Self-assembled, covalently linked, hollow phthalocyanine nanospheres†
Raghunandan
Hota‡
a,
Kangkyun
Baek‡
a,
Gyeongwon
Yun
a,
Youngkook
Kim
a,
Hyuntae
Jung
b,
Kyeng Min
Park
a,
Eunjin
Yoon
c,
Taiha
Joo
c,
Juseok
Kang
d,
Chan Gyung
Park
d,
Su Mi
Bae
e,
Woong Shick
Ahn
e and
Kimoon
Kim
*ab
aCenter for Smart Supramolecules, Department of Chemistry, Division of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), San 31, Hyoja-dong, Nam-gu, Pohang, Kyungbuk 790-784, Republic of Korea. E-mail: kkim@postech.ac.kr; Fax: +82-54-279-8129; Tel: +82-54-279-2113
bSchool of Interdisciplinary Bioscience and Bioengineering, POSTECH, Republic of Korea
cDepartment of Chemistry, POSTECH, Republic of Korea
dDepartment of Materials Science and Engineering, POSTECH, Republic of Korea
eDepartment of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul, 137-701, Republic of Korea
First published on 9th October 2012
Abstract
A rational design and synthesis of covalently linked Pc nanospheres with a very thin shell and hollow interior, composed of approximately 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pc units on average, was demonstrated through thiol–ene “click” chemistry without using any templates or emulsifiers. The ZnPc nanospheres allow post-synthetic modification to improve their dispersibility in aqueous solution without altering the morphology of the nanospheres or the properties of ZnPc cores. More importantly, the ZnPc nanospheres showed higher singlet oxygen generation efficiency and in vitro phototoxicity than monomeric Pc molecules, suggesting that ZnPc nanospheres are potentially useful as a PS for PDT. We anticipate that the ZnPc nanospheres would allow other post-synthetic modifications such as the introduction of targeting ligands to deliver the nanospheres to specific target sites and perform a dual chemo- and photodynamic therapy by the encapsulation of therapeutic agents. The easy synthesis of a hollow spherical framework with a high Pc content, coupled with facile post-synthetic modification may allow Pc nanospheres to be a versatile platform for a diverse range of medical and non-medical applications.
000 Pc units on average, was demonstrated through thiol–ene “click” chemistry without using any templates or emulsifiers. The ZnPc nanospheres allow post-synthetic modification to improve their dispersibility in aqueous solution without altering the morphology of the nanospheres or the properties of ZnPc cores. More importantly, the ZnPc nanospheres showed higher singlet oxygen generation efficiency and in vitro phototoxicity than monomeric Pc molecules, suggesting that ZnPc nanospheres are potentially useful as a PS for PDT. We anticipate that the ZnPc nanospheres would allow other post-synthetic modifications such as the introduction of targeting ligands to deliver the nanospheres to specific target sites and perform a dual chemo- and photodynamic therapy by the encapsulation of therapeutic agents. The easy synthesis of a hollow spherical framework with a high Pc content, coupled with facile post-synthetic modification may allow Pc nanospheres to be a versatile platform for a diverse range of medical and non-medical applications.
Introduction
Phthalocyanines (Pcs) have received much attention as molecular materials because of their excellent electronic and optical properties.1–7 One of the promising applications of Pcs is their use in medicine as a photosensitizer (PS) for photodynamic therapy (PDT). Since Pcs strongly and selectively absorb light in the range of 600–800 nm, they allow a high penetration depth of light in normal tissue while minimizing the risk and complications such as burning.8–11 A key challenge in this application is, however, the highly hydrophobic nature of Pcs and their propensity to form aggregates in aqueous media which reduces their therapeutic activity. Various types of nanocarriers, such as micelles,12–15 liposomes,16,17 and nanoparticles,18–22 have thus been used to overcome this problem, and to prepare a stable dispersion of Pc in aqueous solution. However, most of them still suffer from shortcomings such as (1) poor loading of Pc (small weight% of Pc in nanocarriers), (2) risk of payload leaking before reaching target cells, and (3) laborious, time-consuming fabrication and encapsulation processes.23–25To address this problem, we considered a rational approach to design a well-defined, covalently bonded nanostructure composed of mainly Pc molecules in which most of the Pc units are located or exposed to the surface. We thought that a nanometer-sized hollow polymer sphere with a single-monomer-thick shell mainly composed of covalently linked Pc units would be an ideal candidate for this goal since such Pc nanospheres would offer facile cellular uptake, enhanced permeability and retention (EPR) properties,26–28 and increased local concentration compared to Pc monomers at therapeutic sites.
