 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the packing of [Cu24(isophthalate)24] cuboctahedra in a triazole-containing metal–organic polyhedral framework†
Yong
Yan
a,
Mikhail
Suyetin
a,
Elena
Bichoutskaia
a,
Alexander J.
Blake
a,
David R.
Allan
b,
Sarah A.
Barnett
b and
Martin
Schröder
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: M.Schroder@nottingham.ac.uk; Fax: +44 (0)115 951 3563
bDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 11th February 2013
Abstract
The highly porous (3,24)-connected framework NOTT-122 incorporates a C3-symmetric angularly connected isophthalate linker containing 1,2,3-triazole rings and shows body-centered tetragonal packing of [Cu24(isophthalate)24] cuboctahedra. This unique packing, coupled with the high density of free N-donor sites, is responsible for the simultaneous high H2, CH4 and CO2 adsorption capacities in desolvated NOTT-122a.
Introduction
Metal–organic frameworks (MOFs) are an emerging class of crystalline materials constructed from metal centers (nodes) bridged by organic linkers (spacers) with a high level of structural modularity and diversity.1 The concept of supramolecular building blocks to engineer 3-dimensional crystalline architectures allows the design and prediction of novel structures.2 Porous MOFs show great promise for potential applications in H2 storage3 and CO2 capture4 owing to their chemical and thermal stability, permanent porosity, and high surface area.5 It is now well established that the high-pressure (>20 bar) H2 or CO2 uptake in MOFs is correlated with the surface area and/or the pore volume of the host, consistent with the physisorption process, while the low-pressure (under 1 bar) adsorption performance is strongly correlated with the strength of interaction between the framework and gas molecules.3 Current synthetic efforts to enhance gas–framework interactions include introduction of open metal sites,3d and adjustment of pore shape and size to enhance overlapping energy potential of the pore walls.3d,e,4b Specifically, decoration of the pores with basic nitrogen-containing groups (such as –NH2,6 triazole,7 and tetrazole8) are effective methods to improve CO2 adsorption uptakes in MOFs, as the heteroatoms can afford large dipole–quadrupole interactions with CO2 molecules.4bOur synthetic strategy focuses on the design and synthesis of metal–organic polyhedral cage materials based upon (3,24)-connected frameworks5b,9,10 possessing high structural stability and high surface area and pore volume.11–14 These ubt-type13,15 frameworks are constructed from {Cu2(RCOO)4} paddlewheels connected by C3-symmetric hexacarboxylate linkers, where three isophthalates are connected through a central core in a planar fashion. All these frameworks consist of the same [Cu24(isophthalate)24] cuboctahedron which is formed by 24 linkers and 12 {Cu2} units. The ubt-type net can be viewed as the face-centered cubic (fcc) packing of cuboctahedra in 3D space. These materials show high H2 uptakes at both low and high pressures attributed to the unique structural feature of the ubt-type network showing close packing of cuboctahedral cages containing 24 exposed Cu(II) sites and extra-high surface area. The {Cu24} cage has strong affinity to gas molecules, which has been confirmed experimentally by neutron powder diffraction studies.12,16 Previous studies revealed that the low-pressure H2 adsorption uptake of these ubt-type frameworks can be enhanced by reducing the distance between the two closest cuboctahedra,10c,12 which can be achieved by employing small-sized hexacarboxylate struts. However, there is a limitation to the size of linker that can be used to form (3,24)-connected fcc-packed cuboctahedra as this type of packing will be forbidden by steric hindrance between two closest cuboctahedra.17 By modifying the topology of the hexacarboxylate linker to give more angular connectivity through the C3-symmetric central core, we argued that tight packing of the cuboctahedra might be modulated to give more densely packed cuboctahedra compared to more common fcc-structures (Fig. 1).17,18
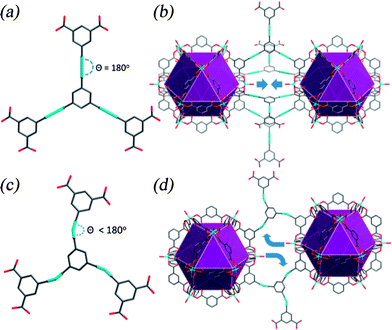 | ||
