 Open Access Article
Open Access Article
DOI:
10.1039/C2SC22137C
(Edge Article)
Chem. Sci., 2013,
4, 1514-1522
Mechanistic insights into the triazolylidene-catalysed Stetter and benzoin reactions: role of the N-aryl substituent†
Received
3rd December 2012
, Accepted 4th January 2013
First published on 4th January 2013
Abstract
The in situ observation, isolation and reversible formation of intermediate 3-(hydroxybenzyl)azolium salts derived from NHC addition to a range of substituted benzaldehydes is probed. Equilibrium constants for the formation of these 3-(hydroxybenzyl)azolium salts, as well as rate constants of hydrogen–deuterium exchange (kex) at C(α) of these intermediates for a range of N-aryl triazolinylidenes is reported. These combined studies give insight into the preference of N-pentafluorophenyl NHCs to participate in benzoin and Stetter reaction processes.
Introduction
N-Heterocyclic carbenes (NHCs) have been widely employed as organocatalysts,1 with the triazolylidene molecular class showing remarkable activity in a diverse range of catalytic processes that proceed through acyl anion,2 azolium enolate,3 azolium homoenolate,4 acyl azolium5 or α,β-unsaturated acyl azolium intermediates.6 Within the triazolylidene family, the N-aryl substituent plays a decisive role in determining catalytic reactivity and selectivity,7 with 2,6-substituted N-aryl units showing unique reactivity profiles.8 For example, N-mesityl (N-Mes) triazolylidenes are preferred for transformations utilising α-functionalised aldehydes,9 while N-pentafluorophenyl (N-C6F5) derivatives usually exhibit increased catalytic activity in Stetter and benzoin processes.10 Insightful studies from Bode have ascribed the N-Mes effect to irreversible addition of the N-Mes substituted NHC to the α-functionalised aldehyde, accelerating the formation of the Breslow intermediate (Fig. 1).11 To date, a mechanistic rationale for the enhanced performance of electron-deficient N-aryl triazolylidenes (N-C6F5 or N-2,4,6Cl3C6H2) in Stetter and benzoin processes has yet to be offered.7
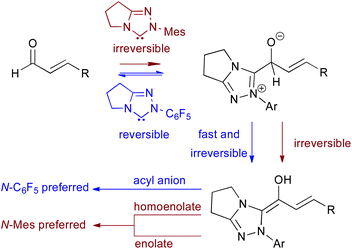 |
| | Fig. 1 Bode's work: the N-Mes effect in NHC-mediated processes. | |
Central to the observed catalytic activity in the benzoin and Stetter reactions is the formation of a common enaminol or Breslow intermediate 4. Nucleophilic addition of NHC 1 to aldehyde 2 gives tetrahedral intermediate 3, with deprotonation at C(α) leading to 4. Onward reaction with an electrophilic Michael acceptor 5, followed by proton transfer and catalyst regeneration, leads to the product 6 (Scheme 1).12 While intermediates of the imidazolinylidene13 and thiazolinylidene14 promoted benzoin reaction similar to 3 have been observed, only limited related studies of triazolinylidene-catalysed reactions have been made.15 Enders and Teles have isolated the 3-(hydroxymethyl)azolium salt addition product of formaldehyde and an NHC,16 but the concentrations of this intermediate during the reaction have not been monitored. Notably, related structural studies of intermediates in the Stetter reaction have not been established, although mechanistic studies from Rovis et al. indicate that proton transfer from the tetrahedral intermediate 3 is a kinetically significant and irreversible step.17
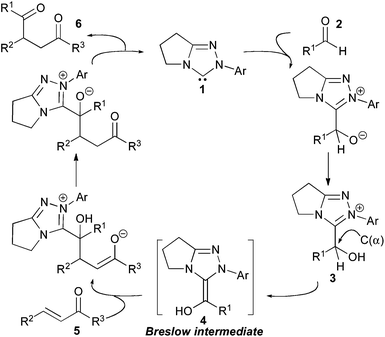 |
| | Scheme 1 Mechanism of the Stetter reaction. | |
As part of our studies regarding NHC-catalysed reaction processes,18 this manuscript describes the in situ observation, isolation and reversible formation of intermediate 3-(hydroxybenzyl)azolium salts derived from NHC addition to aldehydes. Equilibrium constants for the formation of these 3-(hydroxybenzyl)azolium salts, as well as rate constants of hydrogen–deuterium exchange (kex) at C(α) of these intermediates for a range of N-aryl triazolinylidenes is reported. These combined studies give insight into the preference of N-C6F5 NHCs to participate in benzoin and Stetter reaction processes.
