Tridentate cobalt complexes as alternative redox couples for high-efficiency dye-sensitized solar cells†
Kais Ben
Aribia
,
Thomas
Moehl
,
Shaik M.
Zakeeruddin
* and
Michael
Grätzel
*
Laboratory for Photonics and Interfaces, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: shaik.zakeer@epfl.ch; michael.graetzel@epfl.ch; Fax: +41 (0)21 693 61 00; Tel: +41 (0)21 693 31 12
First published on 15th October 2012
Abstract
Cobalt terpyridine complexes could rival the classical triiodide/iodide redox couple as efficient alternative redox couples for dye-sensitized solar cells. Cobalt bis(2,2′,6′,2′′-terpyridine) complexes enable systematic tuning of the redox potential by variation of the substituents on the terpyridine ligand to optimize the open circuit voltage and the overall photovoltaic performance of the device. A [CoII(Cl-terpy)2](TFSI)2/[CoIII(Cl-terpy)2](TFSI)3 based electrolyte in combination with the Y123 donor–π–acceptor dye yielded a solar to electric power conversion efficiency (PCE) of 8.7% under standard air mass 1.5 global (AM 1.5G) simulated sunlight. Transient photocurrent and photovoltage decay as well as electrochemical impedance spectroscopy was used to rationalize the differences in the photovoltaic parameters of devices with iodide/triiodide and cobalt complex based redox electrolytes.
Introduction
Since the original publication from O'Regan and Grätzel1 work on the dye-sensitized solar cells (DSCs) has been highly active to understand and optimize each component of the DSC to enhance the power conversion efficiency (PCE). The redox electrolyte is a unique feature and a main component of such solar cells. The redox potential of the electrolyte determines, in conjunction with the conduction band position of the mesoporous metal oxide, the maximum achievable open circuit voltage (VOC). The iodide/triiodide couple is the most investigated redox system for DSCs. Devices based on the iodide/triiodide redox electrolyte possess intrinsically relative low open circuit voltages around 700 mV, due to the low-lying redox potential of the electrolyte. A large energy difference between the dye HOMO level and the redox energy level of the electrolyte is the reason for this low VOC. For efficient dye regeneration, 200 mV driving force is sufficient, and implies a loss of 0.5 V in the open circuit voltage. In recent years there has been an increase in the research activity on alternative redox electrolytes that have redox potential values higher than the commonly used iodine/triiodide redox system. New redox systems with a higher redox potential used in DSCs include e.g. ferrocene,2 organic radicals3 and thiols.4 Amongst these different redox shuttles, cobalt complexes with bidentate ligands (2,2-bipyridine or 1,10-phenanthroline) have shown outstanding performances, superseding in several cases the power conversion efficiencies reached by their iodine-based counterparts.5–19One of the advantages of cobalt-based redox electrolytes is the lower absorption in the visible range compared to the triiodide species,20,21 and that they show no corrosive behaviour towards metal contacts, which is important for durability of DSC devices for commercial applications.22 Another attractive feature of cobalt complexes is the facile tuning of their redox potential by varying the ligands with different electron withdrawing or donating groups. Depending on the HOMO level of the dye, the redox potential of the cobalt electrolyte can be tuned to the required range, in order to have sufficient driving force for the dye regeneration and to enable maximum open circuit voltages. Numerous cobalt complex-based electrolytes have been reported with bidentate and tridentate polypyridyl complexes with a wide range of redox potentials and were successfully applied in DSCs to obtain higher VOC and PCEs. Recently, using a [CoII/III(2,2′-bipyridine)2](B(CN)4)-based redox electrolyte in conjunction with a donor–π–acceptor zinc porphyrin co-sensitized with Y123 dye, a PCE of 12.3% was obtained.23
In this study we prepared two tridentate cobalt complexes, [CoII/III(terpy)2](B(CN)4)2/3 and [CoII/III(Cl-terpy)2](TFSI)2/3 (TFSI = bis(trifluoromethanesulfonyl)imide) for redox electrolytes and compared the photovoltaic performance with an iodide/triiodide based electrolyte. The origin of the increase in VOC for the devices with cobalt redox electrolytes is explained by transient photocurrent and photovoltage decay and electrochemical impedance spectroscopy measurements.