| Fig. 1 (a) Hexacarboxylate linker with co-planar linearly connected isophthalates. (b) Two close packed {Cu24} cuboctahedra within a ubt-type network aligned along the same axis. There has to be a minimum distance between these two polyhedra due to the steric hindrance from the two axial water molecules. (c) Hexacarboxylate linker with co-planar angularly connected isophthalate units of lower symmetry; (d) two near neighbour {Cu24} cuboctahedra packed in a distorted manner due to the angular hexacarboxylate linker. The framework constructed from the angular hexacarboxylate linker has the tendency to form a more close packed bct-ubt net compared to the more common fcc-ubt network. | ||
We report the introduction of five-membered heterocycles into the C3-symmetric hexacarboxylate linker to introduce an angular character to the external arms between the terminal isophthalates and the central core. Additionally, heterocycles can potentially provide enhanced binding sites for CO2 molecules.4b The 1,2,3-triazole rings have been incorporated previously into MOFs, including a flexible framework MIL-112, but no porosity was observed for this system.19 Most recently, the triazole group has been incorporated into porous organic polymers20 using “click” chemistry.21 Herein, we report the use of the 1,2,3-triazole-containing linker 5,5′,5′′-(4,4′,4′′-(benzene-1,3,5-triyl)tris(1H-1,2,3-triazole-4,1-diyl))triisophthalic acid (H6BTTI)22 to construct a highly porous (3,24)-connected polyhedral framework (denoted NOTT-122) incorporating {Cu2} paddlewheel units and having a high density of accessible nitrogen-donor sites,23 and achieving high adsorption capacities for both H2 and CO2.
Results and discussion
H6BTTI was obtained in good yield via Cu(I)-catalyzed 1,3-cycloaddition of 1,3,5-triethynylbenzene with 5-azidoisophthalic acid (Scheme 1; see ESI†). Solvothermal reaction of H6BTTI with Cu(NO3)2·2.5H2O in DMF (DMF = N,N-dimethylformamide) in the presence of HCl afforded truncated octahedron-shaped blue-green crystals of NOTT-122 in high yield. The complex [Cu3(BTTI)(H2O)3]·10DMF·10H2O crystallizes in tetragonal space group I4/m with large unit cell [a = b = 30.926(5) Å, c = 45.103(7) Å]. Single crystal X-ray diffraction confirms‡ that NOTT-122 is a (3,24)-connected network in which a cuboctahedron (Cage A) is constructed by 12 {Cu2(COO)4} paddlewheels and 24 isophthalates from 24 different (BTTI)6− units, and each linker acts as a 3-connected node connected to 3 cuboctahedra. The face of the (BTTI)6− unit has a distorted hexagonal geometry and the overall structure is based upon body-centered tetragonal (bct) packing of cuboctahedra in 3D space (Fig. S7†). This is of lower symmetry compared to the more usual fcc-packing in a ubt net. Two other types of polyhedral cages, a distorted truncated tetrahedron (Cage B) and a distorted truncated octahedron (Cage C) are observed in this structure, and the three types of cages tessellate in the same way as in the ubt-type network. One Cage B is connected to 4 cuboctahedra and one Cage C is connected to 6 cuboctahedra, by sharing the triangular and square windows of Cage A, respectively. The ratio of the different cages (Cage A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B:C) is 1:2:1. Taking into account the van der Waals surface of the cage interior, the largest spheres that can fit within these cavities have diameters of 12.2 Å and 19.3 Å for Cage B and Cage C, respectively. The length of the largest cage in the framework can reach ca. 3.2 nm and lies within the mesoporous regime. This structural analysis reveals that the bct-packing is another type of efficient tiling of cuboctahedra for creating hierarchically assembled cavities. It is worth noting that both Cage B and C have a remarkably high density of nitrogen-donor sites on the pore walls (Fig. 2), and the apertures which access the empty space in these cavities are densely decorated with uncoordinated triazole-rings. In contrast, the triazole groups in most other triazolate-bridged frameworks are coordinated to metal ions by donating one or two nitrogen atoms of the heterocycle,24,25 thus drastically reducing the number of accessible nitrogen-donor sites within the pores.
