig. S3 (ESI†). In fact, the overall dispersion stability benefits from the presence of water as it enforces nanographene/porphyrin interactions.
Initial insights into the graphite exfoliation came from Raman spectroscopy performed with G6a/b and G7a/b. Laser excitation at, for example, 532 nm is in resonance with 6a/b and 7a/b and with nanographene. Owing to the quantitative quenching of the porphyrin fluorescence in G6a/b and G7a/b – vide infra – the Raman peaks of 6a/b and 7a/b and nanographene evolve – see Fig. S4 (ESI†). In particular, at 1096 (δ(Cβ–H)), 1122, 1194, 1220 (ν(Cα–N)), 1463 (ν(Cα–Cβ) + δ(Cβ–H)), 1481 and 1509 cm−1 (ν(Cα–Cm)) porphyrin-centered Raman peaks are observed.48 Interesting is the fact that G7a and G7b gave rise to much better results in terms of exfoliating graphite into few layer graphene than G6a and G6b. This conclusion came from Raman spectra that revealed 2D/G ratios between 0.45 and 1 as well as FWHM values that ranged from 46 to 94 cm−1. Responsible for the aforementioned trend is the fact that porphyrin metallation to afford 6a and 6b leads to an increase in steric demand and, in turn, to a weaker interaction with the graphene surface.49 Despite the successful graphite exfoliation and porphyrin immobilization, the 2D/G ratios suggest a limited extent of doping, which is in line with a recent report on porphyrin interactions with the basal plane of graphene.50
The Raman spectrum of G7b confirms the presence of turbostratic and electronically decoupled single layer graphene – Fig. 3. The confinement is affirmed by the symmetric 2D-band, with a 2D/G ratio of around 1 and a FWHM of 49 cm−1 when fit by a single Lorentzian. The strong D-band centered at 1299 cm−1, which is absent in the Raman features of natural graphite, is indicative of a diminished flake size and increased edge sites contributing to the double resonant Raman effect.
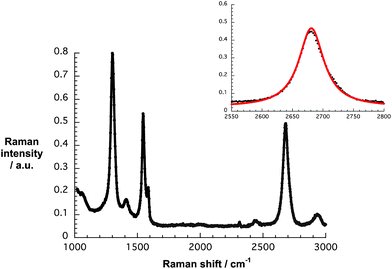 | ||
| Fig. 3 Raman spectrum of G7b drop-coated onto silicon oxide wafers upon laser excitation at 532 nm. The inset displays the experimental 2D peak (black) and its single Lorentzian fit (red). | ||
Additional experiments focused on screening different solvent mixtures with respect to graphite exfoliation starting with pure THF, to which increasing amounts of water were subsequently added – vide supra. The best results in terms of graphite exfoliation and nanographene functionalization were obtained in pure THF. Raman spectra gave 2D/G ratios in the range from 0.5 to 0.6 and a FWHM of 94 and 82 cm−1. Nevertheless, greater dispersion stabilities and greater graphene enrichments resulted in THF–water (1:1 v/v). The presence of even more water does not lead to any significant difference in terms of exfoliation efficiency, not taking dispersion stabilities into account. In fact, the Raman assays suggest that the porphyrin aggregates are difficult to dissemble.
AFM images shed light on the nanographene/porphyrin flake height and size distribution. The graphene flakes were about 2–6 nm in height – note that a calibrated single layer is with this apparatus in the range of 1 nm – owing to the presence of strongly folded and intertwined sheets. Fig. 4 illustrates that the lateral sizes are up to 500 nm. Self-aggregation, re-aggregation, and the rolling up of graphene sheets should be considered in the interpretation of the AFM height profiles.