Experimental
Composition of the redox electrolytes
Two one-electron transfer redox couple systems with different redox potential were used (see Fig. 1): [CoII(terpy)2](B(CN)4)2/[CoIII(terpy)2](B(CN)4)3 and [CoII(Cl-terpy)2](TFSI)2/[CoIII(Cl-terpy)2](TFSI)3. The synthesis of these complexes is explained in detail in the ESI.† The composition of the cobalt-based electrolytes was optimized by variation of the concentrations of the Co2+ and Co3+ complexes. The concentration of the additives (0.1 M Li+ and 0.2 M tert-butylpyridine) was kept constant. All the electrolytes were freshly prepared before fabricating the devices and the compositions are listed in Table 1.![Structure of [CoII/III(terpy)2] (left) and [CoII/III(Cl-terpy)2] (right).](/image/article/2013/SC/c2sc21401f/c2sc21401f-f1.gif) | ||
| Fig. 1 Structure of [CoII/III(terpy)2] (left) and [CoII/III(Cl-terpy)2] (right). | ||
| Electrolyte | Complex | [Co3+]/M | [Co2+]/M | [Li+]/M | [TBP]/M | Redox potential/mV |
|---|---|---|---|---|---|---|
| E1 | [CoII/III(terpy)2](B(CN)4)2/3 | 0.02 | 0.2 | 0.1 | 0.2 | 441 |
| E2 | [CoII/III(terpy)2](B(CN)4)2/3 | 0.04 | 0.2 | 0.1 | 0.2 | 459 |
| E3 | [CoII/III(terpy)2](B(CN)4)2/3 | 0.07 | 0.2 | 0.1 | 0.2 | 473 |
| E4 | [CoII/III(terpy)2](B(CN)4)2/3 | 0.10 | 0.2 | 0.1 | 0.2 | 482 |
| E7 | [CoII/III(Cl-terpy)2](TFSI)2/3 | 0.07 | 0.2 | 0.1 | 0.2 | 603 |
| E17 | [CoII/III(terpy)2](B(CN)4)2/3 | 0.07 | 0.1 | 0.1 | 0.2 | 491 |
| E18 | [CoII/III(terpy)2](B(CN)4)2/3 | 0.07 | 0.3 | 0.1 | 0.2 | 463 |
Redox potentials of the cobalt complexes and diffusion coefficients in the electrolyte
The redox potentials of the above two cobalt complexes were measured in a glove-box under an argon atmosphere using thermally deposited Pt on FTO as a working electrode and two Pt wires as counter and reference electrode, respectively. 0.1 M TBAPF6 (tetrabutylammonium hexafluorophosphate) in acetonitrile was used as a supporting electrolyte. Cyclic voltammetry (CV) experiments were performed at a scan rate of 5 mV s−1. The redox potential was determined using the ferrocene–ferrocenium redox couple (E0(Fc/Fc+) = 0.63 V vs. NHE) as an internal calibration.The diffusion coefficient of Co3+ was determined by CV using an electrochemical cell consisting of two identical platinized FTO conducting glasses (TEC7, Pilkington). The FTO glass was platinized by thermal deposition from a 3 mM H2PtCl6 solution in isopropyl alcohol with heating at 410 °C for 15 min. The two electrodes were sealed with a square shape Surlyn frame (70 μm thick) having an inner area of 36 mm2. Electrolytes were introduced into these sealed devices by vacuum filling through the back hole made by a sandblast machine. The hole was then closed with a Bynel disk and a thin glass to avoid leakage of the electrolyte. CVs were performed between −0.9 and +0.9 V with a scan speed of 5 mV s−1. DCo3+ was calculated by using the equation:16
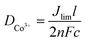
Device assembly
State-of-the art double layer mesoporous TiO2 films (4.μm; 20 nm particles (DSL 18NR-T, DYESOL) plus 4.5 μm; 400 nm light scattering particles (HPW-400NRD, CCIC)) were screen printed onto FTO conducting glass (Solar-4 mm, Nippon Sheet Glass Co, Ltd.) and heated, following our reported procedure.24 The TiO2 films (area: 0.283 cm2) were sensitized by immersing them into a solution of the Y123 dye (0.1 mM) in tert-butyl alcohol–acetonitrile (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) for 16 h at room temperature. The composition of the electrolytes is shown in Table 1. A platinized FTO conducting glass (TEC7, Pilkington) was used as counter electrode. The devices were sealed as for the electrochemical cells for the DCo3+ determination but using a 25 μm Syrlin. An antireflection film (ARCTOP, Mihama Co.) was attached on top of the photoanode side.