:B:C) is 1:2:1. Taking into account the van der Waals surface of the cage interior, the largest spheres that can fit within these cavities have diameters of 12.2 Å and 19.3 Å for Cage B and Cage C, respectively. The length of the largest cage in the framework can reach ca. 3.2 nm and lies within the mesoporous regime. This structural analysis reveals that the bct-packing is another type of efficient tiling of cuboctahedra for creating hierarchically assembled cavities. It is worth noting that both Cage B and C have a remarkably high density of nitrogen-donor sites on the pore walls (Fig. 2), and the apertures which access the empty space in these cavities are densely decorated with uncoordinated triazole-rings. In contrast, the triazole groups in most other triazolate-bridged frameworks are coordinated to metal ions by donating one or two nitrogen atoms of the heterocycle,24,25 thus drastically reducing the number of accessible nitrogen-donor sites within the pores.
 | ||
| Scheme 1 Synthesis of H6BTTI. | ||
 | ||
| Fig. 2 The crystal structure of NOTT-122 showing a (3,24)-connected network. The framework is decorated with a high density of N-donor sites. | ||
The total accessible volume of NOTT-122 after removal of the guest and coordinated water molecules is estimated to be 74% using the PLATON/VOID routine,26 and the calculated density of the desolvated framework is 0.589 g cm−3. Thermogravimetric analysis (TGA) confirmed that the framework is thermally stable up to 270 °C. The phase purity of the bulk sample of NOTT-122 was confirmed by powder X-ray diffraction (PXRD). The acetone-exchanged sample was degassed under dynamic vacuum at 110 °C for 12 hours to afford the activated sample of NOTT-122a. The PXRD profiles of the activated NOTT-122a indicate that the framework retains its crystallinity, reflecting a highly robust structure attributed to the (3,24)-connected net (Fig. S9†).
N2 sorption at 77 K in NOTT-122a (Fig. S12†) exhibits a reversible type-I sorption profile with small changes in slope in the pressure region P/P0 0.03–0.1 indicating the sequential filling and desorption of gas within micro- and meso-porous cavities. The apparent Brunauer–Emmett–Teller (BET) surface area is 3286 m2 g−1, and the calculated pore size, derived from the N2 isotherm and based on the nonlocal density functional theory (NLDFT) model, is distributed between 10–15 Å, and 15–30 Å (Fig. S14†), consistent with the single crystal structure analysis. NOTT-122a has a total pore volume of 1.41 cm3 g−1 calculated from the N2 isotherm data.
The gravimetric H2 sorption isotherms for NOTT-122a were recorded at 77 and 87 K up to 20 bar (Fig. 3a and S15†). Significantly, NOTT-122a can adsorb 2.61 wt% of H2 at 1 bar and 77 K [wt% = 100(weight of adsorbed H2)/(weight of host material)]. To date, there are only a handful of MOFs with H2 uptakes exceeding 2.6 wt% under the same conditions.3 The H2 capacity of NOTT-122a is higher than all the (3,24)-connected ubt-type frameworks NOTT-112 to NOTT-116,12–14 NOTT-119,11 PCN-61,10 and NU-1005b which show H2 uptakes in the range of 1.44–2.42 wt% at 1 bar and 77 K. The isosteric heat of adsorption (Qst) for H2 has an estimated value of 6.0 kJ mol−1 at zero coverage (see ESI†), consistent with H2 binding to the open Cu(II) sites. The remarkable low-pressure H2 adsorption performance is attributed to the combined effects of the close bct-packing of the cuboctahedra with exposed Cu(II) sites and the high concentration of nitrogen-donating sites in the structure. Theoretical calculations predict that N-containing aromatic rings can enhance the affinity to H2 molecules..27 Due to the large BET surface area and pore volume, NOTT-122a has a total H2 adsorption capacity of 7.0 wt% (70 mg g−1) at 77 K and 20 bar, among the highest reported to date for MOFs under the same conditions. Thus, NOTT-122a is a new member of the family of the (3,24)-connected Cu-based polyhedral structures showing high H2 sorption capacities at both low and high pressures.