We recently discovered the spontaneous formation of polymer nanocapsules in solution by the thiol–ene photopolymerization of dithiol and allyloxycucurbit[6]uril (allyloxyCB[6]), a cucurbit[6]uril (CB[6]) derivative with twelve allyloxy groups decorating the periphery of the rigid framework of CB[6].29 This discovery led us to develop a new strategy for the one-pot, direct synthesis of hollow polymer nanocapsules with a thin shell (one or two monomers-thick) without using any template or pre-organized structure, which appears to be applicable to any flat, diskshaped monomer with multiple polymerizable groups at the periphery.29–33 Using the same general strategy, we decided to synthesize covalently linked Pc nanospheres with a hollow interior. Herein, we report the rational design and synthesis of covalently linked Pc nanospheres with a very thin shell and hollow interior, composed of approximately 12000 Pc units on average, without using any templates or emulsifiers. The dispersibility of the nanospheres in aqueous media can be improved by simple postsynthetic modification. Furthermore, in vitro experiments demonstrated that the Pc nanospheres show higher PDT activity than a conventional monomeric Pc-based PDT photosensitizer.
Results and discussion
Synthesis and characterization of phthalocyanine nanospheres
We synthesized covalently linked Pc nanospheres with a hollow interior using the general strategy employed for polymer nanocapsules.29–33 We first synthesized zinc Pc monomer 1 (Scheme 1), having eight “arms” with one terminal olefin group on each. However, initial attempts to synthesize Pc nanospheres by reacting 1 with 1,2-ethanedithiol 2 under a typical thiol–ene photopolymerization condition failed as Pc decomposed under UV (254 nm or 300 nm) irradiation. Subsequently, we discovered that thermal polymerization of 1 and 2, initiated by azobisisobutyronitrile (AIBN) successfully produced Pc nanospheres. In a typical experiment, heating a mixture of 1 and 2 in a 1:32 ratio (olefin:thiol = 1:8) in a 1:4 (v/v) mixture of ethanol and dimethylsulfoxide (DMSO) in the presence of 10 mol% of AIBN at 70 °C for 6 h followed by dialysis produced ZnPc nanosphere 3a with an average diameter of 210 ± 70 nm (measured by SEM, see below) in 67% yield based on 1. The size of the polymer nanosphere could be tuned by changing the composition of the reaction medium (Fig. S2†). For example, polymerization of 1 and 2 in pure DMSO and a 3:7 (v/v) mixture of ethanol and DMSO produced Pc nanospheres with an average diameter of 600 ± 100 nm and 170 ± 70 nm, respectively.
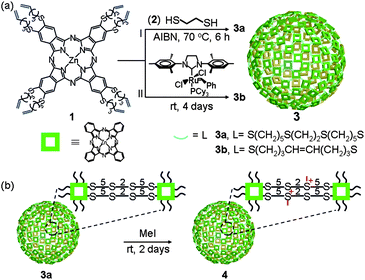 | ||
| Scheme 1 Schematic representation. (a) Direct synthesis of Pc nanospheres through thiol–ene “click” reaction 3a and olefin cross-metathesis reaction 3b. (b) Post-synthetic modification of the Pc nanosphere to enhance its dispersibility in aqueous media. | ||
Alternatively, polymerization of 1 by a olefin cross-metathesis reaction catalyzed by the 2nd generation Grubbs' catalyst produced similar ZnPc nanospheres. For example, stirring a solution of 1 in a 1:4 (v/v) mixture of ethanol and DMSO in the presence of 5 mol% of [1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)(tricyclohexylphosphine)ruthenium at room temperature for 4 days produced 3b with an average diameter of 230 ± 70 nm in 28% yield (Fig. S3†). Because of the low yield, which may be due to partial decomposition of the catalyst in the reaction medium,34 we decided to use the thermal thiol–ene polymerization to synthesize Pc nanospheres for further studies.