:50 v/v) for 16 h at room temperature. The composition of the electrolytes is shown in Table 1. A platinized FTO conducting glass (TEC7, Pilkington) was used as counter electrode. The devices were sealed as for the electrochemical cells for the DCo3+ determination but using a 25 μm Syrlin. An antireflection film (ARCTOP, Mihama Co.) was attached on top of the photoanode side.
Photovoltaic characterization
Photovoltaic measurements employed a 450 W xenon light source (Osram XBO 450). The incident light intensity was regulated to the AM 1.5G solar standard by using a reference Si photodiode equipped with a colour-matched filter (KG-3, Schott) to reduce the mismatch in the region of 350–750 nm between the simulated light and AM 1.5G to less than 4%. The incident photon to current conversion efficiency (IPCE) was measured as a function of wavelength by using a 300-W xenon lamp (ILC Technology), which was focused through a Gemini-180 double monochromator (Jobin Yvon) onto the photovoltaic cell under test. A computer-controlled monochromator was incremented through the spectral range (300–900 nm) to generate a photocurrent action spectrum with a sampling interval of 10 nm and a current sampling time of 4 s. The active area of the devices for these measurements is 0.16 cm2, defined by a black tape mask for excluding any diffused light.Transient photovoltage/photocurrent and EIS measurements
The transient photovoltage measurement setup is similar to the description by O'Regan et al.25,26 The cell was illuminated under open-circuit conditions with a white light bias at various light intensities, and a pulse from a red LED is used to slightly increase the electron concentration in the oxide generating a small increment in the open circuit voltage. After the pulse a photovoltage decay is registered and fitted by a monoexponential decay:| V(t) = V0 + Aexp(−t/τe) |
EIS measurements were performed by an Autolab PGSTAT30 (EcoChemie B.V) in the frequency range between 1 MHz and 0.1 Hz for forward-bias potentials between 0 and 1.1 V (with a 10 mV sinusoidal ac perturbation) in 50 mV steps. The resulting impedance spectra were analysed with the ZView software (Scribner Associate) on the basis of a two-channel transmission line model.27,28 Especially the latter publication of Fabregat et al. in 2011 shows the validity of the transmission line model applied in conjunction with the other circuit elements (counter electrode RC element, the Warburg diffusion element and the RC element for the interface of the photoanode TCO (or underlayer)/electrolyte interface as well as the series resistance accounting for e.g. cable connections or the resistivity of the substrate). In case an ideal capacitor did not lead to sufficient fitting results it was substituted by a CPE (Constant Phase Element) but keeping the exponent between 0.8 and 1.0.
Results and discussion
The UV-visible absorption spectra of the reduced [CoII(terpy)2](B(CN)4)2 and the oxidized [CoIII(terpy)2](B(CN)4)3 show that the absorption maxima at 440 nm have molar extinction coefficients of 966 and 503 L Mol−1 cm−1, respectively. The molar extinction coefficients for [CoII(Cl-terpy)2](TFSI)2 and [CoIII(Cl-terpy)2](TFSI)3 are 849 and 395 L Mol−1 cm−1, respectively (Fig. S2, ESI†). Due to the lower molar extinction coefficient of the cobalt complexes in the visible region compared to the triiodide ion (ε = 2145 L Mol−1 cm−1, 440 nm), it is advantageous to use cobalt-based redox systems as electrolytes, especially for DSC designs with illumination through the counter electrode side.As mentioned, the iodide/triiodide based redox system has a low redox potential (E0(I3−/I−) = 0.35 V in acetonitrile) which can be only slightly modified. However, in the case of the cobalt complex based electrolytes, depending on the basicity of the coordinating ligand to the metal centre, the redox potential can be easily tuned. One can introduce electron withdrawing or donating substituents on the organic ligands to vary the redox potential to match with our experimental requirements. The redox potential of [CoII(terpy)2](B(CN)4)2 and [CoII(Cl-terpy)2](TFSI)2 measured by CV are 500 and 630 mV vs. NHE, respectively. The redox potential of the [CoII(Cl-terpy)2](TFSI)2 complex is 130 mV more positive than the [CoII(terpy)2](B(CN)4)2 complex. Substitution of the central pyridine proton of the terpyridine ligand with chloride, which is an electron-withdrawing group, leads to an increase in the redox potential of the cobalt complex.
