 Open Access Article
Open Access ArticleQuantification of solvent effects on molecular recognition in polyhedral coordination cage hosts†
Martina
Whitehead
,
Simon
Turega
,
Andrew
Stephenson
,
Christopher A.
Hunter
* and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: c.hunter@sheffield.ac.uk; m.d.ward@sheffield.ac.uk
First published on 10th April 2013
Abstract
A water-soluble cubic coordination cage (Hw) has been prepared, which is isostructural with a previously reported organic-soluble cage (H) apart from the hydroxy groups on the external surface which render it water-soluble. These two cages act as hosts for small organic molecules which bind via a combination of (i) hydrogen-bonding interactions with specific sites on the internal surface of the cages; (ii) non-polar interactions such as aromatic and van der Waals interactions between aromatic rings in the guest and the cage internal surface; and (iii) solvophobic interactions. By comparing ΔG° values for guest binding in water (using Hw) and MeCN (using H), and using pairs of related guests that differ in the presence or absence of an aromatic ring substituent, it is possible to construct thermodynamic cycles that allow quantification of the solvophobic contribution to binding. Specifically, this is the difference between the solvophobic contributions to ΔG° in water and MeCN associated with desolvation of both guest and the internal surface of the cage when complexation occurs. A highly consistent value of ca. −10 kJ mol−1 is determined for this solvophobic contribution to ΔG° associated with the aromatic ring in water compared to MeCN, which correlates very well with what would be expected based on the free energy changes associated with transfer of toluene from MeCN to water. Thus, all three contributions to guest binding listed above can be separately quantified. The ability to prepare related pairs of guests with the presence or absence of a wide range of substituents provides a potentially general way to quantify the solvophobic contributions to guest binding of these substituents.
Introduction
Desolvation plays an important role in solution phase recognition processes.1 In host–guest equilibria, the non-covalent interactions that the solutes make with solvent in the free state are often similar in magnitude to the interactions between host and guest in the bound state.2 In complexation processes involving non-polar solutes in polar solvents, desolvation can be the most important thermodynamic contribution to binding.3 However, the solvent is always an intrinsic part of the system, and so the thermodynamic contribution of desolvation is usually difficult to dissect experimentally, and computational techniques for accurate solvent modelling are still at an early stage of development. We have developed an electrostatic solvent competition model that makes it possible to estimate solvent effects on polar H-bonding interactions between individual functional groups.4 However, the effects of desolvation on the interactions of extended molecular surfaces are still poorly understood at a quantitative level.Given the importance of solvent effects on self-assembly and molecular recognition, a means of quantifying solvent effects associated with specific molecular substituents or functional groups is clearly desirable, and we present here a method based on a comparison of guest binding in the cavities of isostructural MeCN-soluble and water-soluble coordination cages which act as hosts. Self-assembled coordination cages are appealing molecular hosts as they have large, well-defined and generally hydrophobic cavities which can stabilise otherwise unstable molecular guests5 and can catalyse reactions whose transition states match the cavity shape and size.6 Here we exploit the ability of one our family of coordination cages7 to bind a range of small organic guests, on which the substituents can be systematically varied, as the basis of a quantitative analysis of solvophobic contributions to molecular recognition.
Results and discussion
The octanuclear coordination cage [Co8L12](BF4)16 (host H; Fig. 1, Scheme 1) is MeCN-soluble,8 and we reported recently binding of organic guests that contain H-bond accepting substituents in the central cavity of the cage.9 The strength of binding correlated well with the H-bond acceptor parameter10β of the guest indicating that the internal surface of the cage contained an H-bond donor site (Fig. 1b). We identified this as a convergent group of CH protons that are located close to a fac tris-chelate metal centre where the metal ion is relatively exposed, leading to a region of high positive electrostatic potential: this part of the cage internal surface acts as an H-bond donor site comparable in strength to a phenol.9b Comparison of the binding affinities of a series of monocyclic guests with the corresponding bicyclic guests – with an additional fused aromatic ring (shaded in Fig. 1b) – allowed us to identify two distinct components to guest recognition by the cage: (i) electrostatic interactions worth 0–10 kJ mol−1 depending on the β parameter of the guest; and (ii) a constant contribution of 10 kJ mol−1 associated with interactions of the fused aromatic ring with the internal surface of the cage.9b16 (H) (from ref. 8). (a) A space-filling view with each ligand coloured separately. (b) A view showing the location of the two H-bond donor sites in the cage, and illustrating the two regions the guest molecule isoquinoline-N-oxide that interact with the cage, via hydrogen-bonding interactions (dotted lines) and non-polar interactions of the cage surface with the secondary aromatic ring (shaded in pink).](/image/article/2013/SC/c3sc50546d/c3sc50546d-f1.gif) | ||