The ZnPc nanosphere 3a was fully characterized by elemental analysis, electron microscopy, and various spectroscopic techniques. Elemental analysis showed that the ratio of 1 and 2 incorporated into 3a is approximately 1:3.5, suggesting that upon reaction with 2, ∼7 terminal olefin groups of 1 formed thioether bridges linking neighbouring ZnPc units to yield a two-dimensional polymer network constituting the shell of the nanosphere. This result also indicates that the ZnPc units constitute approximately 33 wt% of the polymer nanosphere, which is, to the best of our knowledge, the highest Pc loading capacity among the nanocarriers reported so far. Solid-state 13C NMR spectroscopy also confirmed the formation of new thioether bridges and disappearance of the terminal olefin groups, suggesting that the monomers are covalently bonded with each other in the nanosphere (Fig. S4†).
The scanning electron microscopy (SEM) images (Fig. 1a) of 3a showed spherical polymer nanospheres with an average diameter of 210 ± 70 nm. The hollow nature of the nanosphere was confirmed by transmission electron microscopy (TEM) (Fig. 1b). High resolution transmission electron microscopy (HR-TEM) studies of 3a after uranyl acetate staining revealed a very thin shell with an average thickness of 0.7 ± 0.2 nm (Fig. 1c), indicating that the shell may be only one or two monomers thick. The high reaction temperature and polar solvent may prevent the aggregation of the Pc monomer during the polymerization step, which may allow the formation of Pc nanospheres with such a thin shell. We confirmed that 3a maintains its spherical shape and size (205 ± 70 nm) in solution (after solvent exchange to THF by dialysis) using cryogenic temperature transmission electron microscopy (Cryo-TEM) (Fig. 1d).
 | ||
| Fig. 1 Electron microscopy images of nanospheres 3a prepared by thermal polymerization of 1 and 2 in 20% EtOH/DMSO. (a) SEM image. (b) TEM image. (c) HR-TEM image showing a thin wall (average thickness: 0.7 ± 0.2 nm) indicated by arrows. (d) Cryo-TEM image in THF. | ||
The FT-IR (Fig. S4†), UV-Vis, and fluorescence spectra of 3a showed characteristic ZnPc-related signals, confirming that the ZnPc units remain intact in the nanosphere. In particular, the absorption spectra (and emission spectra as well) (Fig. 2a) of 1 and 3a in DMSO revealed no significant difference in absorption (and emission) maxima except for a small decrease in absorbance in the nanosphere. No peaks arising from J- or H-type aggregates35 were evident. Taken together, these spectroscopic data suggest that 3a has an almost single-monomer-thick ZnPc shell, which is consistent with the extremely thin shell observed by HR-TEM (Fig. 1c). A modelling study suggested that the shell of a ZnPc nanosphere with a diameter of 210 nm is composed of approximately 12000 ZnPc units.
