 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Practical, highly stereoselective allyl- and crotylsilylation of aldehydes catalyzed by readily available Cinchona alkaloid amide†
Yuan
Huang
,
Licheng
Yang
,
Panlin
Shao
and
Yu
Zhao
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Republic of Singapore. E-mail: zhaoyu@nus.edu.sg
First published on 5th June 2013
Abstract
We have demonstrated that bidentate Lewis base catalysts can be constructed based on the Cinchona alkaloid structure that promote highly stereoselective reactions of allyl- and crotyltrichlorosilane with aromatic as well as aliphatic aldehydes (90–99% ee, >98% diastereoselectivity). The catalysts are available in a one-pot procedure in >70% yield from cheap starting materials and promote the allylation reactions at ambient temperature. Gram scale reactions with catalyst recovery and reuse showcased the practicality of the catalytic system.
Introduction
Cinchona alkaloids and their derivatives have played a significant role as a privileged scaffold in asymmetric catalysis.1 The strongly basic quinuclidine nitrogen can effectively serve as a ligand for metal catalysis2 or as a Brønsted/Lewis base in organocatalytic reactions.3 By incorporating into the catalyst structure another H-bond donor4 or a metal-based Lewis acid,5 structures such as I, II and III (Scheme 1a) have proven powerful bifunctional catalysts for various organic transformations. Surprisingly, the construction of a simple bidentate ligand or catalyst, employing the quinuclidine nitrogen and another Lewis basic donor moiety (as shown in Scheme 1b), has remained elusive in asymmetric catalysis.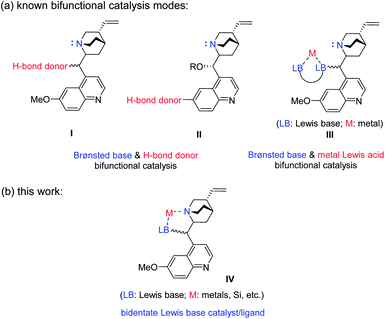 | ||
| Scheme 1 Catalyst design and application. (a) Known catalyst design based on Cinchona alkaloids. (b) Proposed new bidentate Lewis base catalyst. | ||
Towards the development of such a catalyst scaffold that will certainly benefit from the readily available, inexpensive Cinchona alkaloids, we chose the addition of allyl- and crotyltrichlorosilane to aldehydes as our model reaction, not only because it is a mechanically well-established reaction that can be catalyzed by a bidentate Lewis base,6 but more importantly, it represented the first catalytic approach to realize predictable, diastereospecific crotylation (Type I allylation,6a where the use of E- or Z-crotylsilane provides anti- or syn-products with >98% fidelity through the closed chair transition state), which is a key requirement in the formation of propionate units that are ubiquitous in polyketide natural products.7 Many catalytic systems have been developed for this reaction, which are dominantly chiral phosphoramides and N-oxides reported from the groups of Denmark and Fu,6d Nakajima et al.,6e,f Malkov and Kočovský et al.,6g,h Hayashi et al.,6i and Snapper and Hoveyda.6j While the great potential of these methods in chemical synthesis has been demonstrated,8 one common limitation is the lack of reactivity for aliphatic aldehydes, with the only exception being the highly stereoselective allyl- and crotylation of aliphatic aldehydes from the Iseki group that required an impractically long reaction time (2–4 weeks).6k In a related area of research, recent work from the Krische group has revolutionized the field of aldehyde allylation that bypasses the use of allylmetal reagents and can be conducted from either the aldehyde or alcohol oxidation level for both aromatic and aliphatic aldehydes;9 by the clever choice of substituted allyl acetates or butadiene, crotylation products with high diastereo- and enantioselectivities can also be accessed.10 However, considering the diastereospecific nature of Type I allylation, where pure anti- or syn-isomers can be accessed simply