In situ NMR studies: 3-(hydroxybenzyl)azolium salt observation and equilibrium values
To demonstrate the varying catalytic activity of a series of N-aryl triazolylidenes, initial in situ1H NMR spectroscopic studies of the Stetter reaction simply monitored the rate of formation of product 15 from 7 with variation of the N-aryl group of the triazolium salt precatalyst (Fig. 2).19 Under catalytic conditions using NEt3 as a base, electron-deficient N-aryl triazolium precatalysts give markedly superior rates of product formation (Ar = C6F5 > 2,4,6-Cl3C6H2 > 4-FC6H4 > Ph > 4-OMeC6H4 > 2,6-OMeC6H3 > Mes).20
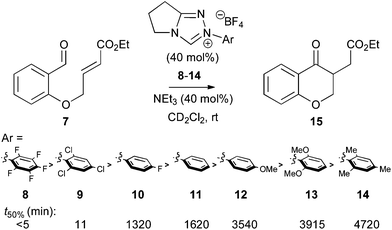 |
| | Fig. 2 Time taken to reach 50% conversion (t50%) of 7 to 15 with variation of N-aryl triazolium substituent. | |
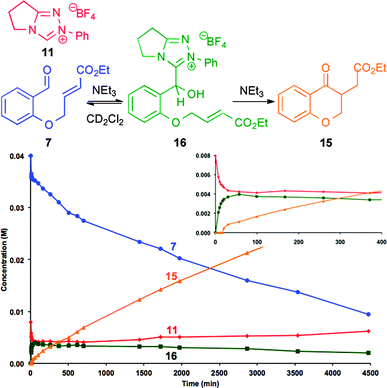
Further studies utilised 1H NMR spectroscopic analysis to follow the course of the Stetter reaction of 7 (0.04 M) employing sub-stoichiometric quantities (0.008 M) of N-phenyl triazolium precatalyst 11. Monitoring the reaction in anhydrous CD2Cl2 using NEt3 (0.008 M) as the base showed the initial rapid appearance of an intermediate 3-(hydroxybenzyl)azolium salt 16, with slower subsequent formation of product 15 over time (Fig. 3).21 Unambiguous structural determination of 16 was obtained by simply mixing equimolar quantities of triazolium precatalyst 11 with 7 and NEt3, giving, after 10 minutes, 16 that could be isolated by silica chromatography in 48% yield (Table 1).22 The tetrahedral C(α) geometry within 16 was confirmed by HSQC correlation, complementing the keto tautomer of the Breslow intermediate characterised by NMR spectroscopic analysis by Berkessel and co-workers, who generated this species using a free isolated NHC in THF.23 Although a small number of related 3-(hydroxybenzyl) thiazolium and imidazolium salts have been prepared by analogous routes,13,14 it is notable that 16 can be isolated from synthetically relevant conditions.
 |
| | Fig. 3 Reaction profile of the Stetter reaction of 7 (0.04 M), catalysed by NHC precursor 11 (0.008 M) with NEt3 (0.008 M). Inset: expansion of initial time period (<400 min). | |
Table 1 Preparation and isolation of 3-(hydroxybenzyl)azolium 16
|

|
| Solvent |
δ
H C(α)Ha |
δ
C C(α)a |
δ
C C(3)a |
|
HSQC correlation (CD2Cl2) δH 6.30–δC C(α) 63.2.
|
| CD2Cl2 |
6.30 (s) |
63.2 |
152.2 |
| DMSO |
6.33 d, (J = 5.4) |
63.2 |
152.0 |
| CD3OD/DCl |
6.36 (s) |
65.0 |
153.7 |
The stability and reversibility of 16 was then probed using control studies that showed tetrahedral intermediate 16 is stable under either acidic or neutral conditions in CD2Cl2. However, treatment of 16 with NEt3 facilitated rapid equilibration to a mixture of aldehyde 7, precatalyst 11 and 16, with relatively slow onwards formation of product 15 (Fig. 4).19 Performing this experiment with 16 (0.008 M) in the presence of additional aldehyde 7 (0.032 M), gave rise to a similar reaction profile to that obtained starting from 7 (0.04 M) and 11 (0.008 M). These observations indicate that 16 is a reversibly formed intermediate from the addition of NHC to the aldehyde, with slower subsequent onwards reaction, presumably through the expected Breslow intermediate, generating product 15.19
 |
| | Fig. 4 Reaction profile of the reaction of N-Ph 3-(hydroxybenzyl)azolium salt 16 (0.02 M) with NEt3 (0.02 M). | |
To probe the generality of these studies, the synthesis and isolation of a range of 3-(hydroxybenzyl)azolium salts was investigated. Treatment of a series of N-aryl triazolium precatalysts and aldehyde 7 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with excess NEt3 gave the corresponding 3-(hydroxybenzyl)azolium salts 17–21 in 16–96% yield (Table 2). Using the N-C6F5 triazolium precatalyst, the desired tetrahedral intermediate could not be isolated due to rapid conversion into product under these reaction conditions, although electron-deficient N-2,4,6-Cl3C6H2 and N-4-FC6H4 analogues 17 and 18 could be isolated and characterised.
:1) with excess NEt3 gave the corresponding 3-(hydroxybenzyl)azolium salts 17–21 in 16–96% yield (Table 2). Using the N-C6F5 triazolium precatalyst, the desired tetrahedral intermediate could not be isolated due to rapid conversion into product under these reaction conditions, although electron-deficient N-2,4,6-Cl3C6H2 and N-4-FC6H4 analogues 17 and 18 could be isolated and characterised.
Table 2 Preparation of 3-(hydroxybenzyl)azolium salts
In situ reaction monitoring of the model Stetter transformation using NHC precursors 9–14, as well as their 3-(hydroxybenzyl)azolium salt products 16–21 was next investigated. In all cases, starting from either the parent azolium salt precatalyst or 3-(hydroxybenzyl)azolium salt, formation of an equilibrium mixture of the corresponding azolium salt, aldehyde 7 and the 3-(hydroxybenzyl)azolium was observed, before relatively slow subsequent onwards reaction to give product 15 (Fig. 5, representative example shown using N-Mes substituted 3-(hydroxybenzyl)azolium salt 21).
 |
| | Fig. 5 Reaction profile of the reaction of N-Mes 3-(hydroxybenzyl)azolium salt 21 (0.02 M) with NEt3 (0.02 M). | |
From the resultant reaction profiles, values for K (the equilibrium constant for the formation of 3-(hydroxybenzyl)azolium salt) were calculated before significant (<5%) product formation (Table 3).24 These results show that reversible addition is observed in this system even with the N-Mes triazolylidene. Notably, significantly larger equilibrium constants are observed using NHC-precatalysts bearing 2,6-substituted N-aryl units (N-Mes, N-2,6-OMeC6H3 and N-2,4,6-Cl3C6H2), whilst electronic variation of the 4-substituent leads to minimal perturbation of K.25 Despite the large equilibrium concentration of the N-Mes and N-2,6-OMeC6H3 3-(hydroxybenzyl)azolium salts, the rates of product formation are slower than with NHC precatalysts 8–12.