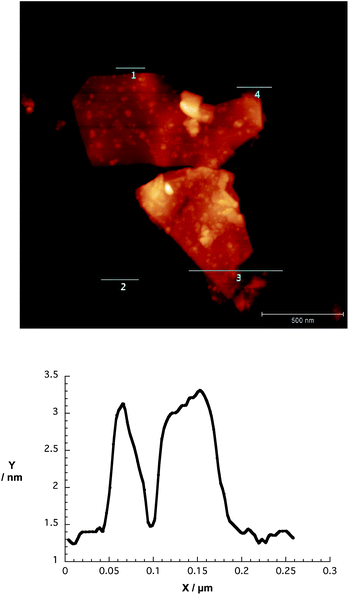 | ||
| Fig. 4 Upper part – AFM image of a spin-coated dispersion of G7b in THF onto silicon oxide wafer. Lower part – corresponding height profile across line 1. Height profiles across lines 2, 3 and 4 are shown in Fig. S5 (ESI†). | ||
Along the same lines, the TEM images reveal folded as well as regularly stacked sheets that reach sizes of up to 1.2 μm2 with around 10% of them exceeding 2.0 μm2 – Fig. 5 and S6.† It is noteworthy that the graphene flakes of sizes below 1 μm2 are more likely to be thinner than the larger graphene flakes – a finding that goes hand-in-hand with the Raman experiments. The use of a contrast aperture enabled us to detect even thinner flakes, which are difficult to see in TEM investigations.51
 | ||
| Fig. 5 TEM image (left) and zoom in (right) of a drop-casted dispersion of G7b in THF onto a lacey carbon grid, with scale bars of 1000 and 250 nm, respectively. | ||
With stable and concentrated dispersions of nanographene/porphyrin hybrids G6a/b and G7a/b in hand, their photophysics were probed. Overall, the absorption spectra of G6a/b and G7a/b lack the distinct and well-resolved features of 6a/b and 7a/b. In particular, the absorption spectra of G7a and G7b in THF reveal weak maxima at 255/421 and 255/417 nm, respectively. The latter feature corresponds to the Soret band of 7a and 7b – Fig. 6. The amount of dispersed graphene was calculated using the absorption coefficient a = 36.0 mL mg−1 cm−1 at 660 nm,52 resulting in concentrations of approximately 0.006 mg mL−1 for G7a and 0.004 mg mL−1 for G7b.18 In contrast, the absorption spectra of G6a and G6b in THF show somewhat stronger features including a 429 nm maximum for G6a and G6b. The Q-bands are only faintly visible in the form of shoulders at 570/617 and 572/622 nm, respectively. For G6a and G6b, the concentrations of dispersed graphene are around 0.001 and 0.002 mg mL−1, respectively. Noteworthy, the initial concentrations of 6a/b and 7a/b were in all of the experiments on the order of 2 × 10−5 M. Concentration losses during the work up procedure cannot be ruled out, especially during the removal of the larger graphite particles after centrifugation. Nevertheless, we correlate the loss in oscillator strength with the strong electronic interaction between 6a/b and 7a/b and the basal plane of graphene.53
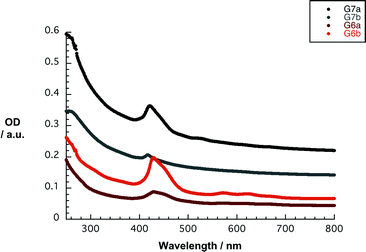 | ||
| Fig. 6 Absorption spectra of G6a/b and G7a/b in THF. The initial concentration of 6a/b and 7a/b was 2 × 10−5 M. The amount of dispersed graphene was calculated using the absorption coefficient a = 36.0 mL mg−1 cm−1 at 660 nm.52 | ||
Turning to THF–water (1:1 v/v), the absorption spectra are nearly featureless. A likely rationale implies higher concentrations of dispersed graphene, namely approximately at 0.013, 0.005, 0.005 and 0.012 mg mL−1 for G7a/b and G6a/b, respectively. Notably, such concentrations are up to a factor of 6 higher than in pure THF – see Fig. S7 (ESI†).
In line with the Raman and absorption studies, the porphyrin fluorescence is strongly quenched in G6a/b and G7a/b reaching up to 88% with respect to 6a/b and 7a/b – see Fig. S8.† The latter is indicative of strong electronic interactions in terms of either electron or energy transfer deactivation of 6a/b and 7a/b in their excited state – vide infra. Interestingly, the fluorescence in THF–water (1:1 v/v) is almost quantitatively quenched with values that reach 95%. The increased hydrophilicity of the mixture seems to favor hydrophobic interactions between the graphene surface and the porphyrins, thus increasing the electronic interaction.