 | ||
| Fig. 3 (a) H2 isotherms for NOTT-122a at 77 K in the pressure range 0 to 20 bar. The inset shows the experimental and simulated H2 isotherms up to 1 bar; (b) experimental and simulated CH4 isotherms for NOTT-122a at both 273 and 298 K; (c) experimental CO2, CH4 and N2 sorption isotherms for NOTT-122a at both 273 and 298 K up to 1 bar; (d) experimental and simulated CO2 isotherms for NOTT-122a in the pressure range 0–1 bar. Adsorption and desorption branches are shown with closed and open symbols, respectively. | ||
The open Cu(II) sites and high porosity also enable high methane storage in NOTT-122a (Fig. 3b). At 20 bar, the total CH4 uptakes of NOTT-122a reach 21.5 wt% [176 cm3(STP) cm−3, STP: standard temperature and pressure] and 16.1 wt% (132.1 cm3 cm−3) at 273 and 298 K, respectively. The volumetric CH4 uptake values for NOTT-122a are comparable to those of other porous MOFs with high CH4 storage such as UTSA-2028 (total 195 cm3 cm−3 at 300 K and 35 bar) and PCN-1129 (excess 171 cm3 cm−3, at 298 K and 35 bar).
The incorporation of accessible polarised 1,2,3-triazole rings in the framework is expected to enhance the affinity to CO2, and indeed at 1 bar, NOTT-122a exhibits remarkable CO2 adsorption capacities of 39.7 wt% (9.02 mmol g−1, 202.1 cm3 g−1, STP) at 273 K and 20.4 wt% (4.63 mmol g−1, 103.8 cm3 g−1, STP) at 298 K (Fig. 3), exceeding the CO2 capacities of most MOFs containing exposed metal sites.4 These values are comparable to the highest values for MOFs such as Mg/DOBDC (35.2 wt%, 296 K),30 Cu–TDPAT17 (44.5 wt%, 273 K; 25.8 wt%, 298 K), and Cu–TPBTM10d (42.6 wt%, 273 K; 23.3 wt%, 298 K). Possessing a similar pore size to another (3,24)-connected framework PCN-6110c,d with ubt-topology, NOTT-122a shows a higher low-pressure (1 bar) CO2 uptake capacity (PCN-61: 25.2 wt%, 273 K; 13.9 wt%, 298 K), which is ascribed to the high nitrogen-content and the different packing mode of the cuboctahedra. NOTT-122a also shows a high CO2 uptake of 25.6 mmol g−1 at 20 bar and 298 K, higher than most other highly porous MOFs such as Cu–TPBTM (23.5 mmol g−1), PCN-61 (21.4 mmol g−1) and PCN-68 (22.1 mmol g−1)10c,d under the same conditions. To evaluate the selective CO2 capture in NOTT-122a, CH4 and N2 sorption isotherms were also measured at ambient temperatures (Fig. 3c). By determining the ratios of the Henry's Law constants from single component isotherms,31 the CO2/N2 adsorption selectivity factors for NOTT-122a are 23.4:1 at 273 K and 14.3:1 at 298 K. The CO2/N2 selectivity in NOTT-122a is higher than both the ubt-type frameworks Cu–TPBTM (∼22:1) and PCN-61 (∼15:1) at 273 K, further suggesting that the polar triazole functionalities have a positive effect on CO2 adsorption. NOTT-122a also shows respectable CO2/CH4 selectivity of 9.2:1 at 273 K and 6.4:1 at 298 K. The analysis of isosteric heats of CO2 adsorption using the virial method reveals a moderate adsorption enthalpy of 24.5 kJ mol−1 at zero coverage, reflecting the expected van der Waals host–guest interactions in this large pore system and consistent with the shallow gradient observed for the CO2 isotherm. Also, no hysteresis is observed in the measured gas isotherms. This is to be expected given the high porosity and high pore volume in NOTT-122.