| Fig. 1 Two views of the parent cubic cage host [Co8L12](BF4)16 (H) (from ref. 8). (a) A space-filling view with each ligand coloured separately. (b) A view showing the location of the two H-bond donor sites in the cage, and illustrating the two regions the guest molecule isoquinoline-N-oxide that interact with the cage, via hydrogen-bonding interactions (dotted lines) and non-polar interactions of the cage surface with the secondary aromatic ring (shaded in pink). | ||
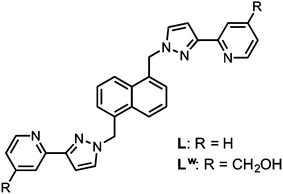 | ||
| Scheme 1 Structural formulae of the ligands used to prepare the host cages. | ||
We have now prepared a water-soluble, isostructural analogue of this coordination cage by attaching hydroxymethyl substituents to the pyridyl C4 sites of the ligand, to give the new ligand Lw (Scheme 1; where ‘w’ denotes ‘water solubilising’). This started from 4-hydroxymethyl-pyridine and required a multi-step sequence of transformations to introduce the 2-acetyl group which is then readily converted to a pyrazole and attached to the naphthalene-1,5-diyl spacer using our normal synthetic methodology;7 full experimental details are in ESI.†
Lw coordinates to Co(II) ions in the same way as does L, with reaction of Co(BF4)2 and Lw in the necessary 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio under solvothermal conditions (see ESI†) giving the cage complex [Co8(Lw)12](BF4)16 (hereafter Hw)‡ which is water-soluble due to the presence of 24 hydroxyl groups on the exterior surface. The internal cavity of the cage is lined with CH and CH2 groups from ligands. 1H NMR and ES mass spectrometric analysis confirmed both the identity of the cage and its integrity in solution. Apart from the changes associated with attachment of –CH2OH substituents at the C4 positions of the pyridyl rings, the paramagnetically-shifted 1H NMR spectrum of [Co8(Lw)12](BF4)16 in D2O (Fig. 2) is similar to that of [Co8L12](BF4)16 in MeCN,8 with the same pattern of paramagnetically shifted signals and peak widths consistent with two independent ligand environments, each having no internal symmetry, arising from the S6 molecular symmetry in solution (see below). If we assume that the methylene protons of the CH2OH groups, which are relatively remote from the Co(II) centres, are obscured by residual solvent peaks in the 0–10 ppm region (which include MeOH from the crystals as well as HOD) then we can clearly identify 40 other independent 1H resonances between +100 and −60 ppm, exactly as required.
:3 ratio under solvothermal conditions (see ESI†) giving the cage complex [Co8(Lw)12](BF4)16 (hereafter Hw)‡ which is water-soluble due to the presence of 24 hydroxyl groups on the exterior surface. The internal cavity of the cage is lined with CH and CH2 groups from ligands. 1H NMR and ES mass spectrometric analysis confirmed both the identity of the cage and its integrity in solution. Apart from the changes associated with attachment of –CH2OH substituents at the C4 positions of the pyridyl rings, the paramagnetically-shifted 1H NMR spectrum of [Co8(Lw)12](BF4)16 in D2O (Fig. 2) is similar to that of [Co8L12](BF4)16 in MeCN,8 with the same pattern of paramagnetically shifted signals and peak widths consistent with two independent ligand environments, each having no internal symmetry, arising from the S6 molecular symmetry in solution (see below). If we assume that the methylene protons of the CH2OH groups, which are relatively remote from the Co(II) centres, are obscured by residual solvent peaks in the 0–10 ppm region (which include MeOH from the crystals as well as HOD) then we can clearly identify 40 other independent 1H resonances between +100 and −60 ppm, exactly as required.
16 in D2O at 90 °C (at lower temperatures the spectrum becomes broader, probably due to slow molecular tumbling in solution).](/image/article/2013/SC/c3sc50546d/c3sc50546d-f2.gif) | ||
| Fig. 2 1H NMR spectrum of [Co8(Lw)12](BF4)16 in D2O at 90 °C (at lower temperatures the spectrum becomes broader, probably due to slow molecular tumbling in solution). | ||
Further, 1H NMR spectra of H and Hw recorded in a common solvent (CD3NO2) confirm the essential similarity of the two complexes (see ESI†). The ES mass spectrum (Fig. 3 and ESI†) shows a clear sequence of peaks for the intact cage associated with varying numbers of anions, i.e. the sequence {[Co8(Lw)12](BF4)16−n}n+ (n = 4–10).