based on the choice of crotylating reagent, chiral Type I reagents (mainly allylborations such as Brown allylation,11 Roush allylation12 and the recent addition of allylsilylation from the Leighton group13) are still commonly used in asymmetric synthesis; a catalytic Type I allylation that can address the limitations of previous systems is therefore still desired.14 Here we present a Cinchona alkaloid amide as a highly efficient and stereoselective catalyst for the allylation and crotylation of a wide range of aldehydes, and in particular, aliphatic aldehydes (95–99% ee). This system also provides significant practical advantages that enable large scale production: the catalyst can be prepared in a one-pot procedure from inexpensive starting materials, can be easily recovered and reused, and promotes the allylation reactions at ambient temperature (instead of low temperatures of −40 to −78 °C for most previous systems).
Results and discussion
We initiated our studies by examining the catalytic activity of a variety of quinine-derived compounds for the addition of allyltrichlorosilane to 1a, the product of which is highly synthetically useful but was not previously available using Lewis base catalysis (Table 1). Quinine 3 and quinine ester 4 that were previously widely used as nucleophilic catalysts3 proved inefficient for our purpose, presumably due to limited Lewis base activation from the monodentate quinuclidine nitrogen (entries 1–2). The well-established bifunctional catalysts4 (Brønsted base coupled with H-bond donor) urea 5 and thiourea 6 were also poor catalysts (entries 3 and 4). Sulfonamide 7 (ref. 15) and phosphoric amide 8 may serve as bidentate Lewis bases, and interestingly we did obtain product enriched in the opposite enantiomer (40% ee with 7 or 8vs. −41% ee by using 5), however the level of efficiency and selectivity were far from satisfactory (entries 5 and 6). To our delight, a simple quinine amide such as 9, that has rarely proved successful in asymmetric catalysis,16,17 provided the desired product with high efficiency and excellent enantioselectivity (entry 7). Evaluation of the electronics of the aryl group (entries 7–9) clearly showed that the amide moiety serves as a Lewis base (instead of a H-bond donor in which case the catalyst would be more effective with an electron-withdrawing substituent installed such as 10), with catalyst 11 possessing a strongly electron-donating dimethylamino group being the optimal catalyst (88% conv., 96% ee). Cinchonidine-derived 12 (only lacking the methoxy substituent on quinoline) provided essentially the same result as 11, suggesting that the quinoline moiety in the catalyst structure is not directly involved in activation of the silylating reagent (entry 10). Compound 13, possessing the analogous chiral amide moiety but lacking quinuclidine, was much less efficient and selective, which further supported our hypothesis of bidentate Lewis base activation of allyltrichlorosilane (entry 11). Finally, as Cinchona alkaloids exist as pseudo-enantiomers, quinidine-derived 14 was also tested, and provided the enantiomeric product ent-2a with the same excellent enantioselectivity (entry 12).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conv.b (%) | eec (%) | Entry | Catalyst | Conv.b (%) | eec (%) |
| a Unless otherwise stated, reactions were run for 24 h. See ESI for details. b Conv. determined by 1H NMR of the crude reaction mixture. c ee determined by HPLC analysis. d Reactions were run for 48 h. | |||||||
| 1d | 3 | 15 | −8 | 7 | 9 | 83 | 95 |
| 2d | 4 | 10 | −5 | 8 | 10 | 35 | 81 |
| 3 | 5 | 30 | −41 | 9 | 11 | 88 | 96 |
| 4 | 6 | 40 | −20 | 10 | 12 | 83 | 96 |
| 5 | 7 | 36 | 40 | 11 | 13 | 35 | 9 |
| 6 | 8 | 45 | 40 | 12 | 14 | 88 | −96 |