To analyse further the gas adsorption properties of NOTT-122a, grand canonical Monte Carlo (GCMC) simulations for H2, CH4 and CO2 adsorption were performed (see ESI†). The simulated isotherms for these three gases are in good agreement with the experimental data (Fig. 3). GCMC simulations for CO2 adsorption at different pressures (10, 50 and 100 kPa) at 273 K have been employed to define potential interactions between CO2 molecules and the framework. Simulations were performed based on a model where the CO2–framework interactions were described by electrostatic and van der Waals potentials. There are three observed preferential binding sites for CO2 in NOTT-122a identified at 10 kPa (Fig. 4a and b): the open Cu2+ site (I), denoted CuA, inside the cuboctahedral cage (Cage A) (another type of Cu2+ ion sited outside of a cuboctahedral cage is denoted CuB); the triangular-shaped window site (II) with the CO2 sites at the center of the cage opening formed by three isophthalate units and three Cu2(COO)4 paddlewheels;32,33 and site (III), located in the corner of the narrow channel in Cage B formed by three peripheral arms from three BTTI6− units, where the CO2 is close to the N1 atom of one triazole ring from the channel wall. These simulations further suggest that the open metal sites and the polar pore environment created by the triazole groups contribute synergically to the CO2 binding in this particular framework. When the pressure is increased to 50 kPa, a significant amount of CO2 was found in the cuboctahedral cage and the narrow channel of the truncated tetrahedral cage, and then population of the small cavity surrounded by four BTTI6− units connecting the B and C cages begins (Fig. 4c and d). At 100 kPa, after occupying the cuboctahedral cage, the narrow triangular pocket and the cavity connecting the Cage B and C, CO2 molecules continue to diffuse into the remaining empty space of the porous structure, revealing a sequential pore filling mechanism of CO2 in this hierarchically assembled structure.
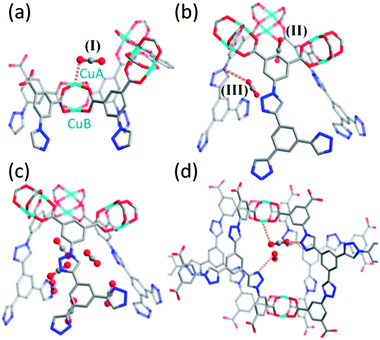 | ||
| Fig. 4 GCMC simulations of CO2 adsorption in NOTT-122a at 10 (a and b) and 50 kPa (c and d). Three likely CO2 adsorption sites in NOTT-122a derived from GCMC at 10 kPa are indicated: site I (CuA center) is shown in (a) and sites (II) and (III) are shown in (b). At 50 kPa, CO2 molecules were found in the narrow channel of Cage B shown in (c) and in the small cavity created by four BTTI6− units connecting Cages B and C (d). | ||
GCMC simulations identified the triazole groups of NOTT-122a as binding sites for CO2 molecules, and a number of configurations occurred regularly in the simulations (Fig. 5). At pressures of 50 kPa and 100 kPa, a chain of CO2 molecules can be formed via CO2–Cu(II), CO2–triazole, and CO2–CO2 interactions, with the triazole ring a particular “hot spot” for binding (Fig. 6).