16 in water, where ‘n’ relates to the number of anions lost and hence the charge in the sequence {[Co8(Lw)12](BF4)16−n}n+. Some high resolution expansions are in ESI.](/image/article/2013/SC/c3sc50546d/c3sc50546d-f3.gif) | ||
| Fig. 3 ES mass spectrum of [Co8(Lw)12](BF4)16 in water, where ‘n’ relates to the number of anions lost and hence the charge in the sequence {[Co8(Lw)12](BF4)16−n}n+. Some high resolution expansions are in ESI.† | ||
Single crystals were obtained from MeOH and an X-ray crystallographic analysis† (Fig. 4) confirmed that the water-solubilised cage Hw is structurally analogous to the parent unsubstituted cage H (see ESI† for specific comparison details) apart from the hydroxymethyl groups on the external surface which lead to extensive hydrogen bonding with water and MeOH molecules. Hw has the expected octanuclear core structure approximating to a cube with (non-crystallographic) S6 symmetry.8,9 As we have observed before, in the solid state the ‘window’ in the centre of each of the cube faces is blocked by a tetrafluoroborate anion (Fig. 4a). Importantly the two binding sites defined by the convergent group of methylene CH protons close to the fac tris-chelate vertices, which provide the internal H-bond recognition sites at either end of a long diagonal, are the same in Hw [Co(3)⋯Co(3′) separation across diagonal, 18.22 Å] as they were in H [Co(2)⋯Co(2′) separation across diagonal, 18.13 Å].8 In the crystal structure of Hw these two sites are each occupied by a water molecule whose O atom defines nicely the position that will be occupied by the H-bond acceptor atoms of the bound guest (Fig. 4c, cf.Fig. 1b), with O(1S)⋯Co(3) being 5.65 Å. The O(1S)⋯H distances to the six methylene protons are in the range 2.95–3.34 Å, and the corresponding O(1S)⋯C (methylene) separations are 3.53–3.67 Å.
16·8H2O·8MeOH (Hw). (a) A view of the complex cation with four of the twelve bridging ligands shown, emphasising the disposition of bridging ligands spanning each edge (C, grey; N, blue; O, red; Co, orange). Also included are those tetrafluoroborate anions that occupy the windows in the centre of each of the cube faces. (b) A space-filling view emphasizing the windows leading to the central cavity, and decoration of the external surface with hydroxyl groups. (c) A view showing the location of water molecules at the hydrogen-bonding sites generated by the two fac tris-chelate vertices [Co(3)].](/image/article/2013/SC/c3sc50546d/c3sc50546d-f4.gif) | ||
| Fig. 4 Three views of the structure of the complex cation of [Co8(Lw)12](BF4)16·8H2O·8MeOH (Hw). (a) A view of the complex cation with four of the twelve bridging ligands shown, emphasising the disposition of bridging ligands spanning each edge (C, grey; N, blue; O, red; Co, orange). Also included are those tetrafluoroborate anions that occupy the windows in the centre of each of the cube faces. (b) A space-filling view emphasizing the windows leading to the central cavity, and decoration of the external surface with hydroxyl groups. (c) A view showing the location of water molecules at the hydrogen-bonding sites generated by the two fac tris-chelate vertices [Co(3)]. | ||
With H and Hw available we could compare binding strengths of differently substituted guests in different solvents by 1H NMR spectroscopic titrations, a process greatly facilitated by the paramagnetism of the host cages which disperses their 1H resonances over a range of 200 ppm, eliminating the problems associated with extensive overlap of signals that occur in the corresponding diamagnetic cages.8,9 The range of guests studied is shown in Scheme 2, and the results of the 1H NMR titration experiments are provided in Table 1.
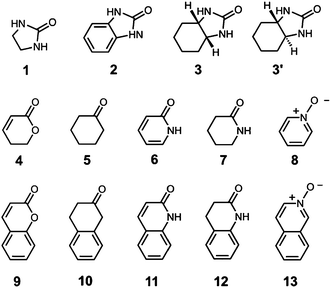 | ||
| Scheme 2 Guests used in this work. | ||
| Guest | β | Binding in H in MeCN | Binding in Hw in water | ||
|---|---|---|---|---|---|
| K/M−1 | ΔG°/kJ mol−1 | K/M−1 | ΔG°/kJ mol−1 | ||
|
a Data for guests 4–13 in MeCN taken from ref. 9b.
