Sequential and click-type postfunctionalization of regioregular poly(3-hexylthiophene) for realization of n-doped multiplet state†
Yuping
Yuan
a,
Wonsung
Choi
b,
Hiroyuki
Nishide
b and
Tsuyoshi
Michinobu
*c
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo, 152-8552, Japan
bDepartment of Applied Chemistry, Waseda University, Tokyo, 169-8555, Japan
cGlobal Edge Institute, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo, 152-8550, Japan. E-mail: michinobu.t.aa@m.titech.ac.jp; Fax: +81-3-5734-3774; Tel: +81-3-5734-3774
First published on 12th September 2012
Abstract
Regioregular poly(3-hexylthiophene) was postfunctionalized by a three-step high-yielding reaction producing an electron-accepting 1,1,4,4-tetracyanobuta-1,3-diene unit at the regioregular position of the thiophene rings. Electrochemical measurements of the postfunctionalized polythiophene derivative showed two one-electron reduction waves with the first reduction potential of −0.66 V (vs. Fc/Fc+). The electrochemically generated poly(anionic radical)s were confirmed by their electron spin resonance (ESR) signals. The magnetic measurements of the poly(anionic radical)s, produced by chemical reduction with cobaltocene or decamethylcobaltocene, suggested the multiplet state (S ∼ 2/2) at low temperatures.
Introduction
Regioregular polythiophene derivatives with a head-to-tail connectivity are an effective π-conjugated platform to realize high-spin organic polyradicals.1 Multiple unpaired electrons which are directly introduced into the regioregular position of the thiophene rings satisfy a non-Kekulé and non-disjoint connectivity, resulting in high-spin ground states.2 The degree of the high-spin states is represented by the spin quantum number (S), which is proportional to the degree of polymerization.This “side-chain polyradical” approach can be applied to most conjugated polymers, and the main advantage of using this approach is the remote ferromagnetic interactions between radicals through the π-conjugated main chain.3 Accordingly, the spin alignment is insensitive to radical defects. It should be noted that the radical defect problem is detrimental to a “main-chain polyradical” approach.4 With this fact in mind, a variety of radical species has been introduced into the side chains of conjugated polymers, and this systematic study reveals the clear relationship between the radical stability and spin exchange interactions. Chemically stable radicals, such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), can easily be handled even under ambient conditions,5 but the spin density, mostly localized on the NO moiety, leads to negligible spin exchange interactions when multiple TEMPO radicals are connected to a single molecule. In contrast, highly reactive radical species, such as carbene, nitrene, and triarylmethine radicals, show large spin exchange interactions in non-Kekulé type polyradicals, but they can survive only at extremely low temperatures due to their limited stability.6 We and others have, based on this relationship, used intermediate radical species with both a relatively high chemical stability and sufficient spin exchange interactions. For example, the half-lives of the neutral radicals of 2,6-di-tert-butylphenoxyl and tert-butylnitroxide at room temperature are in the order of days, and consequently, they can be handled without special caution. Their non-Kekulé type oligo- and polyradicals with a stilbene or bithiophene spacer showed intramolecular spin exchange interactions with the ferromagnetic coupling constant (2J) of about 70 cm−1.7 Other radical species adopted as the spin source of high-spin organic polyradicals according to the “side-chain polyradical” guideline are cationic radicals of aromatic amines (aminium cationic radicals).8 The high stability of the charged radicals may be achieved by an electrostatic repulsion. The robust high-spin ground states of the non-Kekulé type oligo- and poly(aminium cationic radical)s were definitely demonstrated by the temperature dependence of the magnetic measurements.9 However, there have, to the best of our knowledge, been few reports on similar polyradical studies using anionic radicals.10 This might be due to the lower chemical stability of anionic species compared to cationic ones, because of the high reactivity of anionic species toward protonic substances. Also, the number of available n-type doping reagents is significantly limited,11 rendering this radical species almost unexplored in the high-spin molecules.
