Porous calcium–manganese oxide microspheres for electrocatalytic oxygen reduction with high activity†
Xiaopeng
Han
a,
Tianran
Zhang
a,
Jing
Du
a,
Fangyi
Cheng
*ab and
Jun
Chen
*ab
aInstitute of New Energy Material Chemistry, College of Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: fycheng@nankai.edu.cn; chenabc@nankai.edu.cn; Fax: +86-22-23506808; Tel: +86-22-23506808
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin, 300071, P. R. China
First published on 27th September 2012
Abstract
A series of calcium–manganese oxides (Ca–Mn–O) were prepared through thermal decomposition of carbonate solid–solution precursors and investigated as electrocatalysts for oxygen reduction reaction (ORR). The synthesized crystalline Ca–Mn–O compounds, including perovskite-type CaMnO3, layered structured Ca2Mn3O8, post-spinel CaMn2O4 and CaMn3O6, presented similar morphologies of porous microspheres with agglomerated nanoparticles. Electrochemical results, surface analysis, and computational studies revealed that the catalytic activities of Ca–Mn–O oxides, in terms of onset potential, reduction current, and transferred electron number, depended strongly on both the surface Mn oxidation state and the crystallographic structures. Remarkably, the as-synthesized CaMnO3 and CaMn3O6 exhibited considerable activity and enabled an apparent quasi 4-electron oxygen reduction with low yield of peroxide species in alkaline solutions, suggesting their potential applications as cheap and abundant ORR catalysts.
Introduction
Achieving efficient catalysis of the sluggish oxygen reduction reaction (ORR) plays an important role in a variety of electrochemical energy conversion and storage technologies including fuel cells and metal–air batteries.1–5 Precious metals such as Pt and Pt alloys are known to exhibit the best overall catalytic performance for the ORR, but their limited availability and high price hinder the widespread practical applications.6–8 Thus, extensive research effort has been dedicated to developing alternative catalysts based on non-noble elements.9–13 Among them, manganese oxides are particularly attractive because of high abundance, low cost, minimum environmental impact, and considerable ORR activity.14–22 Additionally, the variable valence of Mn (e.g., +2, +3, +4, +6 and +7) and structural flexibility of oxides (e.g., available in binary oxide type or capable of incorporating another metal to form composite oxide structures such as perovskite and spinel) offer opportunity yet challenges to tailor the physical–chemical and catalytic properties of the manganese oxide family.23,24Manganese oxides can also serve as electrocatalysts for the oxygen evolution reaction (OER). In Nature, photosystem II (PS II) has the unique capability to oxidize water to molecular oxygen via the oxygen-evolving complex (OEC), which contains a cubane-like μ-oxido-Mn4Ca cluster housed in a special protein environment.25 Inspired from the biological OEC active site, much interest has been focused on functional Mn-based molecular mimics for the OER.26 As there are parallels between abiotic and biotic MnOx structures,27 substantial progress has also been made in inorganic nanoscaled binary manganese oxides such as Mn2O3 nanostructured films,19 nanocrystalline λ-MnO2,28 and Mn oxide clusters.29 Interestingly, recent studies demonstrated that incorporation of Ca into binary manganese oxides significantly improved their water oxidation activities.30,31 Considering the inherent connection between the oxygen electrochemistry of ORR and OER, the Ca–Mn oxides might also act as efficient catalysts for the ORR. However, to the best of our knowledge, this has not been systematically investigated so far.
Aiming at developing new ORR catalysts composed of earth abundant and inexpensive elements, we report in this study the preparation of a series of calcium–manganese oxides (CaMnO3, Ca2Mn3O8, CaMn2O4 and CaMn3O6) and the detailed investigation of their electrocatalytic properties. This mixed metal oxide series are featured by different chemical compositions, crystal structures and oxidation states. The synthesis of these Ca–Mn–O compounds was achieved through a simple calcination route using Ca1−xMnxCO3 solid–solution precursors. The parallel formation of highly crystalline Ca–Mn–O porous microspheres with similar textures allowed us to systematically investigate the structural and compositional effect on electrochemical properties. In addition, the ORR and OER catalytic activities of the synthesized Ca–Mn–O compounds were compared with MnOx and benchmark Pt/C catalysts in alkaline conditions. Both experimental results and theoretical study demonstrated that the surface Mn oxidation state and crystal structure are influential factors determining the activity of Ca–Mn–O electrocatalysts.