Table 3 Equilibrium constants of 3-(hydroxybenzyl)azolium saltsa
|
|
Ar |
K (M−1) |
|
Starting concentrations: 7 0.04 M, NHC 0.008 M, NEt3 0.008 M.
|

|
2,4,6-Cl3C6H2 |
336 |
| 4-FC6H4 |
28 |
| Ph |
27 |
| 4-OMeC6H4 |
31 |
| 2,6-OMeC6H3 |
1283 |
| Mes |
140 |
Whilst this proved instructive, the effect of the N-C6F5 substituent upon the equilibrium values could not be evaluated within this system due to rapid onwards reactivity to product. Given the typical recalcitrance of 2-substituted benzaldehydes to participate in homo-benzoin processes,10c,10d a series of model 3-(hydroxybenzyl)azolium salts 22–27 were prepared from 2-methoxybenzaldehyde, allowing an evaluation of their K values (Table 4).26 Using precatalysts 8, 9 and 11–14 a similar trend was observed, with 2,6-substituted N-aryl NHCs yielding significantly larger K values. The use of N-C6F5 precatalyst 8 also led to a high K value, suggesting that both steric effects from 2,6-substituents and electron-withdrawing electronic effects within the N-aryl substituent lead to increased K values. This increase in K reflects the relative stabilities of the respective 3-(hydroxybenzyl)azolium salt in comparison to the starting materials. Simplistically, we assume that 2,6-N-aryl substitution within the NHC forces the N-aryl ring to adopt an essentially orthogonal orientation with respect to the triazole ring.27 Furthermore, in all 3-(hydroxybenzyl)azolium salts the N-aryl group presumably adopts a non-coplanar conformation with the triazole in order to minimize 1,2-steric interactions, with orthogonality enforced with 2,6-N-aryl substitution. 3-(Hydroxybenzyl)azolium salt formation may then be favoured for 2,6-N-aryl substituted NHCs due to better accommodation of the 3-(hydroxybenzyl) substituent by the N-aryl unit. Furthermore, the reverse process may then deviate from expected leaving group ability, being more favourable for non-2,6-N-aryl substituted NHCs. Further work will generate additional mechanistic insight by quantifying the rates of the individual processes.
Table 4 Equilibrium constants of 3-(hydroxybenzyl)azolium saltsa
|
|
Ar |
K (M−1) |
|
Starting concentrations: 2-methoxybenzaldehyde 0.01 M, NHC 0.002 M, NEt3 0.002 M.
|

|
C6F5 |
521 |
| 2,4,6-Cl3C6H2 |
601 |
| Ph |
56 |
| 4-OMeC6H4 |
46 |
| 2,6-OMeC6H3 |
741 |
| Mes |
143 |
Rates constants of exchange (kex) for 3-(hydroxybenzyl)- or 3-(methoxybenzyl)azolium salts
Given the proposed kinetic relevance of the deprotonation step to generate the Breslow intermediate in the Stetter reaction,17,28 the relative rate constants of deuterium exchange (kex, s−1) at the C(α)–H position were investigated by 1H NMR spectroscopy using either KOD solutions or triethylamine buffers in D2O/CD3OD. For Stetter derived 3-(hydroxybenzyl)azolium salts 16–21, C(α)H exchange could only be monitored alongside competitive dissociation to aldehyde and NHC,29 while attempted O-methylation of 16 led to extensive decomposition. To circumvent these issues a series of alternative model 3-(hydroxybenzyl)azolium salts was prepared by triazolinylidene addition to benzaldehyde and a number of substituted benzaldehyde derivatives (Table 5). Only low isolated product yields of 3-(hydroxybenzyl)azolium products were obtained for triazolinylidene addition to either benzaldehyde or 4-substituted benzaldehydes (<10%), with products 28–31 isolated by preparative LC. Despite these poor yields, crystallisation from DCl/D2O allowed unambiguous structure determination of chloride salt 41 by X-ray crystal structure analysis (Fig. 6).30 NHC addition to 2-(benzyloxy)benzaldehyde gave higher isolated yields and showed increased stability to chromatographic purification, furnishing reasonable to excellent isolated yields (up to 94%) of aldehyde–NHC addition products 32–38. This synthetic route was extended to morpholine-containing NHC precursors, giving 39, while a chiral NHC precursor gave 40 as a 75:25 mixture of diastereoisomers in an excellent 99% yield.31 3-(Hydroxybenzyl)azolium salts 28, 32, 33, 35, 37, 38 and 40 were subsequently O-methylated32 to facilitate an evaluation of their rates of deuterium exchange.