Finally, to characterize and to quantify the electronic interactions between graphene and the porphyrins in terms of energy and/or electron transfer, we performed transient absorption experiments with G6a/b and G7a/b in THF and in THF–water (1:1 v/v). In G6a/b and G7a/b, the presence of nanographene evokes subtle changes when compared to 387 nm excitation experiments with 6a/b and 7a/b. For example, all of the transient absorption changes are governed early on by nanographene related features, that is, optical phonons that propagate within the two-dimensional carbon crystal. To this end, minima are discernable in the visible range between 450 and 750 nm and in the near-infrared range between 800 and 1500 nm for G6a/b and G7a/b. In the visible, for example, a transient minimum at 495 nm, which is formed instantaneously, red-shifts to 685 nm and intensifies throughout the first 1.5 ps before it drops in intensity. In parallel, a maximum at 475 nm develops. Similarly, in the near-infrared, a transient minimum at 1180 nm evolves, which also red-shifts as a function of time, in addition to a 860 nm maximum. Such spectral dissimilarities result from changing electron–phonon couplings as they evolve from different stacking of the porphyrins onto the basal plane of graphene. A rapid transformation (3.6 ± 0.2 ps) results in G7a and G7b in the formation of a new transient species in the 450–800 nm range with distinct maxima at 490 and 625 nm as well as minima at 660 nm – Fig. 7. In the 800–1500 nm range, the new transient involves broad bands that maximize at 890 and 1090 nm. Of key importance is the resemblance of these attributes with those of the one-electron oxidized radical cation of 7a and 7b, as they were determined in pulse radiolytic investigations.
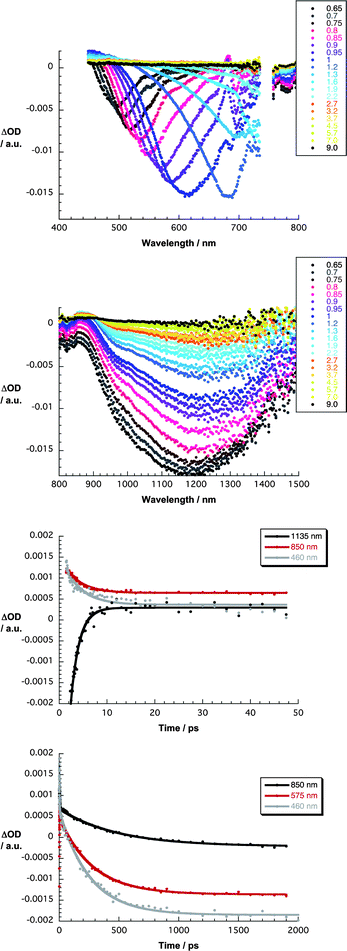 | ||
| Fig. 7 Upper part – differential absorption spectra (visible) obtained upon femtosecond pump–probe experiments (387 nm) of G7a in THF with several time delays between 0.6 and 9.0 ps at room temperature. Upper central part – differential absorption spectra (near-infrared) obtained upon femtosecond pump–probe experiments (387 nm) of G7a in THF with several time delays between 0.6 and 9.0 ps at room temperature. Lower central part – time absorption profiles at 460, 850 and 1135 nm monitoring the charge separation. Lower part – time absorption profiles at 460, 575 and 850 nm monitoring the charge recombination. | ||
For G6a and G6b, the corresponding transformation (2.4 ± 0.1 ps) leads to features that include maxima at 490, 535 and 690 nm as well as minima at 575 and 625 nm – Fig. S9 (ESI†). Similarly, the near-infrared range is important, which immediately after the photoexcitation shows phonon related bleaching in the form of minima around 1225 nm and porphyrin centered maxima at 860 nm for G7a and G7b and at 870 nm for G6a and G6b. Here, new features are seen during the transient decay/formation, namely maxima at 890 and 1025 nm for G7a and G7b as well as 905 and 1010 nm for G6a and G6b. These features imply a rapid charge transfer, that is, resulting in an oxidized porphyrin and new conduction band electrons in the nanographene. Multi-wavelength analyses of these newly developed charge transfer states reveal their metastable character with lifetimes in THF of 250 ± 20 ps for G7a, 260 ± 20 ps for G7b, 390 ± 50 ps for G6a, and 420 ± 50 ps for G6b. The shorter lifetime in the former two when compared to the latter two is ascribed to the weaker interactions of the more hydrophilic zinc porphyrins as well as the fact that the more planar free-base porphyrins 7a/b better immobilize onto the basal plane of the graphene. The observation that tighter nanographene/porphyrin interactions lead to shorter lifetimes is reinforced in THF–water (1:1 v/v), in which the lifetime values are 130 ± 20 ps for G7a and G7b and 190 ± 20 ps for G6a and G6b – Fig. S10 and S11 (ESI†).