Experimental section
Materials synthesis
Samples of CaMnO3, Ca2Mn3O8, CaMn2O4 and CaMn3O6 were synthesized by simply firing the corresponding Ca1−xMnxCO3 (x = 0.5, 0.6, 0.67, and 0.75) solid–solution precursors (see ESI†, Fig. S1) in air.32 The Ca–Mn–O compounds were obtained by heating the solid–solution carbonate precursors in recrystallized alumina boats in air. Calcination parameters: 900 °C, 5 h for CaMnO3, 700 °C, 1 h for Ca2Mn3O8, 800 °C, 1 h for CaMn3O6, and 950 °C, 10 h for CaMn2O4. The benchmark Pt/C catalyst (30 wt% Pt nanoparticles supported on Vulcan XC-72 carbon powders, ESI†, Fig. S2) was prepared following the procedures of our previous report.33Materials characterization
Chemical composition of the as-synthesized samples was determined by atomic adsorption spectrometry (AAS, Hitachi 180-80 spectrophotometer) and inductively coupled plasma-atomic emission (ICP) spectrometer (ICP-9000, Thermo Jarrell-Ash). The oxidation state of Mn was analyzed by a chemical titration: samples were dissolved in dilute H2SO4 and H3PO4 solution with an excess of Fe(NH4)2(SO4)2, and titrated with calibrated KMnO4 solution. Structures and morphologies were characterized by powder X-ray diffraction (XRD, Rigaku D/max-2500 X-ray diffractometer, Cu Kα radiation), scanning electron microscopy (SEM, FEI Nanosem 430), and transmission electron microscopy (TEM, Philips Tecnai F20) equipped with energy dispersive spectroscopy (EDS). X-Ray photoelectron spectroscopy (XPS) data were collected using a Perkin Elmer PHI 1600 ESCA system.Electrochemical tests
Electrochemical measurements were conducted using a standard three-compartment electrochemical cell. A Pt foil and a saturated calomel electrode (SCE) were used as the reference electrode and counter electrode, respectively. The working electrode was a catalyst-coated glass carbon (GC) substrate, which is confined in a rotating disk electrode (RDE, diameter 4 mm) or a rotating ring-disk electrode (RRDE, Pt ring and GC disk, disk diameter 4.2 mm). The thin catalyst layer was obtained by naturally drying a catalyst ink, which was produced by ultrasonically dispersing a mixture of Ca–Mn–O sample, carbon powders (Carbot Vulcan XC-72) and neutralized Nafion solution (5 wt%, Sigma-Aldrich) in isopropyl alcohol. The oxide samples were mixed with carbon at a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mass ratio to overcome electronic conductivity limitation. Catalyst loading is approximately 0.09 and 0.12 mg cm−2 for RDE and RRDE, respectively.
:7 mass ratio to overcome electronic conductivity limitation. Catalyst loading is approximately 0.09 and 0.12 mg cm−2 for RDE and RRDE, respectively.
All tests were performed at room temperature on a computer-controlled potentionstat/galvanostat workstation. The supporting electrolyte comprising 0.1 M aqueous KOH solution was purged with Ar or O2 (Air Product, purity 99.995%) for at least 30 min prior to testing and maintained under Ar or O2 atmosphere during the test. In RDE measurement, the working electrode was scanned at a rate of 5 mV s−1 in the potential range of 0.1 to −0.6 V (ORR) or 0.4–0.9 V (OER) versus SCE. In RDE measurement, the ring potential was set at 0.5 V versus SCE. Unless stated, all potentials were reported with reference to the reversible hydrogen electrode (RHE) potential scale. In 0.1 M KOH solution, the potential of SCE was calibrated as +0.990 V with respect to RHE.34 All the ORR curves obtained in O2-saturated solutions were corrected by subtracting the background capacitive current obtained in an Ar-saturated electrolyte.
Results
Structures and morphologies
Fig. 1 illustrates the crystal structures of carbonate calcite and composite oxides with increasing Mn:O ratio. The solid–solution calcite adopts the trigonal structure with space group of R-3c (no. 167).35 In calcite, the Ca/Mn cations and carbonate groups are arranged in a distorted halite structure, in which cations are coordinated by six oxygen anions and each oxygen is bonded to two cations (Fig. 1a). The orthorhombic perovskite structure of CaMnO3 is based on a framework of corner-sharing MnO6 octahedra (Fig. 1b). The Mn(IV) cations locate at the center of the MnO6 octahedra while each Ca2+ ion occupies the inter-octahedral sites and is surrounded by 12 oxygen anions.36 The monoclinic layered structure of Ca2Mn3O8 can be described as infinite sheets of Mn3O84− held together by prismatically coordinated Ca2+ ions, which separate the edge-sharing MnO6 octahedral sheet (Fig. 1c).37 Both CaMn3O6 and CaMn2O4 are built up with double chains of edge-sharing MnO6 octahedra (Fig. 1d and e). The corner-linked double chains create the six-sided tunnels for Ca2+ ions to reside in. Compared to the post-spinel phase of CaMn2O4, one third of the Ca position remains vacant in the tunnels of CaMn3O6, which can be viewed as an equivalent notation of Ca2/3Mn2O4.38 The structural diversity of perovskite, layer and post spinel phases of the present Ca–Mn–O series is a great motivation for us to investigate their electrolytic properties.