|
|||||||

The evaluation of reaction parameters showed that DIPEA was necessary for the reaction to take place. THF was the optimal solvent in terms of reactivity as well as enantioselectivity. The optimal reaction conditions can be employed to produce a wide range of homoallylic alcohols in excellent enantioselectivity (Table 2). The reactions were carried out with 11 or 14 that yielded both antipodes of the products in comparable excellent enantioselectivity. The enantioselectivities for allylations of aliphatic aldehydes are uniformly high (95–99%). It is noteworthy that various functional groups such as ethers and silyl ethers (entries 1–3), esters (entry 4) as well as N-heterocycles (entry 5) are all well-tolerated, in addition to simple alkyl and alkenyl aldehydes (entries 6–10). This unprecedented scope bodes well for application in complex natural product synthesis. The allylation of β-chiral aldehyde 1o was also examined (Scheme 2). While allylation of 1o (96% ee) catalyzed by 11 provided 2o with 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 diastereomeric ratio, suggesting >98% selectivity for the installation of the new stereogenic center, the other diastereomer epi-2o could be obtained with a high diastereomeric ratio of 97.5:2.5 from the reaction catalyzed by 14. Aromatic aldehydes also work under the same conditions to yield products with enantioselectivities ranging between 90 and 94% ee (entries 11–14, Table 2).
:2 diastereomeric ratio, suggesting >98% selectivity for the installation of the new stereogenic center, the other diastereomer epi-2o could be obtained with a high diastereomeric ratio of 97.5:2.5 from the reaction catalyzed by 14. Aromatic aldehydes also work under the same conditions to yield products with enantioselectivities ranging between 90 and 94% ee (entries 11–14, Table 2).
|
|
|||
|---|---|---|---|
| Entry | Product 2 | With 11 | With 14 |
| Yield (%); ee (%) | Yield (%); ee (%) | ||
| a All reactions were carried out at ambient temperature for 24 h. The yields are isolated yields based on the average of two runs. See ESI for details. b 20 mol% catalyst was used. c Toluene was used as the solvent that provided higher conversion for aromatic aldehydes. | |||
| 1 |

|
83; 96 | 86; −97 |
| 2 |

|
89; 99 | 82; −98 |
| 3 |

|
70; 95 | 73; −96 |
| 4 |

|
90; 98 | 88; −98 |
| 5 |

|
75; 95 | 80; −96 |
| 6 |

|
80; 96 | 74; −96 |
| 7 |

|
76; 96 | 71; −95 |
| 8b |

|
76; 97 | 70; −98 |
| 9 |

|
78; 97 | 85; −96 |
| 10 |

|
83; 96 | 80; −96 |
| 11c |

|
77; 90 | 75; −91 |
| 12c |

|
83; 92 | 90; −92 |
| 13c |

|
77; 94 | 80; −93 |
| 14c |

|
86; 93 | 89; −92 |

 | ||
| Scheme 2 Allylation of chiral aldehyde. | ||
More importantly, we demonstrated that our catalytic system can be applied to the crotylation of aliphatic aldehydes with high enantioselectivity as well as reliable diastereospecificity, characteristic of Type I allylation. As shown in Table 3, with the use of either E- or Z-crotyltrichlorosilane 15 (each prepared in one step from commercially available starting materials),18 the alcohol products 16 of the two ether-containing substrates were obtained with excellent ee as well as high dr (>99% transfer of the geometry of crotylsilane to the product diastereomeric ratio). In contrast, the classical chiral Lewis acid-catalyzed addition of allylic organometallic reagents (Si, Sn, B) to aldehydes (Type II allylation; open transition state) provides a mixture of diastereomers, predominantly syn-isomer, independent of starting allylic geometry, while the addition of allylic organometallic reagents (Cr, Zn, In) generated in situ from the corresponding allylic halides catalyzed by chelating ligands (Type III allylation) yields predominantly the anti-isomer regardless of starting allylic geometry.6a
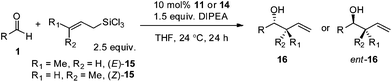