b For very weakly bonding guests (K ∼ 1 M−1) particular care was taken with the measurements. These were all fast-exchange systems for which K was calculated by fitting Δδ values on host signals to 1:1 binding isotherms. Precautions included (i) repeating each measurement several times with independent samples; (ii) using long accumulation times during spectra acquisition to give good signal:noise and baseline separation of the signals selected; (iii) ensuring that the titration was continued to give ≈50% of bound host; and (iv) fitting several different signals from the host in each titration experiment.
|
|||||
| 1 | 8.5 | 30 ± 7 | −9.4 ± 0.6 | 1 ± 1b | 0 ± 2 |
| 2 | 8.5 | 2700 ± 1000 | −19.6 ± 0.9 | 3100 ± 400 | −19.9 ± 0.3 |
| 3 | 8.5 | 28 ± 2 | −8.3 ± 0.2 | 73 ± 10 | −10.6 ± 0.3 |
| 3′ | 8.5 | 94 ± 10 | −11.3 ± 0.3 | 150 ± 40 | −12.4 ± 0.7 |
| 4 | 5.3 | 1 ± 1a,b | 0 ± 2 | Not soluble | |
| 5 | 5.3 | Too weak to measure | 2 ± 1b | −2 ± 1 | |
| 6 | 8.3 | 33 ± 1a | −8.7 ± 0.1 | 1 ± 1b | 0 ± 2 |
| 7 | 8.3 | 14 ± 1a | −6.5 ± 0.2 | 3 ± 1b | −2.5 ± 0.8 |
| 8 | 9.8 | 74 ± 7a | −10.7 ± 0.2 | 1 ± 1b | 0 ± 2 |
| 9 | 5.3 | 78 ± 20a | −10.8 ± 0.6 | 7600 ± 900 | −22.2 ± 0.3 |
| 10 | 5.3 | 28 ± 4a | −8.3 ± 0.4 | Not soluble | |
| 11 | 8.3 | 600 ± 90a | −15.9 ± 0.4 | 9100 ± 2000 | −22.6 ± 0.5 |
| 12 | 8.3 | 880 ± 300a | −16.8 ± 0.8 | 6700 ± 3000 | −21.8 ± 1.1 |
| 13 | 9.8 | 2100 ± 700a | −19.0 ± 0.8 | 3100 ± 400 | −19.9 ± 0.3 |
An illustrative example is provided by comparison of the binding affinities of two guests, 2-imidazolidone 1 and 2-hydroxybenzimidazole 2 (which exists as the keto tautomer illustrated in Scheme 2), with H in MeCN and with Hw in water. Both guests have a urea-type carbonyl group, which is a good hydrogen-bond acceptor, but guest 2 has an additional fused aromatic ring. By analogy with the previous experiments in MeCN, we would expect these guests to bind in the central cavity such that the oxygen H-bond acceptor is located close to one of the fac tris-chelate termini of the cage where the electrostatic potential is most positive, and the presence of the fused aromatic ring in 2 should result in an increase in the stability of the complex compared with 1.9b
The association constants measured by 1H NMR titrations in MeCN for complexes H·1 and H·2 are 30 M−1 and 2700 M−1 respectively, giving ΔG° values of −8.4 and −19.6 kJ mol−1 for guest binding. For the smaller guest 1, the exchange of free and bound host and guest signals is fast on the 1H NMR timescale, so the equilibrium constant was determined by fitting the changes in chemical shift of the host signals to a 1:1 binding isotherm. For the larger guest 2, the exchange of free and bound host and guest signals is slow on the 1H NMR timescale, so integration of distinct signals for free H and the complex H·2 was used to directly determine the equilibrium constant at each point in the titration (Fig. 5). Note that in some cases slow tumbling of the guest inside the cavity can reduce the symmetry of the cage such that a single signal for the free cage is split into several components on guest binding [e.g.Fig. 5(a), spectrum (iii)].9b
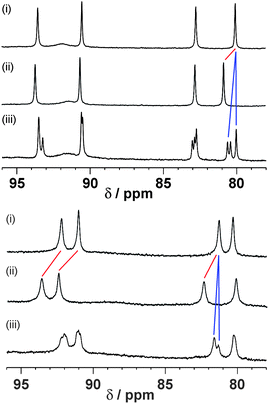 | ||
| Fig. 5 Representative examples of fast-exchange and slow-exchange guest binding on the 1H NMR timescale at 400 MHz. Top: Binding in host H (0.2 mM) in CD3CN: (i) free host; (ii) effect of addition of guest 1 (230 mM) showing the steady shift of host signals due to fast exchange; (iii) effect of addition of guest 3′ (0.8 mM) showing formation of separate signals for free and complexed host due to slow exchange. Bottom: Binding in host Hw (0.2 mM) in D2O: (i) free host; (ii) effect of addition of guest 1 (1100 mM) showing the steady shift of host signals due to fast exchange; (iii) effect of addition of guest 3′ (11 mM) showing formation of separate signals for free and complexed host due to slow exchange. | ||
For H·1, the major contribution to binding comes from the H-bonding interaction between the oxygen atom of the guest and the H-bond donor site of H.9b For 2, there is an additional contribution of −11 kJ mol−1 to ΔG° provided by interactions of the fused aromatic ring with the walls of the host cavity (some combination of van der Waals, CH⋯π and solvophobic interactions). In water, the association constants for formation of the corresponding complexes Hww·1 and Hww·2 are 1 M−1 and 3100 M−1 respectively, giving ΔG° values of 0 and −19.9 kJ mol−1. In water, solvation of polar functional groups competes with the H-bonding contribution to guest binding, and the stability of the Hww·1 complex in water is significantly lower than the stability of the corresponding H·1 complex in MeCN. In contrast, the stability of the Hww·2 complex in water is comparable to the stability of the H·2 complex in MeCN, with the fused aromatic ring of guest 2 providing an additional −20.5 kJ mol−1 to ΔG° in water compared with the value of −11.2 kJ mol−1 measured in MeCN. Given that interactions of this aromatic ring with the interior surface of the cage will be the same in either solvent, this difference of 9 ± 1 kJ mol−1 provides a measure of the additional solvophobic contribution associated with replacing MeCN as solvent by water, which compensates for the loss of polar interactions in the Hww·2 complex.