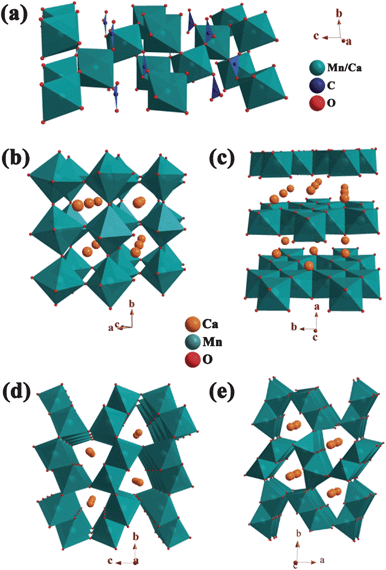 | ||
| Fig. 1 Schematic crystal structures of (a) Ca1−xMnxCO3 calcite, (b) CaMnO3, (c) Ca2Mn3O8, (d) CaMn2O4 and (e) CaMn3O6. | ||
The XRD patterns of the CaxMn1−xCO3 precursors could be readily indexed to a calcite-type structure (ESI†, Fig. S1). Fig. 2 displays the XRD patterns and corresponding Rietveld refinement of the obtained samples after firing the precursors. No peak from other phases was detected, indicating the formation of high-purity oxides. The four oxides could be readily assigned to orthorhombic CaMnO3 (Joint Committee on Powder Diffraction Standards, JCPDS card no. 76-1132), monoclinic Ca2Mn3O8 (no. 73-2290), orthorhombic CaMn2O4 (no. 70-4889) and monoclinic CaMn3O6 (no. 31-0285), respectively. The Rietveld refinement of CaMnO3 gave calculated cell parameters of a = 5.270 Å, b = 7.447 Å and c = 5.287 Å, in good agreement with the standard values. Similarly, the refined results of Ca2Mn3O8, CaMn2O4 and CaMn3O6 matched quite well with the experimental data. The EDS elemental analysis confirmed the corresponding Ca:Mn compositional ratio of each synthesized oxide (ESI†, Fig. S3).
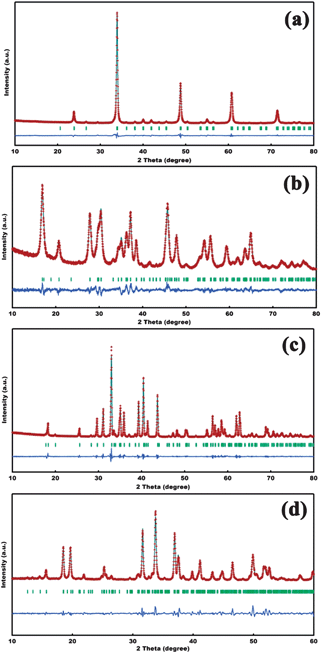 | ||
| Fig. 2 Rietveld refined XRD patterns of the synthesized (a) CaMnO3, (b) Ca2Mn3O8, (c) CaMn2O4 and (d) CaMn3O6, with experimental data (red dots), calculated profile (cyan lines), allowed positions of Bragg reflection (green vertical bars) and difference curves (blue lines). | ||
The morphology and microstructure of the synthesized Ca–Mn–O compounds were analyzed by SEM and TEM imaging. The four samples presented similar shape of hierarchical microspheres, which consisted of aggregated nanoparticles (see typical SEM images in Fig. 3a, d, g and j). All oxide products maintained essential spherical shape of the carbonate precursors, with diameters of 1.0–3.0 μm. Compared to the dense calcite precursors (ESI†, Fig. S4), the product spheres were porous. This was possibly due to two aspects: the phase transition from low-density carbonate to high-density oxides accompanying the release of CO2 from precursor decomposition, and the Ostwald ripening of aggregated particles as previously described in the formation of perovskite microstructures.39 The disparity in surface morphology of obtained oxides could be ascribed to the difference in synthetic conditions. For Ca2Mn3O8, the low calcination temperature and short heating time favored the formation of smaller nanoparticles, which aggregated into loose microspheres. In comparison, the relatively elevated temperature and prolonged time resulted in compact CaMn2O4 microspheres with more smooth surfaces and less porosity.
 | ||
| Fig. 3 SEM and TEM micrographs of synthesized CaMnO3 (a–c), Ca2Mn3O8 (d–f), CaMn2O4 (g–i) and CaMn3O6 (j–l) samples. | ||
The formation of Ca–Mn–O microspheres with aggregated nanoparticles was also clearly revealed by typical TEM observation (Fig. 3b, e, h and k), which showed pores within each sample. The difference in porosity and primary particle sizes among the four hierarchical microspheres coincided with SEM images. To gain structural information, we performed HRTEM imaging on edges of isolated nanoparticles (Fig. 3c, f, i and l). All oxide products indicated high crystallinity with clear distinguished lattice fringes. Taking CaMnO3 as an example, the measured neighboring interlayer distance was consistent with the spacing between the (121), (002), and (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1) planes, confirming the XRD analysis. The well-defined points in the corresponding fast Fourier transform (FFT) diffraction pattern (inset of Fig. 3c) conformed to the orthorhombic crystal structure of perovskite CaMnO3. Likewise, the measured interlayer distances and FFT patterns of Ca2Mn3O8, CaMn2O4 and CaMn3O6 matched the neighboring separations of their related planes and the allowed Bragg diffraction of their phases, respectively.
1) planes, confirming the XRD analysis. The well-defined points in the corresponding fast Fourier transform (FFT) diffraction pattern (inset of Fig. 3c) conformed to the orthorhombic crystal structure of perovskite CaMnO3. Likewise, the measured interlayer distances and FFT patterns of Ca2Mn3O8, CaMn2O4 and CaMn3O6 matched the neighboring separations of their related planes and the allowed Bragg diffraction of their phases, respectively.