Practical, scalable allylation and crotylation
It is noteworthy that the current catalytic system is simple to apply at ambient temperature using a readily available catalyst, and commercial reagents (allyltrichlorosilane, DIPEA, etc.) as received from popular vendors without further purification. As stated earlier, the catalyst can be easily prepared from inexpensive starting materials via a one-pot procedure that includes the previously reported Mitsunobu reaction of quinine with diphenylphosphoryl azide followed by Staudinger reaction to yield 9-amino-9-deoxy-quinine,19 and finally acylation using commercially available 4-(dimethylamino)benzoyl chloride to yield the amide catalyst (Scheme 3a). The yield for the one-pot procedure, after a single purification by silica gel chromatography, was over 70%. To further showcase the utility of the system, gram-scale allylation of 1a was carried out that yielded 2a with comparable chemical yield and enantioselectivity to the small scale reactions (Scheme 3bvs.Table 2). The selectivity of this system is not sensitive towards concentration or heat transfer (as it is carried out at ambient temperature) so scaling up was straightforward. Although a relatively high catalyst loading of 10 mol% is required for the reaction, the catalyst could be easily recovered nearly quantitatively. When the recovered catalyst was used for another gram-scale crotylation of 1a, alcohol 16b was obtained in high diastereo- and enantioselectivity (Scheme 3c).
Mechanistic considerations
It has been showcased by the Denmark group that a Lewis base can liberate a chloride ion from SiCl4 or allyltrichlorosilane to form a silicate intermediate (17 in Scheme 4a).20 In the case of aliphatic aldehydes, however, this chloride adds to the aldehyde to form the corresponding α-chloro silyl ether 18 that is presumably responsible for the lack of allylation reactivity for such substrates. In our case, it was also observed that catalyst 11 binds to SiCl4 to liberate a chloride that quickly adds to aliphatic aldehydes to generate the α-chloro silyl ethers (>60% conv. in 10 min). On the other hand, the related product was not clearly observed when we mixed 11 with allyltrichlorosilane and aliphatic aldehyde (Scheme 4). This is in large contrast to the control experiment using HMPA, which promotes this undesired reaction with both SiCl4 and allyltrichlorosilane. This may be due to the relatively lower Lewis basicity of our catalyst compared to HMPA, which is a fortunate character for the success of aliphatic aldehyde allylation. | ||
| Scheme 4 Exploration of chloride ion liberation and catalyst modification. | ||
It is noteworthy that the secondary amide moiety in our catalyst may react with allyltrichlorosilane in the presence of DIPEA to generate the corresponding O-silyl imidate, in which process the chloride ion that is liberated from the silane will be sequestered as part of the DIPEA·HCl salt (and thus avoid the undesired α-chloro silyl ether formation). As stated earlier, DIPEA was found to be essential for the allylation reaction to proceed to high conversion. Preliminary NMR studies by mixing the catalyst, silane, and DIPEA in a 1:1:1 ratio were inconclusive as a complex mixture was formed; however, the resulting mixture was shown to be catalytically active. We have further tested the activity of catalyst 19 with a methylated amide moiety, which, under otherwise identical conditions, led to only <10% conv. to the allylation product with <5% ee (Scheme 4b). While the steric hindrance of this catalyst may certainly contribute to this low activity and selectivity, it provides support for the O-silyl imidate formation from catalyst 11 and 14. More extensive mechanistic studies as well as calculations will be carried out to further elucidate the nature of the active catalytic species as well as the turnover of the “anionic” catalyst.