Recently, a new efficient synthetic method for constructing n-dopable units was developed during the course of our studies on novel donor–acceptor systems.12 This is based on high-yielding addition reactions of strong acceptor molecules, such as tetracyanoethylene (TCNE) and 7,7,8,8-tetracyanoquinodimethane (TCNQ), to electron-rich alkynes.13 For example, the [2 + 2] cycloaddition of the electron-deficient olefin of TCNE to electron-rich alkynes, followed by a cycloreversion, produced donor-substituted 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) chromophores. When alkynes are substituted by aromatic amines, this reaction quickly proceeds under mild conditions without any side reactions and byproducts. Thus, this class of reactions is currently regarded as metal-free click chemistry,14 and the concept of this chemistry has been applied to the syntheses of functional polymers.15 The TCBD moiety is a highly twisted acceptor unit, stabilizing the corresponding anionic radical. The chemically reduced anionic radicals of some small molecular weight TCBDs were indeed detected by electron spin resonance (ESR).16
We now report the first application of the TCBD anionic radical to high-spin polyradicals. The polyradical precursor was elegantly synthesized by a three-step high-yielding postfunctionalization reaction, starting from the regioregular poly(3-hexylthiophene) (P3HT). The TCBD moieties, densely appended to the regioregular position of the thiophene rings, were reduced by both electrochemical and chemical methods. The significant stability at room temperature and multiplet ground state of the generated poly(anionic radical) were demonstrated by ESR and superconducting quantum interference device (SQUID) measurements.
Results and discussion
P3HT modification
The regioregular P3HT was synthesized by the quasi-living Grignard metathesis (GRIM) polymerization reaction.17 The molecular weight (Mn) and polydispersity (Mw/Mn) of P3HT, determined by gel permeation chromatography (GPC) using THF as the eluent, were 9600 and 1.59, respectively. The regioregularity of P3HT was evaluated from the 1H NMR spectrum by carefully comparing the relative integration ratio of the signals at ca. 2.80 [Head-to-Tail (H–T)] and 2.58 [Head-to-Head (H–H)] ppm (Fig. 1a). A high regioregularity of 98.5% was shown. Taking into account the average molecular weight (about 55 mer), the H–H connectivity exists only at the polymer terminal.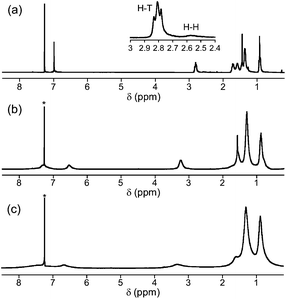 | ||
| Fig. 1 1H NMR spectra of (a) P3HT, (b) P1, and (c) P2 in CDCl3 at 20 °C. The residual CHCl3 peak is marked. Inset: Enlarged peaks ascribed to the α-methylene protons of P3HT are used to estimate the content of Head-to-Tail (H–T) and Head-to-Head (H–H) connectivities. | ||
A dialkylaniline-activated alkyne group was introduced into the side chains of P3HT by the sequential postfunctionalization protocol developed by Holdcroft et al. (Scheme 1).18 This was because the GRIM polymerization of 3-alkyne-substituted thiophene derivatives is terminated by the formation of a stable alkyne–Ni complex at the propagating site.19 A slight excess of N-bromosuccinimide (NBS) was added to a CHCl3 solution of P3HT, quantitatively yielding P3HT–Br. The disappearance of the thiophene protons in the 1H NMR spectrum suggested the occurrence of perfect bromination. This was followed by the Stille coupling reaction with (C6H13)2NPhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSn(C4H9)3, providing the novel regioregular polythiophene derivative P1 with side chain electron-rich alkynes in 98% yield. The successful quantitative introduction of the side chain alkynes was confirmed by 1H NMR, IR, GPC, and elemental analyses. The 1H NMR spectrum of P1 showed a set of new peaks ascribed to the added side chain moieties (Fig. 1b). The aromatic protons of the dihexylaniline group were detected at 6.53 and 7.30 ppm. In addition, the α-methylene peaks detected at 2.6–2.8 ppm for P3HT clearly shifted to 3.25 ppm. This broad peak is overlapped with the α-methylene peak of the dihexylaniline group. In the IR spectrum, a weak alkyne vibration peak appeared at 2195.9 cm−1. The molecular weight increase and elemental analysis data also supported the quantitative substitution by the alkyne unit (see ESI†). It should be noted here that the Sonogashira coupling of P3HT–Br with (C6H13)2NPhCCH resulted in gelation of most of the product, probably due to undesired side reactions. It was expected that the use of the iodo group instead of the bromo group could solve this problem. However, the iodination of P3HT using N-iodosuccinimide did not proceed.