Electrocatalytic results
To evaluate the ORR catalytic activities of the Ca–Mn–O microspheres, electrochemical tests were carried out using rotational disk electrodes in 0.1 M KOH solution saturated with Ar or O2 (ESI†, Fig. S5). Compared with the featureless, negligible reduction current in Ar-saturated electrolyte, the detected current at O2 atmosphere was attributed to the catalytic oxygen reduction. The linear sweeping voltammograms (LSVs) of all oxides displayed similar shape, with two regions of potential–current response. Scanning the potential cathodically, the reduction proceeded under mixed kinetic-diffusion controlled regime (0.9–0.6 V), where the detected currents increased rapidly. At potentials below ∼0.6 V, current plateaus were observed, with the appearance of diffusion-limiting currents (id). The reduction currents increased with rising rotation rates as a result of faster oxygen flux to electrode surface. Fig. 4a compares the LSVs of oxide microspheres, GC support, Vc-72 carbon additive and benchmark Pt/C at 1600 round per minute (rpm). Among the synthesized Ca–Mn–O series compounds, the level of catalytic activities follows an order of CaMnO3 > CaMn3O6 > CaMn2O4 ≈ Ca2Mn3O8, based on their onset potentials (ESI†, Fig. S6) and id values. Note that neat carbon obviously contributes to the ORR kinetics, consistent with reported results in alkaline media.40 Since CaMn2O4 and Ca2Mn3O8 do not show significant activity relative to neat carbon, we next focus on the highly active CaMnO3 and CaMn3O6.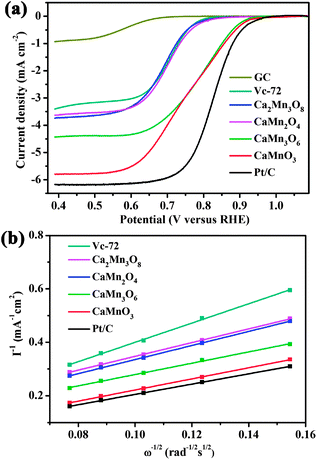 | ||
| Fig. 4 (a) LSVs of CaMnO3, Ca2Mn3O8, CaMn2O4, CaMn3O6, GC, Vc-72 and Pt/C recorded at 1600 rpm in O2-saturated 0.1 M KOH solution. (b) K–L plots of different catalysts at 0.5 V. | ||
The rotational speed-dependent current can be theoretically applied to construct Koutechky–Levich (K–L) plots (i−1versus ω−1/2) according to the K–L equation:
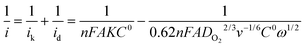 | (1) |
To further analyze the electrocatalytic ORR pathways, the Ca–Mn–O microspheres were further investigated using the rotating ring-disk electrodes (RRDE). Fig. 5a show the polarization curves recorded on the ring and disk. The shape of voltammetry on the disk and the catalytic trend (in terms of both potential and current) were consistent with RDE measurement. Intermediate peroxide species generated from the ORR electrocatalysis on the catalyst disk is detected on the Pt ring. Based on the disk and ring currents (id and ir), the yield of peroxide species (yperoxide, defined as percentage of formed peroxides with respect to the total oxygen reduction products) and the transferred electron number (n) can be determined according to the following equations:
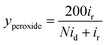 | (2) |
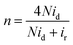 | (3) |
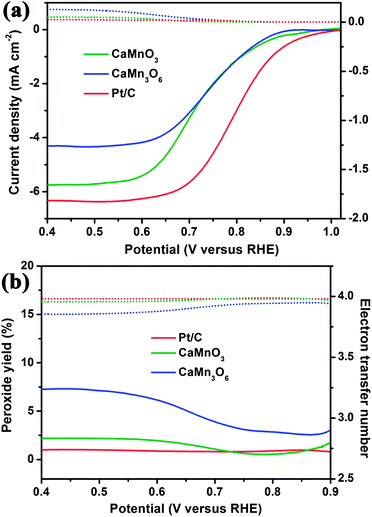 | ||
| Fig. 5 Percentage of peroxide (solid line) and the electron transfer number (dotted line) of CaMnO3, CaMn3O6 and Pt/C at different potentials. | ||
Besides high activity, Ca–Mn–O microspheres also exhibited respectable catalytic stability in alkaline electrolyte. In a continuous polarization period of 60000 s, the ORR currents maintained 93.6% and 93% of the initial values for CaMnO3 and CaMn3O6, respectively (Fig. 6). The gradual decay of reduction current might be partly due to insufficient gas flux, and the slow catalyst peeling off the electrode surface during the prolonged test with applied constant electrode rotation. Irrespective of this possibility, CaMnO3 and CaMn3O6 exhibited slightly better activity retention as compared to the Pt/C benchmark (92.2%). Activity degradation of Pt/C catalyst in alkaline electrolytes has also been observed previously and ascribed to dissolution/aggregation of Pt nanoparticles and corrosion of carbon support.22,34 The morphologies of Ca–Mn–O oxides may provide some merit against particulate agglomeration. Furthermore, the electrochemical durability was also confirmed by elemental analysis that indicated negligible leaching of Ca and Mn cations in the electrolytes after cycles of voltammetry test (ESI†, Fig. S7).