The conformational analysis of related Cinchona alkaloid amides both in solution and in the solid state has been performed by the Brunner group using NMR, X-ray as well as molecular orbital calculations during their studies of enantioselective decarboxylation reactions using these catalysts as chiral Brønsted bases.16 These studies showed that these molecules prefer the open conformation,21 where the quinuclidine nitrogen points away from the quinoline unit, and in turn, towards the 9-amide moiety. In particular, the calculated minimum energy conformation of the protonated cinchonine amide 20 (Scheme 5a) possesses a H-bond interaction between the amide oxygen and the ammonium hydrogen.16b These data pointed to the possibility of 9-amide and quinuclidine serving as a bidentate catalyst. We have also carried out kinetic studies of our catalytic system, which suggested that the allylation reaction is first-order dependent on the catalyst (see ESI† for details), lending further evidence for the bidentate nature of our catalyst. Based on the above rationale, we propose the transition state model (with catalyst 11) in Scheme 5b.
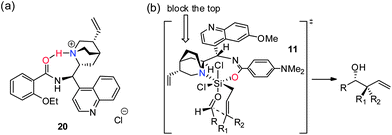 | ||
| Scheme 5 Proposed reaction transition state model. | ||
On the basis of the principles and mechanistic studies presented by the Denmark group22a and others,22b,c the aldehyde was placed trans to chloride to increase its electrophilicity; the allyl group, on the other hand, would coordinate trans to the strongly Lewis basic quinuclidine nitrogen, rendering it more nucleophilic. While the quinoline moiety points away to the back, the quinuclidine moiety effectively blocks the top of the complex so that the aldehyde is placed underneath. Allylation/crotylation through the chair like Zimmerman–Traxler transition state23 then provides the desired product in excellent enantioselectivity and predictable, perfect diastereoselectivity.
Conclusions
In conclusion, we have demonstrated, for the first time, the utility of bidentate Lewis base catalysts constructed from Cinchona alkaloids in the highly stereoselective allyl- and crotylsilylation of aldehydes. The catalytic procedure provides a practical, scalable preparation of various homoallylic alcohols that are useful building blocks in organic synthesis. Current efforts in these laboratories are focused on detailed mechanistic studies to further elucidate the origin of the asymmetric induction, further extending the synthetic utility of the system to allyl/crotylation of chiral aldehydes and application of this family of catalysts to other important organic transformations.Acknowledgements
We are grateful for the generous financial support from the Singapore National Research Foundation (NRF Fellowship) and the National University of Singapore.Notes and references
- For selected reviews, see: (a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS; (b) K. Kacprzak and J. Gawroñski, Synthesis, 2001, 961 CrossRef CAS; (c) H. Li, Y. Chen and L. Deng, in Privileged Chiral Ligands and Catalysts, ed. Q. Zhou, Wiley, 2011, p. 361 Search PubMed.
- For selected reviews, see: (a) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS; (b) R. R. Deshmukh, D. H. Ryu and C. E. Song, in Cinchona Alkaloids in Synthesis and Catalysis, ed. C. E. Song, Wiley-VCH, 2009, p. 73 Search PubMed.