The DCo3+ values of the cobalt complexes used in this study are in the order of 10−6 cm2 s−1, which is in agreement with the reported values (Table S1, ESI†).13,15 The diffusion coefficient for the CoIII(terpy)2 complex is almost double (∼6 × 10−6 cm2 s−1) compared to the value of the CoIII(Cl-terpy)2 complex (∼3 × 10−6 cm2 s−1). Previous investigations show that the diffusion coefficients of cobalt polypyridyl complexes are in general lower than that of a triiodide ion (∼2 × 10−5 cm2 s−1) due to their bulkier size. This could lead to mass transport problems because of the diffusion limitations of the electrolyte at higher illumination intensities.
In DSCs the open circuit voltage is determined by the difference between the Fermi level of the semiconductor and the redox potential of the electrolyte. Increasing the concentration of the oxidized species in the electrolyte will increase the redox energy of the redox electrolyte and should subsequently increase the VOC. However, increasing the concentration of the oxidized form of the redox couple in the electrolyte also increases the dark current, which would lead to a decrease in VOC. The higher concentration of Co3+ in the electrolyte will also influence the FF due to the lower diffusion and charge transfer resistance. Using the [CoII/III(terpy)2](B(CN)4)2/3 salts six electrolytes were formulated (see Table 1) and used in DSCs. In the E1–E4 series the concentration of Co2+, TBP and LiClO4 are kept constant, varying only that of Co3+. The photovoltaic parameters of the devices fabricated with electrolytes E1–E4 are listed in Table 2. Increasing the Co3+ concentration decreases the VOC due to the enhanced dark current while the FF and the JSC are increased.
| Electrolyte | Light intensity (mW cm−2) | V OC (mV) | J SC (mA cm−2) | FF | PCE (%) |
|---|---|---|---|---|---|
| E1 | 10 | 828 | 1.28 | 0.71 | 8.0 |
| 50 | 877 | 6.80 | 0.68 | 8.0 | |
| 100 | 893 | 13.04 | 0.64 | 7.4 | |
| E2 | 10 | 811 | 1.32 | 0.71 | 8.2 |
| 50 | 868 | 7.04 | 0.70 | 8.4 | |
| 100 | 886 | 13.68 | 0.67 | 8.2 | |
| E3 | 10 | 761 | 1.40 | 0.74 | 8.4 |
| 50 | 840 | 7.46 | 0.71 | 8.7 | |
| 100 | 866 | 13.92 | 0.70 | 8.4 | |
| E4 | 10 | 760 | 1.35 | 0.74 | 8.1 |
| 50 | 845 | 7.22 | 0.71 | 8.5 | |
| 100 | 872 | 13.52 | 0.69 | 8.1 | |
| E7 | 10 | 812 | 1.33 | 0.73 | 8.5 |
| 50 | 895 | 7.16 | 0.71 | 8.9 | |
| 100 | 922 | 13.7 | 0.68 | 8.7 | |
| E17 | 10 | 775 | 1.34 | 0.73 | 8.0 |
| 50 | 844 | 7.10 | 0.73 | 8.6 | |
| 100 | 864 | 13.53 | 0.71 | 8.4 | |
| E18 | 10 | 736 | 1.36 | 0.75 | 8.0 |
| 50 | 820 | 7.34 | 0.71 | 8.4 | |
| 100 | 850 | 14.31 | 0.69 | 8.4 | |
| I−/I3− | 9 | 673 | 1.22 | 0.69 | 6.4 |
| 50 | 734 | 6.71 | 0.66 | 6.4 | |
| 100 | 753 | 13.10 | 0.65 | 6.4 |
The highest power conversion efficiency (PCE) of 8.4% was reached with the E3 electrolyte under an illumination of AM 1.5G. The incident photon-to-current conversion efficiency (IPCE) of the devices exhibit high values (80–90%) over the range 400–580 nm (Fig. 2, inset). The JSC value obtained from integrating the product of the IPCE spectrum with the AM 1.5G spectral solar photon flux was within 4% of the measured JSC value.