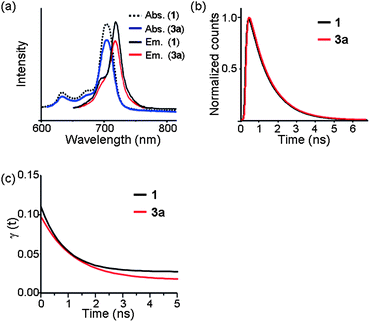 | ||
| Fig. 2 Spectroscopic data of 1 and 3a in DMSO. (a) Absorption and emission (excitation at 640 nm) spectra. (b) Normalized fluorescence decay curves and (c) fitted fluorescence anisotropy decay curves at 720 nm with the excitation at 640 nm. | ||
To investigate the nature of ZnPc in the ZnPc nanosphere in more detail, the time-resolved fluorescence (TRF) and TRF anisotropies of 1 and 3a were measured by a time-correlated single photon counting (TCSPC) method.36 TRFs of 1 and 3a give the same lifetime of 1.15 ± 0.02 ns within experimental uncertainty (Fig. 2b). Anisotropies of 1 and 3a start from 0.1 at time zero, and show exponential decays to give time constants of 880 ± 160 ps and 1220 ± 220 ps, respectively (Fig. 2c). This is consistent with the theoretical expectation: for a doubly degenerate emitting state such as the S1 state of Pc, the initial anisotropy should be 0.7 and decay to 0.1 within the electron dephasing time, which is much faster than the time resolution in this work.37 It is surprising, however, that the anisotropy of 3a decays in about 1 ns. Anisotropy decay arises due to the rotational diffusion and/or energy transfer to the radiative state within the same spectral window. The anisotropy decay of 1 is due to the rotational diffusion, which is consistent with the rotational diffusion time of similar sized molecules in DMSO.38
However, since the size of 3a is much bigger than that of 1, the anisotropy decay of 3a must arise from the energy transfer between the adjacent ZnPc units in the nanosphere. Considering the thickness of shell, as well as the lack of significant changes in absorption maxima and calculated distance between the neighbouring ZnPc units (∼3.3 nm), the energy transfer may occur laterally through a two dimensional polymer network of the shell. We are currently investigating details of the energy transfer phenomenon on the shell of the nanosphere.
Measurement of singlet oxygen generation efficiency
To compare the photosensitizing ability of a monomeric ZnPc and the ZnPc nanosphere, singlet oxygen generation (SOG) of 1 and 3a was studied using 1,3-diphenylisobenzofuran (DPBF) in DMF, or singlet oxygen sensor green (SOSG) reagent in PBS buffer as described in the literature.39,40 In DMF, 1 and 3a (both solutions contain the same amount of Pc) showed almost the same singlet oxygen generation quantum yield (0.52 and 0.5, respectively) suggesting that most of the ZnPc units of 3a behave like a monomer (as opposed to an aggregate). In PBS buffer containing 0.5 wt% pluronic F127 surfactant, however, 3a shows almost seven times higher SOG efficiency than 1 (Fig. 3), reflecting a lower propensity of 3a to aggregate in aqueous solution compared to 1, presumably due to the unique nanosphere structure.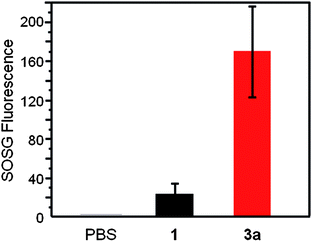 | ||
| Fig. 3 Singlet oxygen generation in the dispersion solution of 1 and 3a in PBS containing 0.5 wt% pluronic F127 surfactant after laser irradiation at 660 nm for 180 s. The singlet oxygen was measured using SOSG reagent. | ||
In vitro cellular uptake measured by flow cytometry
We also investigated the cellular uptake of the ZnPc nanospheres. For this study, a perylene-based fluorescent dye 5 used as a fluorescent probe§ was encapsulated into 3a to produce 5@3a by following the procedure reported earlier.29 The internalization of 5@3a into human oral cancer KB cells was monitored by confocal laser scanning microscopy. Facile internalization of 5@3a was observed at 37 °C, whereas no significant internalization was observed in the control experiments carried out with 5 only, or with 5@3a at 4 °C as illustrated in Fig. 4. These results were also confirmed by flow cytometry (Fig. S6†). Our observations suggested that the cellular uptake of 3a presumably occurred via endocytosis.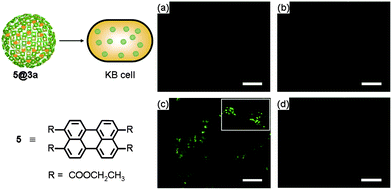 | ||
| Fig. 4 Fluorescence microscopic studies for cellular uptake of 3a encapsulating fluorescent dye 5 into KB cells. KB cells treated with (a) none, (b) only 5, (c) 5@3a at 37 °C, and (d) 5@3a at 4 °C. | ||