- For selected reviews, see: (a) S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. Mcdaid and L. Deng, Acc. Chem. Res., 2004, 37, 621 CrossRef CAS; (b) C. Palomo, M. Oiarbide and R. Lopez, Chem. Soc. Rev., 2009, 38, 632 RSC; (c) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229 CrossRef CAS; (d) E. M. O. Yeboah, S. O. Yeboah and G. S. Singh, Tetrahedron, 2011, 67, 1725 CrossRef CAS. For pioneering work on Cinchona alkaloid-catalyzed highly enantioselective reaction via Lewis base catalysis, see: (e) S.-K. Tian and L. Deng, J. Am. Chem. Soc., 2001, 123, 6195 CrossRef CAS.
- S. J. Connon, Chem. Commun., 2008, 2499 RSC.
- L. Stegbauer, F. Sladojevich and D. J. Dixon, Chem. Sci., 2012, 3, 942 RSC.
- For selected reviews, see: (a) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS; (b) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS; (c) A. V. Malkov and P. Kočovský, Eur. J. Org. Chem., 2007, 29 CrossRef CAS. For representative examples, see: (d) J. Fu and S. E. Denmark, J. Am. Chem. Soc., 2001, 123, 9488 CrossRef; (e) M. Nakajima, M. Saito, M. Shiro and S. Hashimoto, J. Am. Chem. Soc., 1998, 120, 6419 CrossRef CAS; (f) M. Nakajima, S. Kotani, T. Ishizuka and S. Hashimoto, Tetrahedron Lett., 2005, 46, 157 CrossRef CAS; (g) A. V. Malkov, M. Orsini, D. Pernazza, K. W. Muir, V. Langer, P. Meghani and P. Kočovský, Org. Lett., 2002, 4, 1047 CrossRef CAS; (h) A. V. Malkov, L. Dufková, L. Farrugia and P. Kočovský, Angew. Chem., Int. Ed., 2003, 42, 3674 CrossRef CAS; (i) T. Shimada, A. Kina and T. Hayashi, J. Org. Chem., 2003, 68, 6329 CrossRef CAS; (j) J. F. Traverse, Y. Zhao, A. H. Hoveyda and M. L. Snapper, Org. Lett., 2005, 7, 3151 CrossRef CAS; (k) K. Iseki, S. Mizuno, Y. Kuroki and Y. Kolmyashi, Tetrahedron Lett., 1998, 39, 2767 CrossRef CAS. The Zhu group reported a catalytic asymmetric allylation of aliphatic aldehydes, but no crotylation reactions were disclosed, see: (l) B. Bai, L. Shen, J. Ren and H. J. Zhu, Adv. Synth. Catal., 2012, 354, 354 CrossRef CAS.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380 RSC.
- (a) S. E. Denmark and J. Fu, Org. Lett., 2002, 4, 1951 CrossRef CAS; (b) S. E. Denmark, C. S. Regens and T. Kobayashi, J. Am. Chem. Soc., 2007, 129, 2774 CrossRef CAS.
- For methodology development, see: (a) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6340 CrossRef CAS; (b) I. S. Kim, M. -Y. Nagi and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891 CrossRef CAS; (c) Y. Lu, I.-S. Kim, A. Hassan, D. J. Del Valle and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 5018 CrossRef CAS; (d) A.-M. R. Dechert-Schmitt, D. C. Schmitt and M. J. Krische, Angew. Chem., Int. Ed., 2013, 52, 3195 CrossRef CAS. For selected applications to complex molecule synthesis, see: (e) P. Harsh and G. A. O’Doherty, Tetrahedron, 2009, 65, 5051 CrossRef CAS; (f) S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559 CrossRef CAS; (g) Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876 CrossRef CAS; (h) Y. Feng, X. Jiang and J. K. De Brabander, J. Am. Chem. Soc., 2012, 134, 17083 CrossRef CAS.
- (a) I. S. Kim, S. B. Han and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2514 CrossRef CAS; (b) J. R. Zbieg, J. Moran and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 10582 CrossRef CAS; (c) X. Gao, H. Han and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 12795 CrossRef CAS; (d) X. Gao, Y. J. Zhang and M. J. Krische, Angew. Chem., Int. Ed., 2011, 50, 4173 CrossRef CAS; (e) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324 CrossRef CAS; (f) E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628 CrossRef CAS.