To further exploit the properties of the porphyrins, the carboxylic groups of 6a/b and 7a/b were probed as an anchor for nanoparticles. In this context, three batches of G7a with increasing loads of TiO2 nanoparticles were prepared by dispersing them with TiO2 nanoparticles in water. The resulting composites were deposited and dried onto copper grids for TEM imaging – Fig. S12 (ESI†). The batch with the highest concentration of TiO2 nanoparticles yielded large TiO2 agglomerates that are adhered to the functionalized nanographene surfaces. A decrease of TiO2 nanoparticle concentrations, on the other hand, results in a more homogenous coverage of the graphene flakes. Most importantly, the presence of TiO2 nanoparticles is restricted exclusively to areas covered with G7a.
Intrigued by the features described for the new nanographene/porphyrin hybrid materials, we decided to investigate their impact on the performance of dye-sensitized solar cell (DSSC) devices. Details regarding device fabrication and characterization are summarized in the Experimental section.
We initially focused our attention on elucidating which type of TiO2 electrode – transparent (TiO2-T) or light-scattering (TiO2-S) – and which sort of suspension – THF–water (1:1 v/v) or THF solvents – were optimal for fabricating DSSCs with the nanographene/porphyrin hybrids G6a/b and G7a/b. To this end, we immersed the previously prepared electrodes coated with thin films of each type of TiO2 into the nanographene/porphyrin dispersions in each solvent for a period of 120 h to ensure sufficient coating of the TiO2 film by the graphene composite. By the naked eye, we observed that the adsorption of the nanographene hybrids and porphyrins from THF–water (1:1 v/v) was poor in comparison with the colorful electrodes obtained from THF suspensions – Fig. 8.
 | ||
| Fig. 8 Left – SEM images of TiO2-T (left) and TiO2-S (right) soaked with G7a THF for 120 h at low (A) and high (B) magnification, respectively. Right – photographs of TiO2-T (left) and TiO2-S (right) soaked with G7a in THF and THF–water (1:1 v/v) for 120 h. | ||
To validate the presence of G6a/b and G7a/b, we performed scanning electron microscopy (SEM) analyses of the different modified electrodes. Importantly, we deduce from the SEM images that the choice of TiO2 electrode is, indeed, a key factor when dealing with such hybrid materials. For instance, as is seen in Fig. 8, the G7a coverage is better in the case of the TiO2-S electrode than that of the TiO2-T electrode. This is presumably due to the larger particle size of the TiO2-S creating a more porous electrode into which the graphene composite can intercalate. Although we performed cross-sectional SEM with the TiO2 electrodes (Fig. S13, ESI†), we have been unable to determine the depth of penetration of the composite into the TiO2-S films. Nevertheless, it is possible to estimate the thickness of the deposited materials as a few hundred of nanometers.
Additionally, probing the modified TiO2 electrodes at high magnification reveals that, to a notable extent, re-aggregation of the graphene flakes takes place during the integration of G7a. Despite the presence of aggregates, their chemical and physical nature is clearly graphene-like rather than graphite-like due to the presence of the porphyrin. This indicates that the nature of the nanographene/porphyrin hybrids is preserved when attaching to the TiO2 interface. To corroborate this notion, we also recorded SEM images for nanographene/porphyrin hybrids G6a/b and G7b leading essentially to the same results – Fig. 9 and S14 (ESI†).