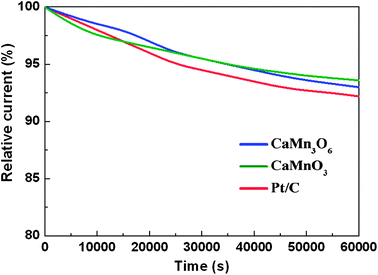 | ||
| Fig. 6 Chronoamperometric curves (percentage of retained current as a function of operation time) of CaMnO3, CaMn3O6, and Pt/C electrodes maintained at 0.8 V versus RHE in O2-saturated electrolyte (0.1 M KOH). | ||
Fig. 7 displays the Tafel plots of CaMnO3, CaMn3O6, and comparative Pt/C in alkaline electrolyte. Kinetic currents derived from the mass-transport correction (ik = (i × id)/(id − i)) were employed to construct the Tafel curves. Two linear regions are observed at low and high overpotentials, respectively. The corresponding slopes are close to −2.303RT/F and −2.303(2RT/F) (i.e., −59 and −118 mV dec−1 at 25 °C). This feature is similar to that previously reported for manganese oxides.15,42 The variation of Tafel slopes indicates a possible change in the ORR reaction mechanism. It has been suggested for copper manganite spinels that at low applied potential the rate determining step (rds) is the conversion of S⋯O2− (S denotes active sites) into an adsorbed peroxide species while at high potential the reduction of the adsorbed O2 to S⋯O2− is rate-limiting.42 The presence of two slopes could be also interpreted by structural effects of the catalyst layer or potential-dependent coverage of adsorbed species on catalyst surface.40 Further elucidation of the underlying mechanism is still required.
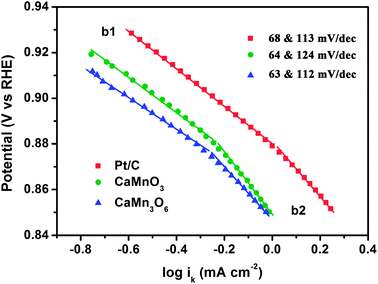 | ||
| Fig. 7 Tafel plots of CaMnO3, CaMn3O6, and Pt/C derived by the mass-transport correction of voltammetry data. | ||
Discussion
The main electrocatalytic characteristics of the Ca–Mn–O series compounds are summarized in Table 1. Comparing the catalytic properties, the four oxides can be divided in two groups of (1) CaMnO3 and CaMn3O6 and (2) Ca2Mn3O8 and CaMn2O4. The first group showed more positive onset and half-wave potentials, larger currents, and higher transferred electron numbers relative to the second group. Remarkably, CaMnO3 was highly active, manifesting electrochemical parameters (apart from lower potentials) close to those of the benchmark Pt/C. Also, CaMn3O6 demonstrated comparable activities to CaMnO3 despite lower reduction current. In contrast, Ca2Mn3O8 and CaMn2O4 exhibited inferior performance. The rising question is what is responsible for the different activities of the Ca–Mn–O series. In regard to the heterogeneous ORR electrocatalysis on transition metal oxides, the catalytic mechanism involves oxygen adsorption, dissociation and reduction on catalyst surface, which is closely correlated with redox reactions of transition metal-containing species.5,14–22 As a result, it is reasonable to hypothesize that chemical composition, elemental valence, crystal structure, and surface state are possible factors determining the catalytic activities.| Catalyst | E onset (V) | E half (V) | I s (mA cm−2) | I m (mA mg−1) | n |
|---|---|---|---|---|---|
| a E onset, Ehalf, Is, Im, and n denote onset potential, half-wave potential, specific current, mass current density and electron transfer number, respectively. Is, Im, and n correspond to values determined at 0.5 V. | |||||
| CaMnO3 | 0.96 | 0.76 | −5.79 | 62.7 | 3.96 |
| Ca2Mn3O8 | 0.85 | 0.70 | −3.54 | 38.3 | 3.53 |
| CaMn2O4 | 0.85 | 0.69 | −3.71 | 40.2 | 3.50 |
| CaMn3O6 | 0.95 | 0.78 | −4.43 | 48.0 | 3.86 |
| Pt/C | 0.99 | 0.84 | −6.17 | 66.8 | 3.98 |
For Ca–Mn–O series oxides, Mn possesses variable valences and thereby more likely acts as the active center. The nominal distribution of valence is Mn(IV), Mn(IV), Mn(III), and 1/3Mn(IV)2/3Mn(III) for CaMnO3, Ca2Mn3O8, CaMn2O4, and CaMn3O6, respectively. The difference in catalytic activities of the series compounds cannot be directly attributed to bulk composition and Mn valence. We performed XPS analysis to characterize the surface state of the oxides as it is a critical factor. The core-level XPS data are shown in Fig. 8 and Table 2. All the Mn 2p spectra (Fig. 8a) displayed two strong peaks centered at about 642 and 653 eV, which were attributed to Mn 2p3/2 and Mn 2p1/2 spin–orbit doublet, respectively. These broad peaks were asymmetrical towards higher binding energy side, agreeing with early studies.43,44 The peak position of Mn 2p3/2 moved from 641.5 eV in CaMn2O4 to 642.0 eV in Ca2Mn3O8. This shift was ascribed to changes in the electrostatic energy at the Mn site, which was driven by the decrease in the 3d count.44