- (a) H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092 CrossRef CAS; (b) H. C. Brown and K. S. Bhat, J. Am. Chem. Soc., 1986, 108, 293 CrossRef CAS.
- (a) W. R. Roush, A. E. Walts and L. K. Hoong, J. Am. Chem. Soc., 1985, 107, 8186 CrossRef CAS; (b) W. R. Roush, K. Ando, D. B. Powers, A. D. Palkowitz and R. L. Halterman, J. Am. Chem. Soc., 1990, 112, 6339 CrossRef CAS.
- (a) J. W. A. Kinnaird, P. Y. Ng, K. Kubota and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 7920 CrossRef CAS; (b) K. Kubota and J. L. Leighton, Angew. Chem., Int. Ed., 2003, 42, 946 CrossRef CAS; (c) B. M. Hackman, P. J. Lombardi and J. L. Leighton, Org. Lett., 2004, 6, 4375 CrossRef CAS; (d) H. Kim, S. Ho and J. L. Leighton, J. Am. Chem. Soc., 2011, 133, 6517 CrossRef CAS; (e) L. M. Suen, M. L. Steigerwald and J. L. Leighton, Chem. Sci., 2013, 4, 2413 RSC.
- The Hall group reported intriguing Brønsted acid-catalyzed allylboration of aldehydes and developed an asymmetric variant catalyzed by a chiral diol that requires multi-step synthesis together with SnCl4: (a) V. Rauniyar, H. Zhai and D. G. Hall, J. Am. Chem. Soc., 2008, 130, 8481 CrossRef CAS. The Antilla group reported chiral phosphoric acid-catalyzed allylboration of aldehydes: (b) P. Jain and J. C. Antilla, J. Am. Chem. Soc., 2010, 132, 11884 CrossRef CAS.
- S. H. Oh, H. S. Rho, J. W. Lee, J. E. Lee, S. H. Youk, J. Chin and C. E. Song, Angew. Chem., Int. Ed., 2008, 47, 7872 CrossRef CAS.
- (a) H. Brunner and P. Schmidt, Eur. J. Org. Chem., 2000, 2119 CrossRef CAS; (b) H. Brunner, P. Schmidt and M. Prommesberger, Tetrahedron: Asymmetry, 2000, 11, 1501 CrossRef CAS; (c) A. E. Taggi, A. M. Hafez, H. Wack, B. Young, D. Ferraris and T. Lectka, J. Am. Chem. Soc., 2002, 124, 6626 CrossRef CAS.
- For multifunctional Cinchona alkaloid-based catalysts bearing amide as a linker structure, see: (a) J.-R. Chen, X.-L. An, X.-Y. Zhu, X.-F. Wang and W.-J. Xiao, J. Org. Chem., 2008, 73, 6006 CrossRef CAS; (b) X.-F. Xiong, Z.-J. Jia, W. Du, K. Jiang, T.-Y. Liu and Y.-C. Chen, Chem. Commun., 2009, 6994 RSC; (c) Q. Zhu and Y. Lu, Angew. Chem., Int. Ed., 2010, 49, 7753 CrossRef CAS; (d) F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710 CrossRef CAS; (e) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS; (f) M. Hayashi, N. Shiomi, Y. Funahashi and S. Nakamura, J. Am. Chem. Soc., 2012, 134, 19366 CrossRef CAS.
- M. Kira, T. Hino and H. Sakurai, Tetrahedron Lett., 1989, 30, 1099 CrossRef CAS.
- V. Benedek, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967 CrossRef.
- (a) S. E. Denmark, T. Wynn and G. L. Beutner, J. Am. Chem. Soc., 2002, 124, 13405 CrossRef CAS; (b) S. E. Denmark and J. Fu, Org. Lett., 2002, 4, 1951 CrossRef CAS.
- G. D. H. Dijkstra, R. M. Kellogg, H. Wynberg, J. S. Svendsen, I. Marko and K. B. Sharpless, J. Am. Chem. Soc., 1989, 111, 8069 CrossRef CAS.
- (a) S. E. Denmark and J. Fu, J. Am. Chem. Soc., 2003, 125, 2208 CrossRef CAS; (b) A. V. Malkov, P. Ramíez-López, L. Biedermannová, L. Rulíšek, L. Dufková, M. Kotara, F. Zhu and P. Kočovský, J. Am. Chem. Soc., 2008, 130, 5341 CrossRef CAS; (c) L. Ducháčková, A. Kadlčíková, M. Kotara and J. Roithová, J. Am. Chem. Soc., 2010, 132, 12660 CrossRef.
- H. E. Zimmerman and M. D. Traxler, J. Am. Chem. Soc., 1957, 79, 1920 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and NMR spectral charts. See DOI: 10.1039/c3sc50973g |
| This journal is © The Royal Society of Chemistry 2013 |