 | ||
| Fig. 9 SEM images of TiO2-S electrodes soaked with G7b (top), G6a (middle) and G6b (bottom) in THF for 120 h at high magnification. | ||
Loading of G6a/b and G7a/b for 120 h onto both TiO2-T and TiO2-S electrodes, produced active devices albeit with low photocurrents – Fig. 10. Not surprisingly, the larger the amount of nanographene composite on the photoanode, the higher the photocurrent with TiO2-S films featuring higher values than TiO2-T films. In order to optimize the adsorption process of the nanographene/porphyrin hybrids on the TiO2-S electrode, we focused our investigations on the impact of different soaking times, that is, 2, 4, 8 and 16 h, on the device performance using both THF and THF–water (1:1 v/v) suspensions. Overall, the best performances were observed for devices using photoanodes prepared from THF composite suspensions (Tables S1 and S2, ESI†). Therefore, we will focus on the trends obtained from these DSSCs. The photovoltaic characteristic trends are given in Fig. 11 and the best DSSC performance values given in Table 1.
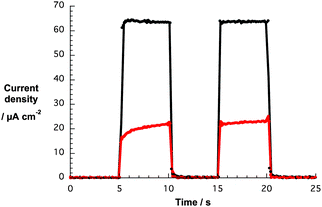 | ||
| Fig. 10 Photocurrent responses vs. time of DSSCs based on TiO2-T (red) and TiO2-S (black) electrodes soaked with G7a in THF for 120 h. | ||
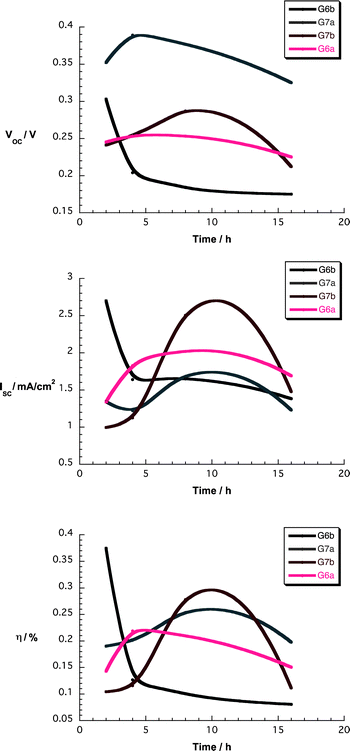 | ||
| Fig. 11 Upper part – Voc as a function of soaking time for devices using nanographene/porphyrin hybrids in THF. Central part – Isc as a function of soaking time for devices using nanographene/porphyrin hybrids in THF. Lower part – η as a function of soaking time for devices using nanographene/porphyrin hybrids in THF. | ||
Overall, the device parameter vs. soaking time dependence is quite similar for all hybrid materials. While open-circuit voltages (Voc) remain nearly constant, short-circuit currents (Isc) increase and reach maximum values at around 8 h for most of the nanographene/porphyrin composites. The impact of the latter is seen when inspecting the efficiencies (η) – Fig. 11. We note a sizeable enhancement of Isc and η in G6a/b and G7a/b devices when compared to those of 6a/b and 7a/b under comparable conditions – Tables S1 and S2 (ESI†). Such a phenomenon is likely to relate to the benefits from the presence of graphene flakes in terms of influencing the injection processes, the recombination processes, and the adsorption behavior on TiO2.
To get insights into the penetration of the nanographene/porphyrin hybrids into the TiO2 network, we reduced the TiO2 film thickness from 8 to 4 μm. Please note that cross sectional imaging was unsuccessful – vide supra. Important is the fact that the trends on the thinner device performance are virtually identical to those previously mentioned – Fig. S15 (ESI†). This leads us to conclude the depth of penetration of the nanographene/porphyrin hybrids is not a limiting factor.
To elucidate the injection mechanism that is operative in nanographene/porphyrin based DSSCs, incident photon-to-current efficiency spectra (IPCE) were recorded. Importantly, the IPCE spectra of Fig. 12 match the absorption spectra of Fig. 6 and, as such, confirm that the porphyrin generates the photocurrent. The overall broadening suggests that the electronic coupling between the porphyrin and graphene is still intact even on TiO2. For comparison, IPCE spectra were taken of devices with graphene and porphyrin only photoanodes. The former is featureless with a maximum of around 0.2% indicating no contribution of this moiety while the latter features a maximum at around 425 nm similar to the nanographene/porphyrin hybrid devices but, in line with the photogenerated currents, a much lower value was noted (Fig. 12). In addition, SEM images show that the adsorption of graphene flakes at these electrodes is rather poor (Fig. S16, ESI†) and, in turn, the photocurrent is very low (3 μm cm−2). In light of the aforementioned, it is safe to assume that graphene on its own plays no significant role in the overall photosensitization process.