Table 5 Synthesis of 3-(hydroxybenzyl)azolium saltsa
|
Experimental conditions: NHC precursor (1 equiv.), aldehyde (1 equiv.), NEt3 (2 equiv.), CH2Cl2, rt, 5 min to 1 h.
|

|
 |
| | Fig. 6 Representation of the X-ray crystal structure of 3-(hydroxybenzyl)azolium chloride 41 (chloride counterion and water of crystallisation not shown for simplicity). | |
Deuterium exchange studies were carried out upon 36 and 38 and 3-(methoxybenzyl)azolium salts 42–48. In all cases deuteroxide catalysed exchange of C(α)–H for deuterium could be monitored without any detritic side reactions (Fig. 7).33 The fastest exchange was observed with electron-deficient N-aryl triazolium derivatives (Ar = C6F5 > 2,4,6-Cl3C6H2 > Ph > 4-OMeC6H4 > Mes > 2,6-OMeC6H3, Table 6).34 For 48, the rate constant of exchange for both diastereoisomers could be evaluated, with the minor diastereoisomer exhibiting an enhanced kex value.35 Within this system, the rate of exchange with variation of N-aryl substitution generally parallels the rate of observed product formation in the Stetter transformation.36 This presumably reflects the increased acidity of C(α)–H of the intermediate 3-(hydroxybenzyl)azolium species with increasing electron-withdrawing N-aryl substituents, and is consistent with rate determining deprotonation of this species, as postulated by Rovis.17
![Deuterium exchange of C(α)H (5.85 ppm) relative to CD3OD (3.31 ppm) of 44 (5 mM) in NEt3 buffer in 6.5 : 1 D2O : CD3OD at pD 10.9 and 25 °C [a] T = 9 min. [b] T = 109 min. [c] T = 252 min.](/image/article/2013/SC/c2sc22137c/c2sc22137c-f7.gif) |
| | Fig. 7 Deuterium exchange of C(α)H (5.85 ppm) relative to CD3OD (3.31 ppm) of 44 (5 mM) in NEt3 buffer in 6.5:1 D2O:CD3OD at pD 10.9 and 25 °C [a] T = 9 min. [b] T = 109 min. [c] T = 252 min. | |
Table 6 Relative acidities of 3-(hydroxybenzyl)azolium salts
|
|
pD |
k
ex (s−1) major |
k
ex (s−1) minor |
|
48
|
10.9 |
1.36 × 10−4 |
1.94 × 10−4 |
Although the formation of the Breslow intermediate is implied by these NMR hydrogen–deuterium exchange studies, its direct observation was not possible due to its transient nature in the protic solvent conditions. The direct isolation of Breslow intermediates such as 4 is a widely recognised challenge, although Jordan and co-workers have characterised an O-protected thiazolinylidene-derived enamine using NMR spectroscopy37 and Goldup has reported the observation of an O-benzylated intermediate in an imidazolinium-mediated transesterification.38 Most recently, Rovis and co-workers have described the isolation of nitrogen analogues of the Breslow intermediate using 2,6-substituted NHC precursors.39 Mayr has similarly reported the isolation and reactivity of a range of O-methylated Breslow intermediates,40 whilst Berkessel has reported the isolation of Breslow intermediates derived from free imidazolinium NHCs and benzaldehydes.41 Despite not being able to observe species such as 4 in these studies, treatment of O-methylated derivative 47 with a 10-fold excess of potassium dimsyl in DMSO allowed the acquisition of a UV-Vis spectrum of the enamine with a characteristic λmax at 380 nm and an extinction coefficient (ε) of 7846 M−1 cm−1, which is consistent with the more extended conjugated system present in 49 relative to the precursor (Fig. 8).42 Under these conditions, this absorbance decayed to zero over ∼20 min, presumably due to reprotonation of 49 by adventitious water. Under analogous conditions, O-methylated derivative 44 exhibited a λmax at 395 nm and an extinction coefficient of 5423 M−1 cm−1.
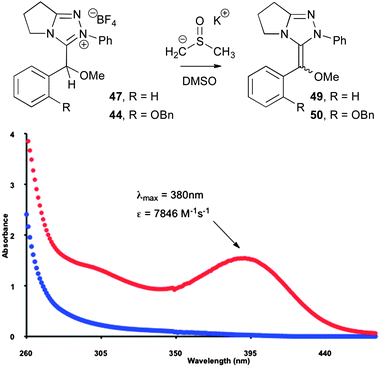 |
| | Fig. 8 UV-Vis spectra of a 0.2 mM solution of 3-(methoxybenzyl)azolium salt 47 (blue) and 49 (red). | |
Hydroxybenzylazolium salts as precatalysts
Consistent with these mechanistic studies, the validity of 3-(hydroxybenzyl)azolium salts 18 and 32 as precatalysts for the Stetter reaction was investigated (Fig. 9). As expected, treatment of 7 with 18 or 32 (20 mol%) and NEt3 (20 mol%) in CH2Cl2 at rt gave 15 in excellent isolated yield, with the rate of product formation using the electron deficient N-C6F5 salt 30 significantly faster than the N-4-FC6H4 salt 18. Notably, no homo- or crossed-benzoin products were observed in the reaction employing either 18 or 32 as precatalyst.