These data can be formalised as a thermodynamic cycle.11–13Fig. 6 illustrates the host–guest and solvation interactions that are important in determining the thermodynamic properties of these complexes. Δ1 is the difference in the free energies of complexation of the mono- and bicyclic guest in water. Δ2 is the difference in the free energies of complexation of the mono- and bicyclic guest in MeCN. Δ3 is the difference between the free energies of complexation of the monocyclic guest in water and in MeCN. Δ4 is the difference between the free energies of complexation of the bicyclic guest in water and in MeCN. ΔΔ (= Δ1 − Δ2 = Δ3 − Δ4) measures the influence of solvent on the interaction of the fused aromatic ring with the walls of the host cavity. The cartoon representation in Fig. 6 shows guest 2 completely filling the cage with no additional solvent molecules encapsulated in this complex; this has not been established experimentally, but the conclusions below are not significantly affected by this assumption.§ The interactions that contribute to the difference in the stabilities of the complexes formed with 1 and with 2 in the same solvent are solvation of the fused aromatic ring of 2, solvation of the cage cavity in the H·1/Hww·1 complexes, interactions of the fused aromatic ring of 2 with the cage walls, and solvent–solvent interactions. In water, this difference is −21 kJ mol−1 (Δ1), and in MeCN, the difference is −11 kJ mol−1 (Δ2). The thermodynamic contributions to Δ1 and Δ2 include identical contacts between the fused aromatic ring of 2 and the internal surface of the cage, and different contributions due to changes in solvation. Thus the change in the contribution to binding of the fused aromatic ring in water compared with MeCN is −9 ± 1 kJ mol−1 (ΔΔ = Δ1 − Δ2 = Δ3 − Δ4).
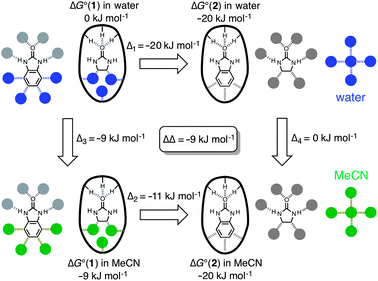 | ||
| Fig. 6 Thermodynamic cycle showing ΔG° values for binding of guests 1 (left) and 2 (right) into a host cage in both MeCN (bottom) and water (top), from which a value for the additional free energy of binding of the fused aromatic ring of 2 in water compared to MeCN can be derived (ΔΔ = Δ1 − Δ2 = Δ3 − Δ4). The bold oval with the three pendant H substituents represents the H-bond donor site of the cages (ref. 9b, Fig. 1b) and the filled circles represent solvent. Non-covalent interactions that cancel out in the thermodynamic cycle are shown in grey, and interactions that contribute to ΔΔ are colored (blue in water, green in MeCN). | ||
The interactions that cancel out in the thermodynamic cycle in Fig. 6 are highlighted in grey, and the interactions that are measured by ΔΔ are coloured. Fig. 7a shows a simplified version of the interactions measured by the thermodynamic cycle (constructed by cancelling out all of the grey interactions in Fig. 6), and demonstrates that the thermodynamic cycle in Fig. 6 provides a measure of the difference between the solvation energies of the fused aromatic ring and the partially filled cage in water and in MeCN. This implies that the behaviour of these systems might be understood based on solvent transfer free energies. Fig. 7b shows that the interactions that determine the free energy of transfer of toluene from water to MeCN are essentially the same as those in Fig. 7a (assuming that the fused aromatic ring of 2 and the internal surface of the cage are both toluene-like with respect to their solvation properties). The free energy change for the transfer of toluene from water to MeCN is −15.9 kJ mol−1.14 Hydrocarbon solvation energies are related to molecular surface area,15 which for toluene is 132.3 Å2.16 In comparison the surface areas of 1 and 2 are 109.2 Å2 and 148.4 Å2 respectively, so the additional surface area of guest that is transferred from solvent to the cage cavity when 2 binds is 39.2 Å2. If we assume that the same internal surface area of the host cage is also desolvated when 2 binds, the total additional surface area that is desolvated when 2 binds compared to binding of 1 is 78.4 Å2.¶ Assuming that the solvation properties of these surfaces are similar to toluene, we can use the solvent transfer free energy data for toluene to estimate an expected value for ΔΔ measured in the thermodynamic cycle (78.4/132.3) × (−15.9) = −9.4 kJ mol−1, in excellent agreement with the value of ΔΔ measured above.