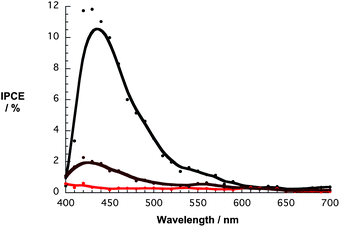 | ||
| Fig. 12 IPCE spectra for DSSCs with G6b (black spectrum), graphene (red spectrum), and 6b (brown spectrum). | ||
Our hypothesis implies that the photoexcited porphyrins inject electrons either directly into the conduction band of TiO2 or indirectly via graphene as a conducting mediator. Both processes are driven by a sizeable thermodynamic driving force as has already been shown.54–56
The competition between the direct and indirect route is seen when comparing G6a/b and G7a/b, in which the former give rise to weaker interactions with the basal plane of graphene – vide supra – and, in turn, favor the direct route. Likewise, comparing G6a/7a with G6b/7b, the presence of two carboxylic groups in the latter shifts the competition to the direct route. Nevertheless, graphene is also known to act as a Schottky barrier,57 which prevents the recombination process that is a typical drawback observed in porphyrin-based DSSCs.54,56
In other words, the presence of graphene seems to be crucial in the overall enhancement of device efficiency by means of affecting the injection process and decreasing the recombination process. Finally, the fact that the chemical and also the physical nature of the nanographene/porphyrin hybrids is clearly graphene-like rather than graphite-like, as revealed by the SEM images (Fig. 8 and 9), cannot be neglected. Thus, its contribution to minimize porphyrin aggregation and thereby to enhance porphyrin regeneration should not be underestimated. To corroborate the latter, we are currently directing our attention to perform femtosecond pump–probe experiments.
Conclusions
In summary, two major milestones in the preparation and use of graphene in solution are provided in this work. Firstly, we have demonstrated the versatility of free-base porphyrins 7a/b to realize nanographene hybrids, while their metalated analogous 6a/b give rise to a much poorer performance in terms of exfoliating graphite. The success has been corroborated by means of Raman, AFM, TEM, and time-resolved pump–probe experiments. Importantly, the latter provides solid evidence for shifting electron density from the porphyrins to nanographene upon excitation. In line with our previous assays, we conclude that the synergy of π–π and electronic interactions is crucial to produce highly stable graphene sheets in solution. Secondly, TEM images of nanographene hybrid, to which TiO2 nanoparticles were grafted, and SEM images of TiO2 electrodes have undoubtedly shown that, even when the porphyrin interacts with the basal plane of graphene, its hydrophilic chain is operative to implement this novel hybrid into solar cells. In this context, the device data prompt to the benefits when the nanographene hybrids are present. Quite likely, aggregation is prevented, which leads to benefits in the regeneration process of the porphyrins, on one hand, and implementation of a Schottky barrier reducing the recombination rate process, on the other hand. These aspects lead to a significant boost of the overall device performance. Specifically, in the optimum photosensitizer uptake for nanographene/porphyrin, the enhancement amounts to a 50% increase of the device efficiency compared to DSSCs constructed from only the respective porphyrins were noted. Currently, we are directing our efforts to further unveil the underlying mechanism of DSSCs that are based on nanographene hybrids. However, it should be noted that the use of nanographene/porphyrin hybrids opens the way for the introduction of other nanoparticles into solar cells.Acknowledgements
We gratefully acknowledge the DFG (GU 517/16-1), ICMM (Interdisciplinary Center for Molecular Materials), the GSMS (Graduate School of Molecular Science), the EAM cluster, and the ZMP (Zentralinstitut für neue Materialien und Prozesstechnik) for financial and intellectual support. Financial support from the Australian Research Council is also gratefully acknowledged. R.D.C. acknowledges the Humboldt Foundation for its support.Notes and references
- T. Braun, A. P. Schubert and R. N. Kostoff, Chem. Rev., 2000, 100, 23–38 CrossRef CAS.