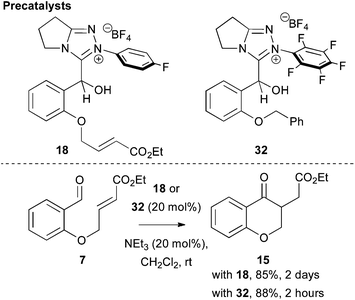 |
| | Fig. 9 Use of 3-(hydroxybenzyl)azolium salts as precatalysts in the Stetter reaction. | |
Similarly, the use of 32 as a precatalyst for the benzoin reaction was probed through performing a crossover reaction with benzaldehyde (Fig. 10). Treatment of 32 (20 mol%) with benzaldehyde and NEt3 (20 mol%) gave preferentially the homo-benzoin product 52, along with only 6% of the crossed benzoin product 53. The small yield of the observed crossed product is consistent with current literature, due to the poor reactivity of the 2-substituted aldehyde as an electrophilic component in benzoin type processes.43
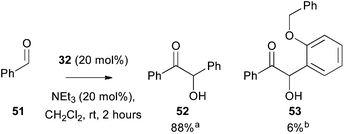 |
| | Fig. 10 Use of 3-(hydroxybenzyl)azolium salt 32 as precatalyst in the benzoin condensation. a Isolated yield. b Approximate yield by 1H NMR. | |
Conclusions
In conclusion, the isolation and in situ spectroscopic observation of aldehyde-NHC addition product intermediates of the Stetter and benzoin reactions is reported. Reaction profiles indicate these intermediates are reversibly formed (irrespective of the N-aryl substitution pattern), followed by relatively slow onwards reaction to yield Stetter product. Estimated K values suggest that the equilibrium constant is affected by both electronic and steric effects of the N-aryl unit, with 2,6- and electron-withdrawing N-aryl substitution resulting in significantly larger K values. By contrast, electronic variation of the 4-substituent leads to minimal perturbation of K. The relative acidities (kex) at C(α) of 3-(methoxybenzyl)azolium salts provided insight into the effect of catalyst architecture on the key Breslow intermediate-forming step. Despite N-2,6-OMeC6H3 and N-Mes NHC precursors 13 and 14 exhibiting enhanced K values (285- and 5-fold vs. N-Ph congener 11 respectively in the Stetter reaction), slow and presumably rate determining deprotonation17 (as indicated by kex) inhibits their use as precatalysts in the Stetter reaction. In contrast N-C6F5 and N-2,4,6-Cl3C6H2 substituted NHCs benefit from both high K values and rapid kex values (21- and 8- fold respectively vs. N-Ph), rationalising why 2,6-electron withdrawing N-substituents are preferred in benzoin and Stetter type processes.44 Further work from our laboratories towards developing a full kinetic understanding of these and other NHC-catalysed reaction processes are underway.
Acknowledgements
We thank the Royal Society for a University Research Fellowship (ADS) and the EPSRC (CJC and RM) for funding, and the EPSRC National Mass Spectrometry Service Centre (Swansea). We would also like to thank James Taylor (ADS) and Stuart Leckie (ADS) for their assistance in the synthesis of triazolium salts.
Notes and references
-
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS;
(b) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS;
(c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
-
(a) H. Vora and T. Rovis, Aldrichimica Acta, 2011, 44, 3–11 CAS;
(b) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS.
-
(a) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 44, 2295–2309 Search PubMed;
(b) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617–1639 Search PubMed.
-
(a) E. Phillips, A. Chan and K. Scheidt, Aldrichimica Acta, 2009, 42, 55–66 CAS;
(b) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336–5346 RSC.
-
(a) N. T. Reynolds, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2004, 126, 9518–9519 CrossRef CAS;
(b) A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905–908 CrossRef CAS;
(c) S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873–3876 CrossRef CAS;
(d) P.-C. Chiang, Y. Kim and J. W. Bode, Chem. Commun., 2009, 4566–4568 RSC;
(e) B. E. Maki, E. V. Patterson, C. J. Cramer and K. A. Scheidt, Org. Lett., 2009, 11, 3942–3945 CrossRef CAS;
(f) B. E. Maki, A. Chan and K. A. Scheidt, Synthesis, 2008, 1306–1315 CAS.
-
(a) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176–14177 CrossRef CAS;
(b) J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 1673–1677 CrossRef CAS;
(c) Z.-Q. Zhu and J.-C. Xiao, Adv. Synth. Catal., 2010, 352, 2455–2458 CrossRef CAS;
(d) S. De Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266–9269 CrossRef;
(e) J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS;
(f) Z.-Q. Rong, M.-Q. Jia and S.-L. You, Org. Lett., 2011, 13, 4080–4083 CrossRef CAS;
(g) Z.-Q. Zhu, X.-L. Zheng, N.-F. Jiang, X. Wan and J.-C. Xiao, Chem. Commun., 2011, 47, 8670–8672 RSC.
- T. Rovis, Chem. Lett., 2008, 37, 2–7 CrossRef CAS.
- X.-L. Huang, L. He, P.-L. Shao and S. Ye, Angew. Chem., Int. Ed., 2008, 48, 192–195 Search PubMed.
- We thank one of the referees of this manuscript who indicated that some exceptions to this generalisation exist. For example the report by Rovis and co-workers of the synthesis of trans-(γ)-lactams using an N-pentafluorophenyl substituted NHC precursor that proceeds via a homoenolate ( X. Zhao, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 12466–12469 Search PubMed ).
-
(a) D. Enders, K. Breuer and J. H. Teles, Helv. Chim.