CSn(C4H9)3, providing the novel regioregular polythiophene derivative P1 with side chain electron-rich alkynes in 98% yield. The successful quantitative introduction of the side chain alkynes was confirmed by 1H NMR, IR, GPC, and elemental analyses. The 1H NMR spectrum of P1 showed a set of new peaks ascribed to the added side chain moieties (Fig. 1b). The aromatic protons of the dihexylaniline group were detected at 6.53 and 7.30 ppm. In addition, the α-methylene peaks detected at 2.6–2.8 ppm for P3HT clearly shifted to 3.25 ppm. This broad peak is overlapped with the α-methylene peak of the dihexylaniline group. In the IR spectrum, a weak alkyne vibration peak appeared at 2195.9 cm−1. The molecular weight increase and elemental analysis data also supported the quantitative substitution by the alkyne unit (see ESI†). It should be noted here that the Sonogashira coupling of P3HT–Br with (C6H13)2NPhCCH resulted in gelation of most of the product, probably due to undesired side reactions. It was expected that the use of the iodo group instead of the bromo group could solve this problem. However, the iodination of P3HT using N-iodosuccinimide did not proceed.
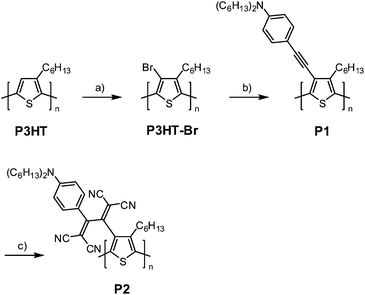 | ||
| Scheme 1 Postfunctionalization of P3HT: (a) NBS, CHCl3, 20 °C, (b) (C6H13)2NPhCCSn(C4H9)3, Pd(PPh3)4, toluene, 100 °C, (c) TCNE, 1,2-dichloroethane, 60 °C. | ||
Postfunctionalization by TCNE
The side chain alkynes of P1 were activated by the dihexylanilino donor. In order to construct the n-dopable TCBD unit, P1 was further converted into P2 by the postfunctional TCNE addition. Previously, similar attempts to add TCNE to the side chain alkynes were successfully performed for various polymers, such as polystyrenes, polyurethanes, aromatic polyamines, and regiorandom polythiophenes, at room temperature within a few minutes.20 However, in this case, the reactivity of P1 was lower than the previous polymers, probably due to steric reasons. Mild heating to 60 °C was necessary to initiate the reaction with TCNE. A more bulky acceptor of TCNQ was not reactive under the same conditions. The addition reaction was monitored by UV-vis-near IR spectroscopy (Fig. 2). Upon the stepwise addition of TCNE to a P1 solution in 1,2-dichloroethane, a new charge-transfer (CT) band appeared at about 600 nm. The intensity of the CT band linearly increased and the position gradually shifted in the longer wavelength direction with the increasing amount of TCNE up to 1.0 equiv. The brown solution color of P1 eventually turned dark green. The presence of isosbestic points suggested progress of the reaction without any side reactions. The addition of excess TCNE did not cause any spectral changes, implying the reaction completion and the high chemical stability of the formed TCBD moieties. The gradual bathochromic shift in the CT band maximum indicated that the polymer energy levels are stepwise lowered by the TCNE addition.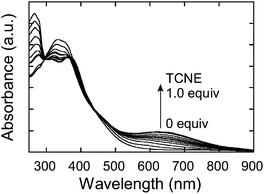 | ||
| Fig. 2 UV-vis-near IR spectral change of P1 (0.162 mM repeat per unit) in 1,2-dichloroethane upon the stepwise addition of TCNE (0.1 equiv. interval) at 60 °C. | ||
Based on the titration experiment, P1 was perfectly postfunctionalized with one equivalent of TCNE, yielding P2. The molecular weight of P2 could not precisely be determined by GPC due to the strong adhesive feature of the TCBD moieties to polystyrene gel columns.21 However, the chemical structure of P2 was reasonably confirmed by 1H NMR, IR, and elemental analyses. In the 1H NMR, the signals ascribed to the side chain protons shifted in a lower magnetic field direction compared to the precursor polymer P1 (Fig. 1c). This result suggests the formation of the electron-accepting TCBD units. The IR spectrum of P2 displayed a strong CN vibration peak at 2209.0 cm−1 instead of the alkyne vibration at 2195.9 cm−1 for P1. Furthermore, the elemental analysis data showed excellent agreement with the theoretical values (ESI†). All these results strongly support the quantitative yields of the three postfunctionalization reactions.