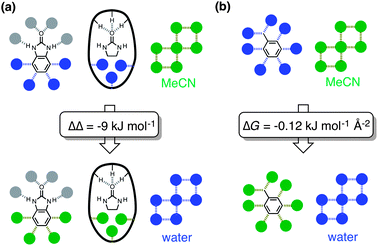 | ||
| Fig. 7 (a) Summary of the interactions measured by the thermodynamic cycle in Fig. 6. The bold oval with the three pendant H substituents represents the H-bond donor site of the cage, and filled circles represent solvent. Interactions that contribute to the value of ΔΔ are colored (blue in water, green in MeCN). (b) Interactions involved in the transfer of toluene from water into MeCN. The free energy of transfer is −15.9 kJ mol−1 (mole fraction standard state), and the surface area of toluene is 132.2 Å2, giving a free energy change per unit surface area of −0.12 kJ mol−1 Å−2. | ||
The same thermodynamic cycle can be applied to a range of different guests, comparing the binding affinities of bicyclic compounds with the corresponding monocyclic compounds that contain the same polar functional group (see Scheme 2). Table 2 summarises the results. The values of ΔΔ show that the presence of a fused bicyclic ring on the guest leads to a substantial increase in binding affinity in water compared to what is observed MeCN (−9 to −16 kJ mol−1). The difference in the surface areas of the mono- and bicyclic guests is similar in all cases, and the analysis of solvent transfer free energies above suggests that the values of ΔΔ in Table 2 reflect an approximately constant hydrophobic contribution to complexation of the fused ring system in all of these complexes. The value of ΔΔ does not depend on whether the ring is aliphatic (3 and 3′) or aromatic (2, 11, 12 and 13). However, the value of Δ1 for the aromatic bicycles is consistently larger in magnitude (−19 to −23 kJ mol−1) than the value for the aliphatic bicycles (−11 to −13 kJ mol−1). This suggests that the aromatic guests make significant additional aromatic interactions with the cage walls that are not possible for the aliphatic guests. The trends observed for Δ1 in water are also found for Δ2 in MeCN, showing that the difference in behaviour is due to host–guest interactions rather than desolvation: the value of Δ2 for the aromatic guests (−7 to −11 kJ mol−1) is consistently larger in magnitude than for the aliphatic guests (0 to −3 kJ mol−1). There are no obvious patterns in the values of Δ3 and Δ4.
| Bicyclic guest | Monocyclic guest | Δ 1 | Δ 2 | Δ 3 | Δ 4 | ΔΔ |
|---|---|---|---|---|---|---|
| a Calculation of ΔΔ values associated with guest pairs 4/9 and 5/10 is not possible as at least one guest was not soluble or did not bind detectably within each pair. | ||||||
| 2 | 1 | −20 | −11 | −9 | 0 | −9 |
| 3 | 1 | −11 | 0 | −9 | 2 | −11 |
| 3′ | 1 | −13 | −3 | −9 | 1 | −10 |
| 11 | 6 | −23 | −7 | −9 | 7 | −16 |
| 12 | 7 | −19 | −10 | −4 | 5 | −9 |
| 13 | 8 | −20 | −8 | −11 | 1 | −12 |
The ΔG° data in Table 1 are presented in a graphical form in Fig. 8. The two solid lines (one for the monocyclic guests and one for the bicyclic analogues) show the ΔG° values for complex formation with H in MeCN. The steep negative gradient of both lines reflects the substantial H-bonding contribution to guest binding in MeCN, which gets stronger as the H-bond accepting (β) value of the guest increases because the solvent is relatively non-competitive. This was discussed in detail earlier.9b The fact that these two lines are approximately parallel with a separation of ca. 10 kJ mol−1 between them reflects the fixed extra contribution to binding of the fused aromatic ring in MeCN (cf. the parameter Δ2 in Fig. 6) whichever H-bonding scaffold it is attached to.