Acta, 1996, 79, 1217–1221 CAS;
(b) D. Enders and U. Kallfass, Angew. Chem., Int. Ed., 2002, 41, 1743–1745 CrossRef CAS;
(c) L. Baragwanath, C. A. Rose, K. Zeitler and S. J. Connon, J. Org. Chem., 2009, 74, 9214–9217 CrossRef CAS;
(d) S. E. O'Toole and S. J. Connon, Org. Biomol. Chem., 2009, 7, 3584–3593 RSC;
(e) D. Enders, K. Breuer, J. Runsink and J. H. Teles, Helv. Chim. Acta, 1996, 79, 1899–1902 CAS;
(f) M. S. Kerr, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2002, 124, 10298–10299 CrossRef CAS;
(g) J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2005, 127, 6284–6289 CrossRef CAS;
(h) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402–10405 CrossRef CAS;
(i) D. A. DiRocco, E. L. Noey, K. N. Houk and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 2391–2394 CrossRef CAS;
(j) T. Jousseaume, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1410–1414 CrossRef CAS;
(k) D. Enders and J. Han, Tetrahedron: Asymmetry, 2008, 19, 1367–1371 CrossRef CAS;
(l) Y. Ma, S. Wei, J. Wu, F. Yang, B. Liu, J. Lan, S. Yang and J. You, Adv. Synth. Catal., 2008, 350, 2645–2651 CrossRef CAS;
(m) I. Piel, M. Steinmetz, K. Hirano, R. Frohlich, S. Grimme and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 4983–4987 CrossRef CAS;
(n) R. Breslow and E. McNelis, J. Am. Chem. Soc., 1959, 81, 3080–3082 CrossRef CAS;
(o) J. C. Sheehan and T. Hara, J. Org. Chem., 1974, 39, 1196–1199 CrossRef CAS;
(p) W. Tagaki, Y. Tamura and Y. Yano, Bull. Chem. Soc. Jpn., 1980, 53, 478–480 CAS;
(q) R. L. Knight and F. J. Leeper, J. Chem. Soc., Perkin Trans. 1, 1998, 1891–1894 RSC;
(r) H. Stetter, Angew. Chem., Int. Ed. Engl., 1976, 15, 639–647 CrossRef.
- J. Mahatthananchai and J. W. Bode, Chem. Sci., 2012, 3, 192–197 RSC.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
-
(a) A. Miyashita, A. Kurachir, Y. Matsuoka, N. Tanabe, Y. Suzuki, K.-i. Iwamoto and T. Higashimo, Heterocycles, 1997, 44, 417–426 Search PubMed;
(b) S. Ohta, S. Hayakawa, K. Nishimura and M. Okamoto, Chem. Pharm. Bull., 1987, 35, 1058–1069;
(c) V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612–1613 RSC;
(d) L.-W. Xu, Y. Gao, J.-J. Yin, L. Li and C.-G. Xia, Tetrahedron Lett., 2005, 46, 5317–5320 CrossRef CAS.
-
(a) R. Breslow and R. Kim, Tetrahedron Lett., 1994, 35, 699–702 CrossRef CAS;
(b) Y.-T. Chen, G. L. Barletta, K. Haghjoo, J. T. Cheng and F. Jordan, J. Org. Chem., 1994, 59, 7714–7722 CrossRef CAS;
(c) M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124–5131 CrossRef CAS;
(d) W. Schrader, P. P. Handayani, C. Burstein and F. Glorius, Chem. Commun., 2007, 716–718 RSC;
(e) M. B. Doughty, G. E. Risinger and S. J. Jungk, Bioorg. Chem., 1987, 15, 15–30 CrossRef CAS;
(f) J. Kiss, R. D'Souza and H. Spiegelberg, Helv. Chim. Acta, 1968, 51, 325–339 Search PubMed;
(g) J. J. Mieyal, G. Bantle, R. G. Votaw, I. A. Rosner and H. Z. Sable, J. Biol. Chem., 1971, 246, 5213–5219 CAS;
(h) A. Takamizawa, Y. Hamashima and H. Sato, J. Org. Chem., 1968, 33, 4038–4045 CrossRef CAS;
(i) Y. Kageyama and S. Murata, J. Org. Chem., 2005, 70, 3140–3147 CrossRef CAS.
-
(a) R. C. Samanta, B. Maji, S. De Sarkar, K. Bergander, R. Frohlich, C. Muck-Lichtenfeld, H. Mayr and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 5234–5238 Search PubMed;
(b) K. J. Hawkes and B. F. Yates, Eur. J. Org. Chem., 2008, 5563–5570 CrossRef CAS;
(c) J. M. Um, D. A. DiRocco, E. L. Noey, T. Rovis and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 11249–11254 CrossRef CAS;
(d) B. Maji, M. Horn and H. Mayr, Angew. Chem., Int. Ed., 2012, 51, 6231–6235 Search PubMed;
(e) G. Doleschall, Tetrahedron Lett., 1980, 21, 4183–4186 Search PubMed;
(f) G. Doleschall, Tetrahedron, 1976, 32, 2549–2558 CrossRef CAS;
(g) G. Doleschall, Tetrahedron Lett., 1975, 16, 1889–1892 Search PubMed;
(h) C. E. I. Knappke, I. I. I. A. J. Arduengo, H. Jiao, J.-M. Neudörfl and A. Jacobi von Wangelin, Synthesis, 2011, 3784–3795 Search PubMed;
(i) C. E. I. Knappke, J. M. Neudorfl and A. J. von Wangelin, Org. Biomol. Chem., 2010, 8, 1695–1705 RSC.
- J. Henrique Teles, J.-P. Melder, K. Ebel, R. Schneider, E. Gehrer, W. Harder, S. Brode, D. Enders, K. Breuer and G. Raabe, Helv. Chim. Acta, 1996, 79, 61–83 CrossRef CAS.
- J. L. Moore, A. P. Silvestri, J. R. de Alaniz, D. A. DiRocco and T. Rovis, Org. Lett., 2011, 13, 1742–1745 CrossRef CAS.