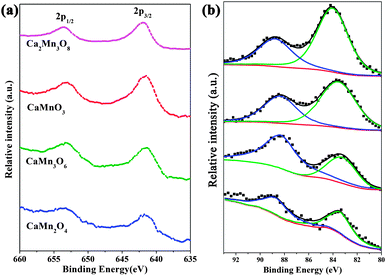 | ||
| Fig. 8 (a) Mn 2p and (b) Mn 3s XPS spectra of synthesized CaMnO3, Ca2Mn3O8, CaMn2O4 and CaMn3O6 microspheres. | ||
| Compound | Mn 2p3/2 (eV) | Mn 3s (eV) | Average oxidation state | ||
|---|---|---|---|---|---|
| Peak 1 | Peak 2 | ΔeV | |||
| a Data of α-Mn2O3, γ-Mn2O3 and β-MnO2 are adapted from ref. 46–48 and listed for comparison. The parenthesis data correspond to the values determined by chemical titration. | |||||
| CaMn2O4 | 641.5 | 88.57 | 83.23 | 5.34 | 2.9 (2.84) |
| CaMn3O6 | 641.6 | 88.30 | 83.25 | 5.05 | 3.4 (3.47) |
| CaMnO3 | 641.7 | 88.45 | 83.60 | 4.85 | 3.8 (3.81) |
| Ca2Mn3O8 | 642.0 | 88.77 | 84.03 | 4.74 | 4.0 (4.00) |
| α-Mn2O3 | 641.9 | 89.1 | 83.9 | 5.2 | 3.0 |
| γ-Mn2O3 | 641.7 | 88.8 | 83.6 | 5.2 | 3.0 |
| β-MnO2 | 642.2 | 89.4 | 84.7 | 4.7 | 4.0 |
Although the chemical shift of Mn 2p qualitatively reflected the oxidation state of Mn,45 the magnitude was quite small to quantitatively deduce the valence. Fortunately, the splitting width of Mn 3s peaks (Fig. 8b) was found to be different among these four manganese oxides. The separation of peak energies (ΔEb) of the Mn 3s increased from ∼4.7 eV for Ca2Mn3O8 to ∼5.3 eV for CaMn2O4, which were close to those expected for Mn(IV) and Mn(III), respectively.46–48 Moreover, there was a linear relationship between the energy separation of 3s splitting and the valence of Mn in the oxides.46–49 Based on this relationship, the mean Mn oxidation states were estimated to be 4.0, 3.8, 3.4 and 2.9 for Ca2Mn3O8, CaMnO3, CaMn3O6 and CaMn2O4, respectively. These surface Mn valences were essentially consistent with but slightly lower than that of the expected values (except Ca2Mn3O8). The lower oxidation state of Mn might originate from the presence of oxygen defects in the oxides synthesized at high calcination temperatures. Chemical analysis of the samples confirmed the slight compositional deviation from the stoichiometry and was consistent with the XPS evaluation (Table 2). Accordingly, the highly catalytically active Ca–Mn–O compounds (CaMnO3 and CaMn3O6) possessed average surface Mn valence between +3 and +4. The pronounced inferior electrocatalytic performance of CaMn2O4 could be attributed to its low reducibility.
Recently, a variety of composite transition metal oxides have been investigated as catalysts for the oxygen electrochemistry.24,50 It has been demonstrated that the intrinsic ORR activity of an oxide is strongly correlated to σ*-antibonding orbital occupancy and metal–oxygen covalency of surface transition-metal cations.24 For Mn-based oxides, the most active catalyst generally has an average valence of Mn(III)–Mn(IV), which affords moderate bond strength between the catalyst surface and the reactant or product. Within this valence range, the compound can be viewed as mixed-valent manganese oxides. Due to the coexistence of multivalent Mn, cation redox reaction, surface charge storage as well as electronic and ionic properties are feasible to be enhanced.51 Additionally, our results suggest that the ORR activity increases with higher Mn oxidation state in the Ca–Mn–O oxides. Previous in situ XANES (X-ray absorption near edge structure) study has also revealed that higher content of Mn(IV) phase in MnOx species was associated with better catalytic performance.52 Unlike noble metal catalysts, transition metal oxides mediate the ORR electrocatalysis through surface redox reactions concomitant with electron transfer to oxygen.5,14–22,24,50 Manganese oxides featured with multiple and high valence may favor oxygen activation and therefore allow better ORR kinetics.