 | ||
| Fig. 5 Possible orientations of CO2 molecules binding at the triazole ring: (a) the CO2 molecule is parallel to the triazole ring, (b) the CO2 molecule interacts with the N-center of the triazole ring and the nearest hydrogen, (c) the CO2 molecule is located above the triazole ring, (d) two CO2 molecules interact simultaneously with the triazole ring. | ||
 | ||
| Fig. 6 Snapshot of the GCMC simulations showing preferential binding sites for CO2 molecules in NOTT-122a: (a) at 50 kPa, (b) at 100 kPa. | ||
Conclusions
In conclusion, we present here the construction of a highly porous (3,24)-connected framework NOTT-122a by using a C3-symmetric angularly connected isophthalate strut bearing high content of free N-donor sites. The bct-packing of the cuboctahedra in NOTT-122a is another type of efficient 3D tessellation of polyhedra, having the advantage of allowing a large number of heterocycles to be sited within distorted hexagonal faces without the necessity of increasing the size of the linker. The bct-packing of {Cu24} cuboctahedra, coupled with the high density of polarizing triazole groups, is responsible for its high H2, CH4 and CO2 uptake, making NOTT-122a one of the rare examples of MOFs showing simultaneous high adsorption performance for these gases. This study also demonstrates that 1,2,3-triazole functionalities can be effectively integrated into porous robust MOFs using “click” chemistry, leading to rigid structures decorated with Lewis basic sites for enhanced interactions with CO2. The GCMC simulation study confirms the combination of effects from open metal sites and accessible triazole rings that affords high CO2 uptakes in NOTT-122a.Acknowledgements
We thank the EPSRC (U.K. Sustainable Hydrogen Energy Consortium, http://www.uk-shec.org.uk/) and the University of Nottingham for funding. We are grateful to the EPSRC-funded National Crystallography Service and Diamond Light Source for data collection on Diamond Beamline I19. M.S. gratefully acknowledges receipt of an ERC Advanced Grant, an EPSRC Programme Grant and a Royal Society Wolfson Merit award.Notes and references
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (b) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef CAS.
- (a) A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117–138 CrossRef CAS; (b) J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC; (c) A. J. Cairns, J. A. Perman, L. Wojtas, V. C. Kravtsov, M. H. Alkordi, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2008, 130, 1560–1561 CrossRef CAS; (d) C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2011, 4, 83–89 CrossRef.
- (a) X. Lin, N. R. Champness and M. Schröder, Top. Curr. Chem., 2010, 293, 35–76 CrossRef CAS; (b) X. Lin, J. Jia, P. Hubberstey, M. Schröder and N. R. Champness, CrystEngComm, 2007, 9, 438–448 RSC; (c) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; (d) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS; (e) D. Zhao, D. Yuan and H.-C. Zhou, Energy Environ. Sci., 2008, 1, 222–235 RSC; (f) M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef; (g) S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487–493 CrossRef CAS; (h) X. Lin, J. Jia, X. B. Zhao, K. M. Thomas, A. J. Blake, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS; (i) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. Brown, J. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS; (j) H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197–3204 RSC; (k) K. Koh, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 4184–4185 CrossRef CAS.
- (a) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS; (b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS; (c) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS; (d) P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. D. Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS; (e) S. Yang, X. Lin, W. Lewis, M. Suyetin, E. Bichoutskaia, J. Parker, C. C. Tang, D. R. Allan, P. J. Rizkallah, P. Hubberstey, N. R. Champness, K. M. Thomas, A. J. Blake and M. Schröder, Nat. Mater., 2012, 11, 710–716 CrossRef CAS; (f) S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS; (g) W. Yang, A. J. Davies, X. Lin, M. Suyetin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, J. E. Parker, C. C. Tang, M. W. George, P. Hubberstey, S. Kitagawa, H. Sakamoto, E. Bichoutskaia, N. R. Champness, S. Yang and M. Schröder, Chem. Sci., 2012, 3, 2993–2999 RSC.
- (a) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS; (b) O. K. Farha, A. Ö. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS.
- (a) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS; (b) S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS.
- (a) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS; (b) J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2009, 131, 5516–5521 CrossRef CAS.
- P. Pachfule and R. Banerjee, Cryst. Growth Des., 2011, 11, 5176–5181 CAS.
- (a) F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS; (b) Y. Zou, M. Park, S. Hong and M. S. Lah, Chem. Commun., 2008, 2340–2342 RSC.