Electrochemistry
The thin films of P1 and P2 were investigated by electrochemistry. Cyclic voltammograms (CVs) of the cast films prepared on a glassy carbon disk electrode were measured in CH3CN with 0.1 M (nC4H9)4NClO4 at 20 °C, and the first oxidation and the first reduction potentials are summarized in Table 1. The precursor polythiophene P1 showed two-step oxidation peaks (Fig. S1a†). The first oxidation occurred at the dihexylaniline moiety, while the second oxidation potential was ascribed to the polythiophene main chain. The irreversible reduction was detected at the peak top value of −2.50 V (vs. Fc/Fc+). Interestingly, the TCNE addition dramatically altered the redox behavior. The postfunctionalized polythiophene P2 displayed only a single oxidation peak, and the peak top potential anodically shifted to 1.02 V (Fig. S1b†). Moreover, two new well-resolved reduction steps appeared at −0.66 and −1.21 V. These reduction potentials are derived from the TCBD moieties. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels of these polymers were calculated from the onset first oxidation and first reduction potentials, respectively, based on the assumption of Fc/Fc+ = −4.80 eV. As listed in Table 1, both the HOMO and LUMO levels were lowered by the postfunctionalization using the TCNE addition reaction. In particular, the LUMO decrease was more significant than the HOMO decrease leading to band gap narrowing from 2.49 eV to 1.25 eV. This dramatic change in the energy levels is consistent with the UV-vis-near IR spectral observations (vide supra).The remarkably facilitated reduction, achieved by the TCNE addition, enabled the efficient generation of the poly(anionic radical)s. First, the poly(anionic radical) of P2 was electrochemically generated in CH2Cl2 with 0.1 M (nC4H9)4NClO4 at 20 °C and in situ detected by ESR. The solution CV of P2 was similar to that of the thin film. When the potentials more negative than the first reduction potential were applied, a weak nine-line ESR signal appeared at g = 2.0029 (Fig. 3). Taking into account the chemical structure, the nine-line splitting is assigned to the hyperfine coupling (hfc) with the four virtually equivalent nitrogen nuclei of the TCBD moiety. The experimental hfc of 0.156 mT and the g value are characteristic of the TCBD-centered anionic radicals.16 The ESR signal intensity increased and its shape became unresolved as more negative potentials were applied. This was due to the formation of polyradicals, which undergo a fast intramolecular spin exchange on the ESR measurement timescale. However, upon the application of potentials more negative than the second reduction of P2, the signal intensity gradually decreased, suggesting the formation of a closed-shell bianion structure.