Next, we consider the effect of crystallographic structure. In the layered structure of Ca2Mn3O8 (Fig. 1c), the edge and corner-sharing MnO6 octahedra sheet (like in birnessite-MnO2) is much denser than that of other compounds, unfavourable for oxygen adsorption. The perovskite CaMnO3, built upon corner-sharing MnO6 octahedra (Fig. 1b), provides (1 × 1) tunnels with comparable size to the O–O bond length (∼0.12 nm) in molecular oxygen.53 Hence, oxygen could be easily accommodated, favouring the cleavage of O–O bond and benefiting the ORR. This speculation explains the positive potential, large current, and high apparent electron transfer number of CaMnO3. For CaMn3O6 and CaMn2O4, the open structures are advantageous to oxygen access, however, this benefit was negated by the large tunnel size. Their activities thus fell between that of Ca2Mn3O8 and CaMnO3. Furthermore, our preliminary density functional theory (DFT) studies indicate that the series Ca–Mn–O compounds lead to different configuration of surface oxygen adsorption and the corresponding strength of oxygen binding coincides with the order of their catalytic activities (ESI†, Fig. S8).
We have also investigated the catalytic activity of the series Ca–Mn–O compounds for the OER (ESI†, Fig. S9). Again, the mixed-valence composite oxides showed superior performance. Interestingly, CaMn3O6 clearly outperformed the other three oxides and the comparative Pt/C, resulting in lower overpotential and higher OER current. In regard to the oxygen electrochemistry on transition metal oxides, the ORR and OER may proceed via identical cycles but through opposite position.24,50 The order of ORR and OER activities for Ca–Mn–O compounds should present dissimilar trends due to a different rds. For multivalent Ca–Mn–O oxides, lower Mn oxidation state leads to higher OER activity, which is consistent with previous reports.54 Regardless of the difference in OER/OER characteristics, CaMnO3 and CaMn3O6 can serve as non-precious bifunctional catalysts because of their prominent performance.
Lastly, the role of Ca in Ca–Mn–O composite oxides deserves to be noted. Our primary electrochemical results revealed that CaMnO3 exhibited more positive reduction potential and larger current than did MnO2, which had similar Mn oxidation state and open tunnel structure (ESI†, Fig. S10). We envisage that Ca plays an important role for the ORR electrocatalysis, although Ca is generally redox inactive. In the PSII OEC center, Ca is identified as an essential cofactor for the OER, probably as a site for the binding and activation of oxygen-containing species.31,55 It is speculated that the incorporation of Ca in manganese oxides may affect behaviours of oxygen adsorption, activation and reduction. Furthermore, calcium cations feasibly stabilize the crystal structure with high-valence Mn, due to its flexible coordination with oxygen in oxide lattice frameworks. This can be validated by the fact that, in the absence of Ca only Mn(II)/Mn(III)-based binary oxides are thermodynamically stable at high temperatures,56 while Ca–Mn–O oxides show chemical and structural robustness in solvothermal systems containing high concentration of KOH.30,57 In contrast to binary manganese oxides that undergo dramatic changes in crystalline and chemical structure upon premature ageing,17 CaMnO3 and CaMn3O6 exhibit remarkable stability in alkaline solution. Therefore, the catalytic durability of Ca–Mn–O microspheres is not surprising.
Conclusions
In summary, a series of porous Ca–Mn–O microspheres were synthesized by calcinating the solid–solution carbonate precursors and systematically investigated as electrocatalysts for the ORR. Catalytic properties of the Ca–Mn–O series compounds were found to be closely correlated with the surface oxidation state of Mn and the crystallographic structures that affects the extent of O2 activation. The perovskite CaMnO3 with open tunnels and multivalence exhibited the highest activities. The current density and electron transfer number were comparable to those of the benchmark Pt/C. Furthermore, CaMn3O6 and CaMnO3 manifested superior OER performance and catalytic durability. The results may shed light on searching for efficient, cheap, and scalable transition-metal-oxide oxygen electrocatalysts that are widely used in metal–air batteries and alkaline fuel cells.Acknowledgements
This work was supported by the National 973 (Grant no. 2011CB935902), 863 (Grant no. 2011AA050704), NSFC (Grant no. 21231005), 111 Project (Grant no. B12015), and Tianjin High-Tech (Grant no. 10SYSYJC27600).References
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS.
- L. Malavasi, C. A. J. Fisher and M. S. Islam, Chem. Soc. Rev., 2010, 39, 4370 RSC.
- P. A. Christensen, A. Hamnett and D. Linares-Moya, Phys. Chem. Chem. Phys., 2011, 13, 5206 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Chem. Soc. Rev., 2010, 39, 2184 RSC.
- D. F. Yancey, L. Zhang, R. M. Crooks and G. Henkelman, Chem. Sci., 2012, 3, 1033 RSC.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS.
- B. Šljukić, C. E. Banks, S. Mentus and R. G. Compton, Phys. Chem. Chem. Phys., 2004, 6, 992 RSC.
- Y. J. Feng and N. Alonso-Vante, Phys. Status Solidi B, 2008, 245, 1792 CrossRef CAS.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238 CAS.
- Z. W. Chen, D. Higgins, A. Yu, L. Zhang and J. J. Zhang, Energy Environ. Sci., 2011, 4, 3167 CAS.