-
(a) K. B. Ling and A. D. Smith, Chem. Commun., 2011, 47, 373–375 RSC;
(b) C. D. Campbell, C. Concellón and A. D. Smith, Tetrahedron: Asymmetry, 2011, 22, 797–811 Search PubMed;
(c) N. Duguet, A. Donaldson, S. M. Leckie, E. A. Kallström, C. D. Campbell, P. Shapland, T. B. Brown, A. M. Z. Slawin and A. D. Smith, Tetrahedron: Asymmetry, 2010, 21, 601–616 CrossRef CAS;
(d) N. Duguet, A. Donaldson, S. M. Leckie, J. Douglas, P. Shapland, T. B. Brown, G. Churchill, A. M. Z. Slawin and A. D. Smith, Tetrahedron: Asymmetry, 2010, 21, 582–600 CrossRef CAS;
(e) J. Douglas, K. B. Ling, C. Concellon, G. Churchill, A. M. Z. Slawin and A. D. Smith, Eur. J. Org. Chem., 2010, 5863–5869 CrossRef CAS;
(f) N. Duguet, C. D. Campbell, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2008, 6, 1108–1113 RSC;
(g) J. E. Thomson, C. D. Campbell, C. Concellon, N. Duguet, K. Rix, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2008, 73, 2784–2791 CrossRef CAS;
(h) C. Concellón, N. Duguet and A. D. Smith, Adv. Synth. Catal., 2009, 351, 3001–3009 CrossRef CAS;
(i) C. D. Campbell, C. J. Collett, J. E. Thomson, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2011, 9, 4205–4218 RSC;
(j) R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421–20432 Search PubMed.
- See ESI† for further details.
- Rovis and co-workers have demonstrated that the rate of product formation in the intermolecular Stetter reaction can be enhanced by the addition of bifunctional additives, assumed to assist in the proton transfer step to generate the Breslow intermediate ( D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402–10405 Search PubMed ). We chose to examine the reaction without such additives in order the aid the simplicity of analysis.
- Reactions were typically followed up to 80% conversion to product 15. See ESI† for full reaction profiles.
- One referee correctly questioned whether 16 (or any other hydroxybenzylazolium salt in this manuscript) was actually the tetrafluoroborate salt as represented, or the corresponding ylide/alkoxide and Et3NH+. While we are unable to distinguish these possibilities in situ, the product 16 isolated after chromatographic purification contained 19F NMR resonances at δF-153.4 and δF-153.5, consistent with it containing a tetrafluoroborate counterion. Similar 19F NMR data was obtained for 18 and 22 and so for simplicity we represent all compounds as the tetrafluoroborate counterion.
- A. Berkessel, S. Elfert, K. Etzenbach-Effers and J. H. Teles, Angew. Chem., Int. Ed., 2010, 49, 7120–7124 CrossRef CAS.
- Values of K were obtained by monitoring the concentrations of the starting materials and 3-(hydroxybenzyl)azolium salt. Agreeable estimates for K could be calculated from reaction profiles starting with 7 (0.04 M), NHC (0.008 M) and NEt3 (0.008 M) or using 7 (0.032 M), 3-(hydroxybenzyl)azolium salt (0.008 M) and NEt3 (0.008 M). See ESI† for full details.
- Glorius postulated that ortho-substituted N-aryl substituents would destabilise the initial tetrahedral adduct formed between an NHC and an aromatic aldehyde due to steric effects (see M. Schedler, R. Frohlich, C. G. Daniliuc and F. Glorius, Eur. J. Org. Chem., 2012, 4164–4171 Search PubMed; N. E. Wurtz, C. G. Daniliuc and F. Glorius, Chem.–Eur. J., 2012, 18, 16297–16301 Search PubMed ). Whilst we believe the enhanced formation of 3-(hydroxybenzyl) azolium salt observed in the case of the N-2,6-disubstituted triazolium catalysts results from a more favourable orientation of the N-aryl ring (due to the presence of two ortho-substituents), more bulky ortho-substituents do not appear to show this effect. Analogous studies of the benzoin condensation using N-Mes and N-(2,6-diisopropylphenyl) imidazolium catalysts resulted in the appearance of hydroxybenzyl adduct only in the case of the mesityl catalyst, whilst no reaction was observed using the more sterically hindered 2,6-diisopropylphenyl catalyst (See ESI† for full details).
-
K values were determined as previously described, see ESI† for full details. During the course of the experiment, none of the crossed benzoin product was observed, presumably due to the poor reactivity of 2-substituted benzaldehydes in the benzoin reaction, consistent with literature studies (see L. Baragwanath, C. A. Rose, K. Zeitler and S. J. Connon, J. Org. Chem., 2009, 74, 9214–9217 Search PubMed; S. E. O'Toole and S. J. Connon, Org. Biomol. Chem., 2009, 7, 3584–3593 CrossRef CAS ).
- Computational and X-ray crystallographic evidence from Mayr indicates that the N-Mes substituent prefers to adopt an almost perpendicular conformation in a range of imidazolium and triazolium carbenes ( B. Maji, M. Breugst and H. Mayr, Angew. Chem., Int. Ed., 2011, 50, 6915–6919 Search PubMed ). Additionally, a study by Cavallo of NHC ligands concluded that the orientation of the N-aryl substituent was determined from the balance between the conjugation energy gained from co-planar geometry and the steric repulsion, with N-Ph substituents co-planar, whilst N-Mes are almost perpendicular to the heterocycle plane ( F. Ragone, A. Poater and L. Cavallo, J. Am. Chem. Soc., 2010, 132, 4249–4258 CrossRef CAS ).