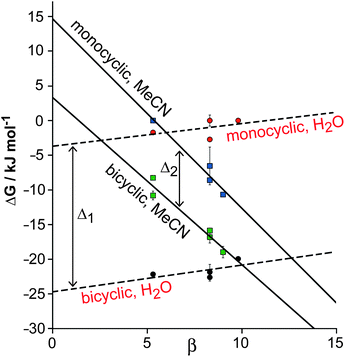 | ||
| Fig. 8 Graphical summary of the binding free energies of isostructural monocyclic guests 4–8 and analogous bicyclic guests 9–13 in cage cavities H (in MeCN) and Hw (in water), to illustrate the contribution to binding of the additional fused aromatic ring in the different solvents. The gap between the solid lines represents parameter Δ2 in Fig. 6; the gap between the dashed lines represents parameter Δ1 in Fig. 6. The difference between these is the ΔΔ value corresponding to the average additional solvophobic contribution to ΔG° arising from the fused aromatic ring of guests 9–13 in water compared to MeCN. | ||
In contrast, the ΔG° values for complex formation of the same two series of guests with Hw in water are shown by the dashed lines. The gradient is slightly positive in both cases, reflecting the fact that the solvent now provides a better H-bonding environment than the cage interior; as β for the guest increases, there is an preference for H-bonding to the solvent rather than the cage. Thus the H-bond component of guest recognition is effectively switched off in water. However the increased separation of ca. 20 kJ mol−1 between the lines for the monocyclic and bicyclic guests (cf. parameter Δ1 in Fig. 6) reflects the presence of additional aromatic interactions with the fused ring (worth −10 kJ mol−1 in MeCN) plus the hydrophobic contribution to binding in water (worth an additional ca. −10 kJ mol−1). This additional hydrophobic contribution to guest binding in water due to the presence of the fused aromatic ring, Δ1 − Δ2, is equivalent to the ΔΔ values measured in the thermodynamic cycle illustrated in Fig. 6 (−9 to −16 kJ mol−1).
Conclusions
In conclusion the availability of a matched pair of host cages – one water-soluble, one MeCN-soluble – together with pairs of guests that differ only in the presence/absence of a fused ring allows three different thermodynamic contributions to guest binding to be disentangled and quantified. Using a single host such as H in one solvent allowed both the hydrogen-bonding contribution and the effect of a fused aromatic ring to be identified.9b The availability of water-soluble Hw allows the hydrophobic contribution associated with binding of the fused ring also to be quantified. The free energy contributions can be measured using either thermodynamic cycles for matched pairs of host and guest, or by the graphical method shown in Fig. 8 for a series of structurally related guests bearing a common substituent. This is potentially a highly versatile way to examine many different types of substituent, as many of the guest types described here can be functionalised with a wide variety of simple substituents (halogen, alkyl, hydroxyl, amine etc.) which will allow the different contributions to binding associated with each of these to be simply determined in different solvents. In addition temperature-dependence of ΔG values will allow enthalpy and entropy contributions to be determined and this will also be the subject of future work.Acknowledgements
We thank EPSRC for financial support, and the EPSRC National Crystallography Service at the University of Southampton for data collection on the crystal of [Co8(Lw)12](BF4)16·8H2O·8MeOH.Notes and references
-
(a) S. B. Ferguson, E. M. Sanford, E. M. Seward and F. Diederich, J. Am. Chem. Soc., 1991, 113, 5410–5419 CrossRef CAS
; (b) H. J. Schneider, R. Kramer, S. Simova and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6442 CrossRef CAS
-
(a) P. W. Snyder, J. Mecinović, D. T. Moustakas, S. W. Thomas III, M. Harder, E. T. Mack, M. R. Lockett, A. Héroux, W. Sherman and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17889 CrossRef CAS
-
(a) N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521 CrossRef CAS
-
(a) J. L. Cook, C. A. Hunter, C. M. R. Low, A. Perez-Velasco and J. G. Vinter, Angew. Chem., Int. Ed., 2007, 46, 3706 CrossRef CAS
-
(a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS
-
(a) C. J. Hastings, D. Fiedler, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 10977 CrossRef CAS
- M. D. Ward, Chem. Commun., 2009, 4487 RSC
- I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS
-
(a) S. Turega, M. Whitehead, B. R. Hall, M. F. Haddow, C. A. Hunter and M. D. Ward, Chem. Commun., 2012, 48, 2752 RSC
-
(a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS
- We note that this type of thermodynamic cycle is analogous to the double mutant cycles used to quantify non-covalent interactions between two substituents within a molecule, or between two functional groups in a supramolecular assembly (ref. 12 and 13). The difference in this case is that one of the two mutations is a change in solvent rather than a change in substituent.