- M. S. El-Deab and T. Ohsaka, Angew. Chem., Int. Ed., 2006, 45, 5963 CrossRef CAS.
- I. Roche, E. Chaînet, M. Chatenet and J. Vondrák, J. Phys. Chem. C, 2007, 111, 1434 CAS.
- A. Debart, A. J. Paterson, J. Bao and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 4521 CrossRef CAS.
- I. Roche, E. Chaînet, M. Chatenet and J. Vondrák, J. Appl. Electrochem., 2008, 38, 1195 CrossRef CAS.
- F. Y. Cheng, Y. Su, J. Liang, Z. L. Tao and J. Chen, Chem. Mater., 2010, 22, 898 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS.
- E. M. Benbow, S. P. Kelly, L. Zhao, J. W. Reutenauer and S. L. Suib, J. Phys. Chem. C, 2011, 115, 22009 CAS.
- F. Y. Cheng, J. Shen, B. Peng, Y. D. Pan, Z. L. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 3517 CrossRef CAS.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546 CrossRef CAS.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55 CrossRef CAS.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444 CrossRef CAS.
- I. Saratovsky, P. G. Wightman, P. A. Pastén, J. F. Gaillard and K. R. Poeppelmeier, J. Am. Chem. Soc., 2006, 128, 11188 CrossRef CAS.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467 CrossRef CAS.
- F. Jiao and H. Frei, Chem. Commun., 2010, 46, 2920 RSC.
- M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233 CrossRef CAS.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330 RSC.
- H. S. Horowitz and J. M. Longo, Inorg. Synth., 1983, 22, 73 CrossRef CAS.
- J. Zhu, Y. Su, F. Y. Cheng and J. Chen, J. Power Sources, 2007, 166, 331 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS.
- B. Fubini and F. S. Stone, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 1215 RSC.
- W. Paszkowicz, J. Pietosa, S. M. Woodley, P. A. Dluzewski, M. Kozlowski and C. Martin, Powder Diffr., 2010, 25, 46 CrossRef CAS.
- G. B. Ansell, M. A. Modrick, J. M. Longo, K. R. Poeppeimeler and H. S. Horowitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1795 CrossRef.
- J. Hadermann, A. M. Abakumov, L. J. Gillie, C. Martin and M. Hervieu, Chem. Mater., 2006, 18, 5530 CrossRef CAS.
- X. Yang, J. Fu, C. Jin, J. Chen, C. Liang, M. Wu and W. Zhou, J. Am. Chem. Soc., 2010, 132, 14279 CrossRef CAS.
- J. Perez, E. R. Gonzalez and E. A. Ticianelli, Electrochim. Acta, 1998, 44, 1329 CrossRef CAS.
- A. J. F. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2000 Search PubMed.
- E. Ríos, S. Abarca, P. Daccarett, H. N. Gong, D. Martel, J. F. Marco, J. R. Cancedo and J. L. Gautier, Int. J. Hydrogen Energy, 2008, 33, 4954 CrossRef.
- M. E. Melo Jorge, A. Correia dos Santos and M. R. Nunes, Int. J. Inorg. Mater., 2001, 3, 915 CrossRef.
- M. Abbate, F. M. F. de Groot, J. C. Fuggle, A. Fujimori, O. Strebel, F. Lopez, M. Domke, G. Kaindl, G. A. Sawatzky, M. Takano, Y. Takeda, H. Eisaki and S. Uchida, Phys. Rev. B: Condens. Matter, 1992, 46, 4511 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184 CrossRef CAS.
- M. Oku, K. Hirokawa and S. Ikeda, J. Electron Spectrosc. Relat. Phenom., 1975, 7, 465 CrossRef CAS.
- V. R. Galakhov, M. Demeter, S. Bartkowski, M. Neumann, N. A. Ovechkina, E. Z. Kurmaev, N. I. Lobachevskaya, Y. M. Mukovskii, J. Mitchell and D. L. Ederer, Phys. Rev. B: Condens. Matter, 2002, 65, 113102 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS.
- M. K. Song, S. Cheng, H. Chen, W. Qin, K. W. Nam, S. Xu, X. Q. Yang, A. Bongiorno, J. Lee, J. Bai, T. A. Tyson, J. Cho and M. L. Liu, Nano Lett., 2012, 12, 3483 CrossRef CAS.
- F. H. B. Lima, M. L. Calegaro and E. A. Ticianelli, J. Electroanal. Chem., 2006, 590, 152 CrossRef CAS.
- Y. Wang and P. B. Balbuena, J. Phys. Chem. B, 2004, 108, 4376 CrossRef CAS.
- M. M. Najafpour, Dalton Trans., 2011, 40, 3793 RSC.
- C. F. Yocum, Coord. Chem. Rev., 2008, 252, 296 CrossRef CAS.
- A. N. Grundy, B. Hallstedt and L. J. Gauckler, J. Phase Equilib., 2003, 24, 21 CAS.
- J. Spooren and R. I. Walton, J. Solid State Chem., 2005, 178, 1683 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization, electrochemical data, and computational details. See DOI: 10.1039/c2sc21475j |
| This journal is © The Royal Society of Chemistry 2013 |