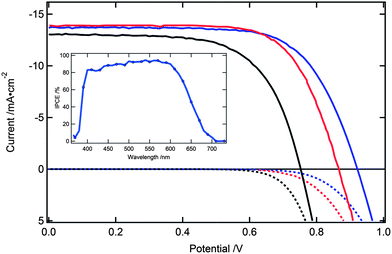 | ||
| Fig. 2 Photocurrent (solid) and dark current (dotted) of DSCs with E3 (red), E7 (blue) and iodine (black) based electrolyte. Inset shows the IPCE spectra of a DSC employing electrolyte E7. | ||
To optimize the concentration of the Co2+ species in the electrolyte, two more electrolytes were tested by varying the Co2+ concentration, i.e. E17 and E18, maintaining the E3 formulation for the other components. From the Nernst equation, increasing the concentration of Co2+ in the electrolyte lowers the redox potential of the electrolyte (Table 1). This results in a lower VOC value for the device containing E18 electrolyte (see Table 2). At the same time a higher concentration of Co2+ in the electrolyte increases the dye regeneration rate so increasing the JSC values (JSC: E17 < E3 < E18. This study shows that for the maximum PCE the optimum concentration of Co2+ is 0.2 M. We selected the optimized E3 electrolyte composition as a benchmark for formulating the electrolyte containing the [CoII/III(Cl-terpy)2] complex (E7). The composition of the E7 electrolyte contains 0.2 M Co2+, 0.07 M Co3+, 0.1 M LiTFSI and 0.2 M TBP in acetonitrile solvent. The photovoltaic characteristics for E7 in Table 2 show similar JSC and FF values as for E3, while the VOC is 56 mV higher, resulting in an overall PCE of the device of 8.7%.
If the redox potential of a redox couple is too high, then the HOMO of the dye cannot be sufficiently regenerated by the reduced form of the cobalt complex and the JSC will be low.19,29,30 Since similar JSC values were obtained with the E3 and E7 electrolytes, this indicates that changing the cobalt redox couple in the electrolyte did not influence on the dye regeneration kinetics.
The increase in VOC is smaller than expected since the redox potential of E7 is 130 mV higher than that of E3. This discrepancy could originate from a shift in the conduction band position of the TiO2 or a change in the recombination rate. EIS analysis was performed to unravel the contribution of the two factors. Values for the main circuit elements, i.e. the charge transfer and transport resistance as well as the chemical capacitance extracted from the measurements are presented in ESI (Fig. S3†). The applied voltage as a function of the density of states (DOS) is plotted in Fig. 3. The DOS is calculated from the chemical capacitance by the formula: DOS = 6.24 × 1018 × C/d(1 − p) where p represents porosity of the TiO2 film, d its thickness and C the chemical capacitance. To reach the same DOS, E7 requires a ca. 110 mV higher voltage than E3, which is close to the difference of the redox potentials of 130 mV between the two electrolytes. Thus there is only a minor difference in the energy level of TiO2 conduction band edge between the E7 and E3 electrolytes. This implies that faster electron recombination with the E7 electrolyte is primarily responsible for the lower than expected VOC.