- P. Verma, P. A. Patni and R. B. Sunoj, J. Org. Chem., 2011, 76, 5606–5613 CrossRef CAS.
- However, the half-life for deuterium incorporation at C(α)H could be
estimated, with fastest exchange observed with electron-withdrawing N-aryl units (Ar = 4-FC6H4 > Ph > 4-OMeC6H4 > Mes). See ESI† for further details.
-
41 and the chloride salts of 29 and 30 (54 and 55 respectively) were characterised by single-crystal X-ray diffraction of their D2O monosolvates which are mutually isotypic (isomorphous).†.
- Treatment of the isolated major diastereosisomer to the reaction conditions led to a 75:25 mix of diastereoisomers, suggesting the obtained dr is a result of a thermodynamic equilibrium.
- 3-(Methoxybenzyl)azolium salts 42–48 were synthesised from methylation of 28, 32, 33, 35, 37, 38 and 40 using diazomethane or TMS-diazomethane. See ESI† for further details. For these compounds, exchange could be monitored without dissociation or decomposition.
- Experiments were carried out at a substrate concentration of 5 mM at 25 °C and I = 1 (KCl) in 6.5:1 D2O/CD3OD. Exchange was monitored for two half lives and a semi-logarithmic plot of the fraction of unexchanged substrate (fs) against time gave kex, the pseudo-first order rate of exchange at that pD. In the case of 36 and 38, measurements at other pD values were not possible due to competing dissociation to aldehyde and NHC at lower pD values and ring opening at higher pD values. See ESI† for full details.
- It is assumed that deprotonation by deuteroxide is significantly slower than deuteration of the Breslow intermediate in all cases so that kex reflects the rate constant for formation of the solvent equilibrated enaminol/enamine. Using stopped flow spectrophotometry Jordan has measured rate constants for the reprotonation of O-methylated thiazolylidene-derived enamine derivatives in the range of 300–540 s−1 ( G. L. Barletta, Y. Zou, W. P. Huskey and F. Jordan, J. Am. Chem. Soc., 1997, 119, 2356–2362 Search PubMed ), which are 107-fold higher than corresponding rate constants for formation of the intermediate at pH 10.5 (∼pD 10.9).
- Upon completion of the deuterium exchange experiment of 48 the C(α)-deuterated 48 formed exhibited a decreased dr, suggesting enamine deuteration is not completely stereoselective.
- The only exception to this is the 2,6-OMeC6H3 substituted analogue 45, which exhibits a lower kex value than 46, despite displaying a faster rate of product formation in the Stetter reaction. We presume that the greatly enhanced K value (Table 3) compensates for the slower rate of deprotonation, giving a faster overall rate of reaction. Jordan has reported a second order rate constant, kHO = 0.019 M−1 s−1 for hydroxide ion deprotonation of the 2-(methoxyphenylmethyl)-3,4-dimethylthiazolium salt to the corresponding enamine using stopped-flow spectrophotometry ( G. L. Barletta, Y. Zou, W. P. Huskey and F. Jordan, J. Am. Chem. Soc., 1997, 119, 2356–2362 Search PubMed ). Using this value, we can calculate a first order rate constant for deprotonation by hydroxide at pH 10.5 (∼pD 10.9) of 8 × 10−6 s−1, which is 3–250-fold smaller than any of the kex values in Table 6 at an equivalent pD.
- F. Jordan, Z. H. Kudzin and C. B. Rios, J. Am. Chem. Soc., 1987, 109, 4415–4416 CrossRef CAS.
- L. Pignataro, T. Papalia, A. M. Z. Slawin and S. M. Goldup, Org. Lett., 2009, 11, 1643–1646 CrossRef CAS.
- D. A. DiRocco, K. M. Oberg and T. Rovis, J. Am. Chem. Soc., 2012, 134, 6143–6145 Search PubMed.
- B. Maji and H. Mayr, Angew. Chem., Int. Ed., 2012, 51, 10408–10412 Search PubMed.
- A. Berkessel, S. Elfert, V. R. Yatham, J. M. Neudorfl, N. E. Schlorer and J. H. Teles, Angew. Chem., Int. Ed., 2012, 51, 12370–12374 Search PubMed.
- This value is consistent with the reported extinction coefficient of 14000 M−1 cm−1 at 410 nm in dioxane/DMSO for the enamine generated from a 2-(methoxyphenylmethyl)-3,4-dimethylthiazolium salt ( G. Barletta, W. P. Huskey and F. Jordan, J. Am. Chem. Soc., 1992, 114, 7607–7608 Search PubMed ).
- None of the homo-benzoin product of 2-(benzyloxy)benzaldehyde was observed over the course of the reaction, consistent with current literature (see S. E. O'Toole, C. A. Rose, S. Gundala, K. Zeitler and S. J. Connon, J. Org. Chem., 2010, 76, 347–357 Search PubMed; M. Y. Jin, S. M. Kim, H. Han, D. H. Ryu and J. W. Yang, Org. Lett., 2011, 13, 880–883 Search PubMed ).
- We are grateful to a referee who pointed out that the electronic determinant of reaction rate in the intramolecular Stetter transformation (superior rates with electon-deficient N-aryl triazolium precatalysts) is in contrast to many other classes of NHC-catalysed reactions. For example, typical reactions that proceed via azolium enolates appear to be dominated by steric constraints. For a computational study see: S. E. Allen, J. Mahatthananchai, J. W. Bode and M. C. Kozlowski, J. Am. Chem. Soc., 2012, 134, 12098–12103 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here. 