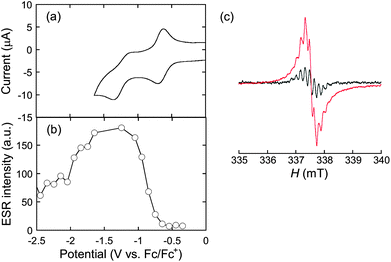 | ||
| Fig. 3 Electrochemical reduction of P2 in CH2Cl2 with 0.1 M (nC4H9)4NClO4 at 20 °C: (a) cyclic voltammogram, (b) ESR signal intensity vs. applied potential, and (c) ESR spectra applied at −0.7 V (black line) and −1.2 V (red line). | ||
Magnetic measurements
With the electrochemical results in mind, we optimized the chemical reduction conditions. Among a list of n-type doping reagents, we selected cobaltocene (Ered = −1.33 V) or decamethylcobaltocene (Ered = −1.94 V), because they possess high reduction potentials and acceptable solubilities in some organic solvents, such as THF,22 allowing the controlled homogeneous one-electron reduction of the repeat unit of P2. Moreover, the cobaltocenium products resulting from the one-electron reduction reactions are 18-electron closed-shell structures, which do not interfere with the magnetic measurements.The chemical reduction of P2 was performed in THF at 20 °C under nitrogen. Similar to the electrochemical reduction, the poly(anionic radical) of P2 exhibited a unimodal ESR signal at g = 2.0029. The polyradical generation was accompanied by a solution color change from deep green to yellowish-green. This color change was monitored by UV-vis-near IR spectroscopy (Fig. S2†). Upon the stepwise addition of the reducing reagents, the CT band (about 630 nm) of P2 gradually decreased. The extent of the decrease was more significant when decamethylcobaltocene was employed, which reflects the stronger reducing power. Although the spectra did not indicate any noticeable side reactions, the maximum spin concentration, estimated from the careful comparison of the ESR intensity to that of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), was 17%. The lifetime of the poly(anionic radical) in THF was estimated from the ESR intensities at 20 °C (Fig. S3†). The polyradical decay clearly followed first-order kinetics, and the half-life was determined to be 26.4 h. This stability is comparable to those of the poly(phenoxyl radical)s and poly(nitroxide radical)s.
The stable poly(anionic radical) of P2 was further subjected to magnetic measurements. The magnetization and static magnetic susceptibility of the poly(anionic radical) were measured by a SQUID magnetometer. To reduce the intermolecular antiferromagnetic interaction, a frozen solvent was used as the matrix. Thus, a THF solution of the poly(anionic radical) was directly transferred to a quartz tube after a small amount of insoluble decamethylcobaltocene, if any, was filtered off. As soon as the tube was sealed off, it was cooled to the measurement temperature range. Plots of the inverse unit molar magnetic susceptibility (1/χmol) versus temperature (T) showed a linear relationship between these parameters, which could be fitted to the Curie–Weiss law with the Weiss constant (θ) of −1.2 K (Fig. S4†). The radical amount or spin concentration was estimated from the saturated magnetization of the weighted polyradical at the applied magnetic field of 7 T and temperature of 1.8 K. The negative θ value reflects the intermolecular antiferromagnetic interactions. Organic polyradicals usually show this behavior at extremely low temperatures, and it is known that this tendency is significant in the case of ionic radicals, such as the aminium cationic radicals.23 This fact became more apparent when the product of χmolT versus T for the polyradical was plotted (Fig. 4). The χmolT values dramatically decreased with the decreasing temperature at <10 K. However, one has to note that the χmolT values in the range of 10–50 K are much higher than the theoretical single paramagnetic spin of 0.375. These values are close to 0.5, which corresponds to the theoretical spin quantum number (S) of 2/2. This result strongly supports the presence of intramolecular ferromagnetic interactions. In other words, an average of two spins generated through the n-doping of a single polythiophene chain ferromagnetically interacted with each other to form a multiplet state.
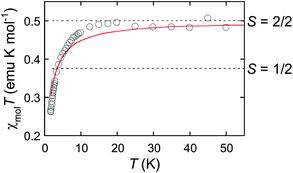 | ||
| Fig. 4 χ mol T vs. T plots of the poly(anionic radical) of P2 with the spin concentration of 0.17. The red line represents the theoretical curve fitted to the Curie–Weiss law with the Weiss constant (θ) of −1.2 K. | ||
Conclusions
The thiophene rings of the regioregular P3HT were successfully functionalized by bromination, Stille coupling, and TCNE-based click chemistry. In particular, the last functionalization enabled the formation of the TCBD-based acceptor unit. Both the electrochemical and chemical reduction produced the corresponding poly(anionic radical) with a half-life exceeding 1 day at 20 °C. The magnetic behavior elucidated by SQUID measurements suggested a ground state multiplet. This is, to the best of our knowledge, the first report about purely organic, high-spin poly(anionic radical)s. In the future, the steric effects of the polythiophene side chains on the multiplet ground states will be investigated by computational calculations.Acknowledgements
This work was supported, in part, by a Grant-in-Aid for Scientific Research from MEXT, Japan, a PRESTO program, JST, and the Iwatani Naoji Foundation.Notes and references
- H. Hayashi and T. Yamamoto, Macromolecules, 1997, 30, 330 CrossRef CAS; C. Xie and P. M. Lahti, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 779 CrossRef; M. Miyasaka, T. Yamazaki, E. Tsuchida and H. Nishide, Macromolecules, 2000, 33, 8211 CrossRef; M. Miyasaka, T. Yamazaki and H. Nishide, Polym. J., 2001, 33, 849 CrossRef; X. Wan, X. Lv, G. He, A. Yu and Y. Chen, Eur. Polym. J., 2011, 47, 1018 CrossRef.