- L. M. A. Bettencourt, D. I. Kaiser, J. Kaur, C. Castillo-Chávez and D. E. Wojick, Scientometrics, 2008, 75, 495–518 CrossRef CAS.
- Advances in Carbon Nanomaterials: Science and Applications, ed. N. Tagmatarchis, Pan Stanford Publishing Pte. Ltd., Singapore, 2012 Search PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS.
- Carbon Nanotubes and Their Applications, ed. Q. Zhang, Pan Stanford Publishing Pte. Ltd., Singapore, 2012 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- P. Sutter, Nat. Mater., 2009, 8, 171–172 CrossRef CAS.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS.
- H. Cao, Q. Yu, R. Colby, D. Pandey, C. S. Park, J. Lian, D. Zemlyanov, I. Childres, V. Drachev, E. A. Stach, M. Hussain, H. Li, S. S. Pei and Y. P. Chen, J. Appl. Phys., 2010, 107, 044310 CrossRef.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef CAS.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308–1308 CrossRef CAS.
- P. Hyesung, A. R. Jill, K. Ki Kang, B. Vladimir and K. Jing, Nanotechnology, 2010, 21, 505204 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- C. Gómez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS.
- C.-J. Shih, A. Vijayaraghavan, R. Krishnan, R. Sharma, J.-H. Han, M.-H. Ham, Z. Jin, S. Lin, G. L. C. Paulus, N. F. Reuel, Q. H. Wang, D. Blankschtein and M. S. Strano, Nat. Nanotechnol., 2011, 6, 439–445 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS.
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- B. Guo, L. Fang, B. Zhang and J. R. Gong, Insci. J., 2011, 80–89 CAS.
- G. L. C. Paulus, Q. H. Wang and M. S. Strano, Acc. Chem. Res., 2012, 160–170 Search PubMed.
- R. S. Swathi and K. L. Sebastian, J. Chem. Sci., 2012, 124, 233–240 CrossRef CAS.
- F. M. Koehler and W. J. Stark, Acc. Chem. Res., 2012 DOI:10.1021/ar300125w.
- S. Jae-Phil, K. DoHyung, C. Minhyeok, L. Takhee, P. Seong-Ju and L. Dong-Seon, Nanotechnology, 2012, 23, 255201 CrossRef.
- B. Lee, Y. Chen, F. Duerr, D. Mastrogiovanni, E. Garfunkel, E. Y. Andrei and V. Podzorov, Nano Lett., 2010, 10, 2427–2432 CrossRef CAS.
- M. Scardamaglia, S. Lisi, S. Lizzit, A. Baraldi, R. Larciprete, C. Mariani and M. G. Betti, J. Phys. Chem. C, 2013, 117, 3019–3027 CAS.
- Z. Yan, Z. Sun, W. Lu, J. Yao, Y. Zhu and J. M. Tour, ACS Nano, 2011, 5, 1535–1540 CrossRef CAS.
- J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53–64 CrossRef CAS.
- K. S. Subrahmanyam, R. Voggu, A. Govindaraj and C. N. R. Rao, Chem. Phys. Lett., 2009, 472, 96–98 CrossRef CAS.
- R. Ishikawa, M. Bando, Y. Morimoto and A. Sandhu, Nanoscale Res. Lett., 2011, 6, 111 CrossRef.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490–13497 CrossRef CAS.
- J. Geng and H.-T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CAS.
- T. Xue, S. Jiang, Y. Qu, Q. Su, R. Cheng, S. Dubin, C.-Y. Chiu, R. Kaner, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2012, 51, 3822–3825 CrossRef CAS.
- A. Hirsch, J. M. Englert and F. Hauke, Acc. Chem. Res., 2013, 46, 87–96 CrossRef CAS.
- M. O. Senge, I. Bischoff, N. Y. Nelson and K. M. Smith, J. Porphyrins Phthalocyanines, 1999, 3, 99–116 CrossRef CAS.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CAS.