Post-synthetic modification of nanospheres
Although 3a has a much lower propensity to aggregate in aqueous solution compared to monomeric Pc molecules, the aqueous dispersion of 3a lacks long-term stability. To increase the dispersibility of 3a in PBS buffer without disturbing the properties of the ZnPc core, we decided to introduce water-solubilizing groups at linker sites by post-synthetic modification. We chose partial alkylation of the thioether units on the linkers to convert them into sulfonium groups (Scheme 1b).41 Treatment of 3a dispersed in 20% EtOH/DMSO with 64 equiv. of MeI at room temperature for two days followed by dialysis produced 4. Successful post-synthetic modification was confirmed by solid-state 13C NMR spectroscopy. It shows several new peaks at 20–50 ppm corresponding to the carbon atoms directly linked to the newly formed sulfonium ion in the linker (Fig. S7†). Elemental analysis of 4 showed that 43% of the thioether groups were converted to alkylmethylsulfonium groups. The morphology of 4, however, remained unaltered even after the MeI treatment as confirmed by TEM images (Fig. S8†).In vitro phototoxicity
To examine the potential utility of the Pc nanospheres in PDT, we prepared 4 dispersed in PBS buffer containing 0.5 wt% pluronic F127¶ and studied its in vitro photodynamic activity. We compared the phototoxicity of 4 and a well-studied zinc phthalocyanine tetrasulfonate (6)42,43 toward human oral cancer KB cells using the MTT assay (Fig. 5). In dark conditions, both monomer 6 and nanospheres 4 were nontoxic in the concentration range of 0.1–10 μM. After irradiation at 660 nm, cell viability decreased with increasing concentrations of the photosensitizers. However, whereas cell viability dropped to only 78% for 6 at 10 μM, it fell to 14% for the same concentration of 4. This result is consistent with the higher SOG efficiency of the Pc nanospheres compared with a Pc monomer in buffer solution. Although further studies are needed to elucidate the origin of the higher in vitro phototoxicity of 4, we hypothesize that the ZnPc nanosphere comprising ∼12000 Pc units may generate a higher concentration of singlet oxygen at the local site, leading to more effective oxidative damage of cancer cells. We note that the spherical shape and nanometer-scale size of 4 may further enhance photodynamic activity in vivo via the EPR effect. In vivo experiments are currently in progress.
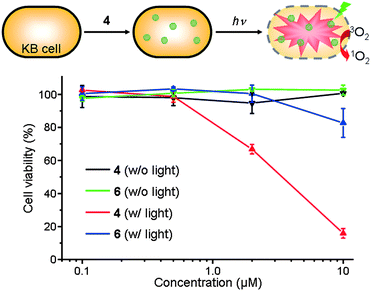 | ||
| Fig. 5 In vitro dark-toxicity and phototoxicity of ZnPc nanosphere 4 and monomeric ZnPc 6 dispersed in PBS solution containing 0.5 wt% pluronic F127 surfactant toward KB cells. | ||
Conclusion
We have demonstrated the design and one-pot direct synthesis of covalently linked ZnPc nanospheres with a very thin shell and hollow interior, composed of approximately 12000 Pc units on average, without using any templates or emulsifiers through thiol–ene “click” chemistry. The ZnPc nanospheres allow post-synthetic modification to improve their dispersibility in aqueous solution without altering the morphology of the nanospheres or the properties of the ZnPc cores. More importantly, the ZnPc nanospheres showed higher SOG efficiency and in vitro phototoxicity than monomeric Pc molecules, suggesting that ZnPc nanospheres are potentially useful as photosensitizers for PDT.
We anticipate that the ZnPc nanospheres would allow other post-synthetic modifications such as the introduction of targeting ligands at the linkers (via covalent attachment) or on the Zn ion (via coordination) to deliver the nanospheres to specific target sites. As described above, the successful encapsulation of a fluorescent dye into the ZnPc nanospheres suggests that therapeutic agents such as doxorubicin can also be encapsulated inside the nanospheres to develop a new dual mode nanocarrier that combines PDT and chemotherapy. The easy synthesis of a hollow spherical framework with a high Pc content, coupled with facile post-synthetic modification may allow Pc nanospheres to be a versatile platform for a diverse range of medical and non-medical applications.