- (a) S. Hong, M. Oh, M. Park, J. W. Yoon, J.-S. Chang and M. S. Lah, Chem. Commun., 2009, 5397–5399 RSC; (b) D. Zhao, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186–9188 CrossRef CAS; (c) D. Yuan, D. Zhao, D. Sun and H.-C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS; (d) B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS.
- Y. Yan, X. Lin, S. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2009, 1025–1027 RSC.
- Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 4092–4094 CrossRef CAS.
- Y. Yan, A. J. Blake, W. Lewis, S. A. Barnett, A. Dailly, N. R. Champness and M. Schröder, Chem.–Eur. J., 2011, 17, 11162–11170 CrossRef CAS.
- Y. Yan, S. Yang, A. J. Blake, W. Lewis, E. Poirier, S. A. Barnett, N. R. Champness and M. Schröder, Chem. Commun., 2011, 47, 9995–9997 RSC.
- O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 515–525 CrossRef.
- V. K. Peterson, Y. Liu, C. M. Brown and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 15578–15579 CrossRef CAS.
- B. Li, Z. Zhang, Y. Li, K. Yao, Y. Zhu, Z. Deng, F. Yang, X. Zhou, G. Li, H. Wu, N. Nijem, Y. J. Chabal, Z. Lai, Y. Han, Z. Shi, S. Feng and J. Li, Angew. Chem., Int. Ed., 2012, 51, 1412–1415 CrossRef CAS.
- R. Luebke, J. F. Eubank, A. J. Cairns, Y. Belmabkhout, L. Wojtasb and M. Eddaoudi, Chem. Commun., 2012, 48, 1455–1457 RSC.
- (a) T. Devic, O. David, M. Valls, J. Marrot, F. Couty and G. Férey, J. Am. Chem. Soc., 2007, 129, 12614–12615 CrossRef CAS; (b) T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef CAS.
- (a) T. Muller and S. Bräse, Angew. Chem., Int. Ed., 2011, 50, 11844–11845 CrossRef CAS; (b) J. R. Holst, E. Stöckel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531–8538 CrossRef CAS; (c) P. Pandey, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Mater. Chem., 2011, 21, 1700–1703 RSC; (d) O. Plietzsch, C. I. Schilling, T. Grab, S. L. Grage, A. S. Ulrich, A. Comotti, P. Sozzani, T. Muller and S. Bräse, New J. Chem., 2011, 35, 1577–1581 RSC.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CAS; (b) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS.
- A. Cadeddu, A. Ciesielski, T. E. Malah, S. Hecht and P. Samorì, Chem. Commun., 2011, 47, 10578–10580 RSC.
- Q. Lin, T. Wu, S.-T. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2012, 134, 784–787 CrossRef CAS.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022–2028 RSC.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS.
- A. L. Spek, PLATON, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef.
- A. Samanta, T. Furuta and J. Li, J. Chem. Phys., 2006, 125, 084714 CrossRef.
- Z. Guo, H. Wu, G. Srinivas, Y. Zhou, S. Xiang, Z. Chen, Y. Yang, W. Zhou, M. O'Keeffe and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 3178–3181 CrossRef CAS.
- X.-S. Wang, S. Ma, K. Rauch, J. M. Simmons, D. Yuan, X. Wang, T. Yildirim, W. C. Cole, J. J. López, A. de Meijere and H.-C. Zhou, Chem. Mater., 2008, 20, 3145–3152 CrossRef CAS.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS.
- M. S. Sun, D. B. Shah, H. H. Xu and O. Talu, J. Phys. Chem. B, 1998, 102, 1466–1473 CrossRef CAS.
- L. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925–17933 CAS.
- H. Wu, J. M. Simmons, G. Srinivas, W. Zhou and T. Yildirim, J. Phys. Chem. Lett., 2010, 1, 1946–1951 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC number for structure of Experimental details, crystallographic parameters, adsorption isotherms, and GCMC calculations. CCDC 906058. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc21769h |
| ‡ Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC 906058. |
| This journal is © The Royal Society of Chemistry 2013 |