- D. A. Dougherty, Acc. Chem. Res., 1991, 24, 88 CrossRef CAS; H. Iwamura and N. Koga, Acc. Chem. Res., 1993, 26, 346 CrossRef; A. Rajca, Chem. Rev., 1994, 94, 871 CrossRef.
- H. Nishide, Adv. Mater., 1995, 7, 937 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503 CrossRef CAS.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339 CrossRef CAS.
- K. Matsuda, N. Nakamura, K. Takahashi, K. Inoue, N. Koga and H. Iwamura, J. Am. Chem. Soc., 1995, 117, 5550 CrossRef CAS; M. Minato and P. M. Lahti, J. Am. Chem. Soc., 1997, 119, 2187 CrossRef; A. Rajca, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 1999, 121, 6308 CrossRef; S. Rajca, A. Rajca, J. Wongsriratanakul, P. Butler and S.-M. Choi, J. Am. Chem. Soc., 2004, 126, 6972 CrossRef.
- H. Nishide, T. Kaneko, T. Nii, K. Katoh, E. Tsuchida and K. Yamaguchi, J. Am. Chem. Soc., 1995, 117, 548 CrossRef CAS; H. Nishide, T. Kaneko, S. Toriu, Y. Kuzumaki and E. Tsuchida, Bull. Chem. Soc. Jpn., 1996, 69, 499 CrossRef.
- T. D. Selby and S. C. Blackstock, J. Am. Chem. Soc., 1998, 120, 12155 CrossRef CAS; R. J. Bushbby and D. Gooding, J. Chem. Soc., Perkin Trans. 2, 1998, 1069 RSC; J. Wu, M. Baumgarten, M. G. Debije, J. M. Warman and K. Müllen, Angew. Chem., Int. Ed., 2004, 43, 5331 CrossRef; G. Zhou, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2007, 129, 12211 CrossRef; R. J. Bushby, N. Taylor and R. A. Williams, J. Mater. Chem., 2007, 17, 955 RSC.
- T. Michinobu, M. Takahashi, E. Tsuchida and H. Nishide, Chem. Mater., 1999, 11, 1969 CrossRef CAS; M. Takahshi, T. Nakazawa, E. Tsuchida and H. Nishide, Macromolecules, 1999, 32, 6383 CrossRef; H. Murata, M. Takahashi, K. Namba, N. Takahashi and H. Nishide, J. Org. Chem., 2004, 69, 631 CrossRef; E. Fukuzaki and H. Nishide, J. Am. Chem. Soc., 2006, 128, 996 CrossRef; H. Murata, D. Miyajima and H. Nishide, Macromolecules, 2006, 39, 6331 CrossRef.
- For triplet bi(anionic radical)s, see: A. Behrendt, C. G. Screttas, D. Bethell, O. Schiemann and B. R. Steele, J. Chem. Soc., Perkin Trans. 2, 1998, 2039 RSC.