- J. Malig, A. W. I. Stephenson, P. Wagner, G. G. Wallace, D. L. Officer and D. M. Guldi, Chem. Commun., 2012, 48, 8745–8747 RSC.
- Q. Wang, W. M. Campbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397–15409 CrossRef CAS.
- M. O. Senge, V. Gerstung, K. Ruhlandt-Senge, S. Runge and I. Lehmann, J. Chem. Soc., Dalton Trans., 1998, 4187–4200 RSC.
- T. Yamamura, S. Suzuki, T. Taguchi, A. Onoda, T. Kamachi and I. Okura, J. Am. Chem. Soc., 2009, 131, 11719–11726 CrossRef CAS.
- G. V. Ponomarev and G. B. Maravin, Khim. Geterotsikl. Soedin., 1982, 1, 59–64 Search PubMed.
- Y. V. Ishkov, Z. I. Khilina, Z. V. Grushevaya and A. M. Shul'ga, Zh. Org. Khim., 1993, 29, 2270–2274 CAS.
- 7a and 7b show higher fluorescence quantum yields in THF–water (1:1 v/v) when compared to measurements in only THF, whereas 6a and 6b reveal lower quantum yields. Increasing the water ratio to 90 or 99% results in strongly broadened absorption features and fluorescence quenching.
- N. C. Maiti, M. Ravikanth, S. Mazumdar and N. Periasamy, J. Phys. Chem., 1995, 99, 17192–17197 CrossRef CAS.
- Comparing the relative lifetime contributions in experiments in THF–water (1:1 v/v) with those in pure THF prompts again to aggregation phenomena that are at force upon adding water. Further increasing the water ratio to 90 or even 99% the low quantum yields made it, however, impossible to measure TCSPC.
- Note, that the fluorescence of G6a/b and G7a/b is strongly quenched, which is in agreement with transient absorption measurements that corroborate an ultrafast electron transfer.
- T. M. Cotton, S. G. Schultz and R. P. Van Duyne, J. Am. Chem. Soc., 1982, 104, 6528–6532 CrossRef CAS.
- H. Li, B. Zhou, Y. Lin, L. Gu, W. Wang, K. A. S. Fernando, S. Kumar, L. F. Allard and Y.-P. Sun, J. Am. Chem. Soc., 2004, 126, 1014–1015 CrossRef CAS.
- J. Geng, B.-S. Kong, S. B. Yang and H.-T. Jung, Chem. Commun., 2010, 46, 5091–5093 RSC.
- It should be noted that the flake size and the flake quality depends strongly on the age of the dispersion at which point it is drop casted onto the TEM grid. In general, the fresher the dispersion the larger the flake size with a significant impact on the overall quality – see Fig. S6 (ESI†).
- Y. Min, S. Zhigang, Z. Xiaojing and M. Shulin, J. Phys. D: Appl. Phys., 2013, 46, 025301 CrossRef.
- Due to a dynamic equilibrium between immobilized and free porphyrins the Soret band did not vanish completely.
- M. J. Griffith, K. Sunahara, P. Wagner, K. Wagner, G. G. Wallace, D. L. Officer, A. Furube, R. Katoh, S. Mori and A. J. Mozer, Chem. Commun., 2012, 48, 4145–4162 RSC.
- Please, note that we were not able to establish the redox properties of the hybrids by electrochemical techniques.
- A. J. Mozer, P. Wagner, D. L. Officer, G. G. Wallace, W. M. Campbell, M. Miyashita, K. Sunahara and S. Mori, Chem. Commun., 2008, 4741–4743 RSC.
- J. Song, Z. Yin, Z. Yang, P. Amaladass, S. Wu, J. Ye, Y. Zhao, W.-Q. Deng, H. Zhang and X.-W. Liu, Chem.–Eur. J., 2011, 17, 10832–10837 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Absorption spectra, fluorescence spectra, fluorescence time decays, Raman spectra, AFM images, AFM height profiles, TEM images, device characteristics, SEM images, tables with performance features. See DOI: 10.1039/c3sc51026c |
| ‡ These authors have contributed equally and should be considered as first authors. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. |
| This journal is © The Royal Society of Chemistry 2013 |