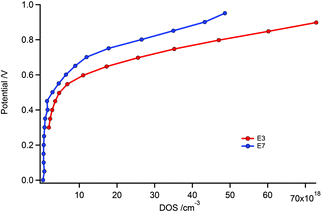 | ||
| Fig. 3 Applied voltages vs. DOS of devices with E3 and E7 electrolytes. | ||
Plotting the electron lifetimes vs. the applied potential can lead to misinterpretation due to the difference in redox potential of the cobalt complexes. Plotting the electron lifetimes vs. DOS or chemical capacitance gives a clearer representation of the comparison. The position inside the DOS can be used as a measure of the Fermi level position of the TiO2 and therefore factoring out the difference in redox potential (see Fig. 4). The DSCs with the E7 electrolyte possesses a lower electron lifetime at the same DOS showing that the recombination rate in DSCs with the [CoII/III(Cl-terpy)2] complex is higher than with that of the [CoII/III(terpy)2] complex. This result shows that the lower than expected VOC in the devices of E7 compared to E3 electrolyte is mainly due to the increase in the recombination rate rather than the shift in the conduction band position of TiO2. Since we obtained similar JSC values with devices made with E3 and E7 electrolytes one can assume that there is no difference in the dye regeneration kinetics of these two devices. On the other hand with the higher energy difference between the conduction band electrons and the redox potential of the E7 electrolyte, a higher driving force for the recombination might be responsible for the lower electron lifetime compared to the E3 device. Similar observations were made with a series of redox electrolytes by Feldt et al.30 although in their case different JSC values were obtained due to less driving force for the dye regeneration process.
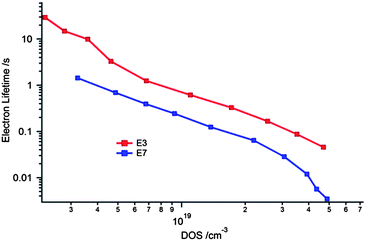 | ||
| Fig. 4 Comparison of electron lifetime vs. DOS of devices with E3 and E7 electrolytes. | ||
To further understand the differences between the iodine and cobalt based electrolytes the photovoltaic performance of a DSC with electrolyte E7 is compared to one with an iodide/triiodide based electrolyte (0.2 M 1,3-dimethylimidazolium iodide (DMII), 0.07 M iodine, 0.1 M LiClO4 and 0.2 M TBP). The iodide/triiodide-based electrolyte has the same concentration of oxidized/reduced species as does the E7 electrolyte as well as the same concentration of additives. According to the Nernst equation the calculated redox potential of I−/I3−-based electrolyte is ∼0.380 V vs. NHE, whereas for the E7 electrolyte, a value of ∼600 mV is calculated, yielding a maximum theoretical difference in redox potential of ∼220 mV between these two electrolytes.
The photovoltaic parameters of the iodide/triiodide based DSC is presented in Table 2 and shows a lower efficiency compared to the E7 electrolyte. Even with the optimized iodide/triiodide electrolyte (Z960) a PCE of only 7.2% was obtained.20 The PCE of the E7 electrolyte outperformed the iodide/triiodide-based electrolytes (of same concentration of oxidized and reduced species) and Z960 electrolyte mainly due to the higher VOC. The observed difference in the VOC of these two devices is 170 mV, which is lower than the difference between the Nernst potentials (220 mV) of the two redox electrolytes. The transient photovoltage and charge extraction (Fig. S6 and S8, ESI†) and the EIS analysis (Fig. S4, S5 and S7, ESI†) of the devices show that the higher recombination rate obtained with the E7 electrolyte containing device than with iodide/triiodide electrolyte device is mainly responsible for the lower VOC values.
It is interesting to note that by comparing the devices made with E3 and the iodide/triiodide based electrolyte that nearly no shift in the conduction band position is observed, and that with the E3 electrolyte, the maximum achievable voltage difference (100 mV) is reached. This indicates that the recombination rate in these two electrolytes is almost the same. This shows that, depending on the cobalt complex ligand and its substituents, the recombination is facilitated or retarded. The overall device performance with cobalt-based electrolytes can be generally further improved by minimizing the FF losses by using alternative counter electrodes in place of Pt, as it is known that the Pt is not a good electro-catalyst for the reduction of cobalt complexes.