- A. Werner, F. Li, K. Harada, M. Pfeiffer, T. Fritz, K. Leo and S. Machill, Adv. Funct. Mater., 2004, 14, 255 CrossRef CAS; P. Wei, J. H. Oh, G. Dong and Z. Bao, J. Am. Chem. Soc., 2010, 132, 8852 CrossRef; N. Cho, H.-L. Yip, S. K. Hau, K.-S. Chen, T.-W. Kim, J. A. Davies, D. F. Zeigler and A. K.-Y. Jen, J. Mater. Chem., 2011, 21, 6956 RSC; P. Wei, T. Menke, B. D. Naab, K. Leo, M. Riede and Z. Bao, J. Am. Chem. Soc., 2012, 134, 3999 CrossRef; S. Guo, S. B. Kim, S. K. Mohapatra, Y. Qi, T. Sajoto, A. Kahn, S. R. Marder and S. Barlow, Adv. Mater., 2012, 24, 699 CrossRef.
- T. Michinobu, J. C. May, J. H. Lim, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross, I. Biaggio and F. Diederich, Chem. Commun., 2005, 737 RSC.
- S.-I. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994 RSC; T. Michinobu, Chem. Soc. Rev., 2011, 40, 2306 RSC.
- K. L. Killops, L. M. Campos and C. L. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS; C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef; A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411 CrossRef; C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- A. Qin, C. K. W. Jim, W. Lu, J. W. Y. Lam, M. Häussler, Y. Dong, H. H. Y. Sung, I. D. Williams, G. K. L. Wong and B. Z. Tang, Macromolecules, 2007, 40, 2308 CrossRef CAS; I. Singh, Z. Zarafshani, J.-F. Lutz and F. Heaney, Macromolecules, 2009, 42, 5411 CrossRef; Y.-G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2009, 42, 7709 CrossRef; A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522 RSC; A. Qin, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2010, 43, 8693 CrossRef; Q. Wei, H. Deng, Y. Cai, J. W. Y. Lam, J. Li, J. Sun, M. Gao, A. Qin and B. Z. Tang, Macromol. Rapid Commun., 2012, 33, 1356 CrossRef.
- M. Kivala, T. Stanoeva, T. Michinobu, B. Frank, G. Gescheidt and F. Diederich, Chem.–Eur. J., 2008, 14, 7638 CrossRef CAS; B. Breiten, Y.-L. Wu, P. D. Jarowski, J.-P. Gisselbrecht, C. Boudon, M. Griesser, C. Onitsch, G. Gescheidt, W. B. Schweizer, N. Langer, C. Lennartz and F. Diederich, Chem. Sci., 2011, 2, 88 RSC.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169 CrossRef CAS; E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526 CrossRef.
- Y. Li, G. Vamvounis and S. Holdcroft, Macromolecules, 2001, 34, 141 CrossRef CAS; Y. Li, G. Vamvounis, J. Yu and S. Holdcroft, Macromolecules, 2001, 34, 3130 CrossRef; Y. Li, G. Vamvounis and S. Holdcroft, Macromolecules, 2002, 35, 6900 CrossRef.
- M. Jeffries-El, G. Sauvé and R. D. McCullough, Adv. Mater., 2004, 16, 1017 CrossRef CAS.
- Y. Li and T. Michinobu, Polym. Chem., 2010, 1, 72 RSC; Y. Li, K. Tsuboi and T. Michinobu, Macromolecules, 2010, 43, 5277 CrossRef CAS; Y. Yuan and T. Michinobu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 225 CrossRef; Y. Li and T. Michinobu, J. Mater. Chem., 2012, 22, 9513 RSC; T. Michinobu, C. Seo, K. Noguchi and T. Mori, Polym. Chem., 2012, 3, 1427 RSC.
- Y. Washino, K. Murata and T. Michinobu, Polym. Bull., 2012, 69, 137 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 887 CrossRef; R. Piskorski and B. Jaun, J. Am. Chem. Soc., 2003, 125, 131120 CrossRef.
- T. Michinobu, J. Inui and H. Nishide, Org. Lett., 2003, 5, 2165 CrossRef CAS; T. Michinobu, J. Inui and H. Nishide, Polym. J., 2010, 42, 575 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, CV of P1 and P2 films, UV-vis-near IR spectra of P2 upon chemical reduction, lifetime evaluation of the poly(anionic radical) of P2 by ESR, and magnetic measurements. See DOI: 10.1039/c2sc21334f |
| This journal is © The Royal Society of Chemistry 2013 |