-
(a) S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172 RSC
- Representative examples:
(a) C. A. Hunter, M. C. Misuraca and S. M. Turega, J. Am. Chem. Soc., 2011, 133, 20416 CrossRef CAS
- V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2007, 3, 2011 CrossRef
- C. A. Hunter, Chem. Sci., 2013, 4, 834 RSC
- The Spartan software package (Spartan 08, Wavefunction Inc) was used to optimise molecular structures using DFT B3LYP with a 6–31G* basis set, and the molecular surface area was calculated using the 0.002 Bohr Å−3 isodensity surface.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC
-
G. M. Sheldrick, SADABS: A program for absorption correction with the Siemens SMART system, University of Göttingen, Germany, 1996 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef
-
(a) M. R. Ams, D. Ajami, S. L. Craig, J. S. Yang and J. Rebek, J. Am. Chem. Soc., 2009, 131, 13190 CrossRef CAS
-
(a)
http://spdbv.vital-it.ch
;
(b) N. Guex and M. C. Peitsch, Electrophoresis, 1997, 18, 2714 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for synthesis and characterisation; comparison of the crystal structures of H and Hw; comparison of the 1H NMR spectra of H and Hw in the same solvent; and high-resolution expansion of some of the ES mass spectral peaks. CCDC 926117. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc50546d |
| ‡ Crystallographic data for [Co8(Lw)12](BF4)16·8H2O·8MeOH: C368H360B16Co8 F64N72O40, M = 8291.68 g mol−1, monoclinic, space group C2/c, a = 27.658(10), b = 39.485(13), c = 42.581(15) Å, β = 106.741(5)°, U = 44531(27) Å3, Z = 4, ρcalc = 1.237 g cm−3, T = 100(2) K, λ(Mo-Kα) = 0.71073 Å, μ = 0.383 mm−1. 111888 reflections with 2θmax = 45° were merged to give 28864 independent reflections (Rint = 0.0748). Final R1 [for data with I > 2σ(I)] = 0.176; wR2 (all data) = 0.477. Data collection was performed by the EPSRC National Crystallography Service at the University of Southampton (ref. 17). Data were corrected for absorption using empirical methods (SADABS) (ref. 18) based upon symmetry-equivalent reflections combined with measurements at different azimuthal angles. The structure was solved and refined using the SHELX suite of programs (ref. 19). The asymmetric unit contains one half of the molecule which lies astride an inversion centre. A combination of disorder of anions/solvent molecules and solvent loss resulted in weak scattering, necessitating use of extensive geometric and displacement restraints to keep the refinement stable. In six of the twelve cases the external hydroxyl O atom was disordered over two sites; these atoms were refined with isotropic displacement parameters, as were all solvent (water and MeOH) molecules. The presence of large regions of diffuse electron density which could not be modelled required use of the SQUEEZE function in PLATON. Full details are in the CIF. |
| § In fact simple molecular volume calculations suggest that there could be some solvent molecules still present in the cavity when bicyclic guests such as 2 bind. As discussed earlier (ref. 9) the cavity volume is 407 Å3, meaning that (on the basis of Rebek's 55% rule, ref. 20) the ideal guest volume should be 224 Å3. Guests 1 and 2 have molecular volumes of 89 and 132 Å3 respectively; H2O and MeCN have molecular volumes of 21 and 53 Å3 respectively. Thus binding of 1 would leave room for 6–7 water or 2–3 MeCN molecules; binding of 2 would still leave room for 4–5 water or 1–2 MeCN molecules. However the effects of these remaining solvent molecules when 1 is replaced by 2 cancel out in the thermodynamic cycles and do not affect the analysis. |
| ¶ For comparison, the solvent-accessible internal surface area of the cubic cage – with the portals occupied by tetrafluorate anions, as per the crystal structures – is 315 Å2, measured using Swiss-PDB viewer (ref. 21). |
| This journal is © The Royal Society of Chemistry 2013 |