Conclusion
Cobalt complexes offer a valuable alternative for the commonly used iodine based redox system in DSCs to achieve high power conversion efficiency. Though the cobalt complexes possess slower diffusion coefficients in the electrolyte than triiodide, they outperform the iodide/triiodide-based electrolytes due to substantially higher VOC values. The two cobalt terpyridyl complexes investigated in this study show the importance of substitution on the ligand coordinated to the cobalt ion in determining the dark current and VOC values of the device due to tuning of the electrolyte energy level. The [CoII/III(Cl-terpy)2](TFSI)2/3 based electrolyte in combination with the Y123 dye yielded an overall PCE of 8.7% at AM 1.5G simulated sunlight with VOC values above 900 mV.Acknowledgements
We are grateful to Dr Tsuguo Koyanagi from JGC C&C (Japan), Dyesol (Australia), Nippon Sheet Glass Co., Ltd, and Mihama Co., for providing the 400 nm sized TiO2 particles, the 20 nm particles (DSL 18NR-T), the FTO glass, and antireflection layer, respectively. We thank Dr Hoi Nok Tsao, Dr Chenyi Yi and Dr Carole Grätzel, for providing TiO2 film, Y123 dye and fruitful discussions, respectively. We thank the Swiss National Science Foundation for the financial support. This work was supported by the ECR advanced grant agreement (no. 247404) under the CE-Mesolight project funded by the European community's 7th FWP.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- T. Daeneke, T. H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 213–215 CrossRef.
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341–346 CrossRef CAS.
- M. K. Wang, N. Chamberland, L. Breau, J. E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS.
- H. Nusbaumer, S. M. Zakeeruddin, J. E. Moser and M. Grätzel, Chem.–Eur. J., 2003, 9, 3756–3763 CrossRef CAS.
- H. Nusbaumer, J. E. Moser, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2001, 105, 10461–10464 CrossRef CAS.
- L. Han, N. Koide, Y. Chiba and T. Mitate, Appl. Phys. Lett., 2004, 84, 2433 CrossRef CAS.
- Y. Bai, J. Zhang, D. F. Zhou, Y. H. Wang, M. Zhang and P. Wang, J. Am. Chem. Soc., 2011, 133, 11442–11445 CrossRef CAS.
- J. Y. Liu, J. Zhang, M. F. Xu, D. F. Zhou, X. Y. Jing and P. Wang, Energy Environ. Sci., 2011, 4, 3021–3029 CAS.
- D. Zhou, Q. Yu, N. Cai, Y. Bai, Y. Wang and P. Wang, Energy Environ. Sci., 2011, 4, 2030–2034 CAS.
- M. Zhang, J. Y. Liu, Y. H. Wang, D. F. Zhou and P. Wang, Chem. Sci., 2011, 2, 1401–1406 RSC.
- Y. Bai, Q. J. Yu, N. Cai, Y. H. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376–4378 RSC.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS.
- H. X. Wang, P. G. Nicholson, L. Peter, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. C, 2010, 114, 14300–14306 CAS.
- P. J. Cameron, L. M. Peter, S. M. Zakeeruddin and M. Grätzel, Coord. Chem. Rev., 2004, 248, 1447–1453 CrossRef CAS.
- A. Hauch and A. Georg, Electrochim. Acta., 2001, 46, 3457–3466 CrossRef CAS.
- Y. R. Liu, J. R. Jennings, Y. Huang, Q. Wang, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 18847–18855 CAS.
- H. Tian and L. Sun, J. Mater. Chem., 2011, 21, 10592–10601 RSC.
- M. J. DeVries, M. J. Pellin and J. T. Hupp, Langmuir, 2010, 26, 9082–9087 CrossRef CAS.
- H. N. Tsao, C. Yi, T. Moehl, J. H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef CAS.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser, C. Yi, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2012, 3, 631 CrossRef.
- B. A. Gregg, S. Ferrere and F. Pichot, Org. Photovoltaics, 2001, 4465, 31–42 Search PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- B. C. O'Regan and F. Lenzmann, J. Phys. Chem. B, 2004, 108, 4342–4350 CrossRef CAS.
- B. C. O'Regan, K. Bakker, J. Kroeze, H. Smit, P. Sommeling and J. R. Durrant, J. Phys. Chem. B, 2006, 110, 17155–17160 CrossRef CAS.
- F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117–131 CrossRef CAS.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Sero and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
- S. A. Sapp, C. M. Elliott, C. Contado, S. Caramori and C. A. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215–11222 CrossRef CAS.
- S. M. Feldt, G. Wang, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2011, 115, 21500–21507 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sc21401f |
| This journal is © The Royal Society of Chemistry 2013 |