Experimental section
Synthesis of ZnPc nanosphere 3a
A mixture of 1 (2.70 mg, 1.96 μmol), 2 (5.20 μL, 62.72 μmol) and AIBN (1.02 mg, 6.27 μmol) was dissolved in a 1:4 (v/v) mixture of ethanol and DMSO at room temperature. After degassing by a freeze–pump–thaw method, the mixture was heated at 70 °C for 3 h. AIBN (1.02 mg, 6.27 μmol) was added again to the solution and the mixture was heated for another 3 h. Finally the product was purified by dialysis against 20% EtOH/DMSO for 2 days. Solvent exchange to methanol by dialysis, evaporation of the solvent, followed by drying in vacuum yielded nanosphere 3a (1.8 mg; 67% based on 1). Elemental analysis calcd for 3a [(C72H88N8S8Zn)1(C1H3S1)7(DMSO)1]n): C 53.16, H 6.33, N 5.90, S 28.71; found: C 52.77, H 6.04, N 6.27, S 28.30%. The elemental analysis data indicates that 3a is composed of 1 and 2 in a ratio of 1:3.5 and contains 67% of the starting material 1.
Measurement of time-resolved fluorescence (TRF) and TRF anisotropies of 1 and 3a in DMSO
Time-resolved fluorescence (TRF) and TRF anisotropy were measured by using a time-correlated single photon counting (TCSPC) technique. The light source was a home-built cavity-dumped optical parametric oscillator (OPO) that gives pulses centered at 1280 nm with a pulse duration of ∼70 fs and a repetition rate of 1 MHz. Pump pulses at 640 nm were generated by the second harmonic generation in a β-barium borate crystal. Fluorescence was collected by a parabolic mirror, dispersed by a monochromator, and detected by a photomultiplier tube (PMH-100, Becker & Hickl). The instrumental response (full width at half-maximum) of the apparatus was 120 ps. Both parallel (Ip) and perpendicular (I⊥) polarization components in reference to the pump polarization were measured to obtain the isotropic component, Iiso = Ip + 2I⊥, and the anisotropy, γ(t) = (Ip − I⊥)/Iiso. The isotropic component is the same as the fluorescence at the magic angle (54.7°), and gives the lifetime of the emitting state.Measurement of singlet oxygen generation quantum yields of 1 and 3a in DMF
Singlet oxygen quantum yield (ΦΔ) determination was carried out in DMF using the steady-state chemical trapping method. Typically, a Pc solution (10 μM, 1 mL, air-saturated DMF) containing DPBF (90 μM) was irradiated with a light (660 nm). The irradiation period was selected in such a way that around 10% of DPBF was bleached in the first cycle. This procedure was repeated 10 times. The decay of the DPBF absorption at 411 nm was measured after each cycle. The singlet oxygen quantum yield was calculated according to the literature.39Singlet oxygen generation of 1 and 3a in PBS buffer
Singlet oxygen generation of 1 and 3a in PBS buffer solution containing 0.5 wt% pluronic F127 surfactant was monitored using singlet oxygen sensor green (SOSG) reagent according to the literature.40 The intensity of the green fluorescence emitted by SOSG upon generation of singlet oxygen species was measured by a spectrofluorometer using excitation/emission of 488/525 nm after laser irradiation at 660 nm for 180 s. The experiment was performed 5 times to obtain standard deviation.Post-synthetic modification of 3a
Methyl iodide (8 μL, 128 μmol) was added to a dispersion of 3a (1.8 mg, 1.3 μmol) in 20% EtOH/DMSO at rt, and incubated for 2 days, followed by dialysis against 20% EtOH/DMSO using a Snakeskin pleated dialysis tubing (MWCO: 8000) producing 4 (1.97 mg, 90%). Elemental analysis calcd for 4 [(C72H80N8S8Zn)1(C2H6S2)7(CH3I)3(DMSO)1]n: C 52.60, H 6.05, N 5.84, S 26.80; found: C 52.12, H 5.55, N 5.83, S 27.32%.In vitro dark-toxicity of 4 and 6 to KB cells
KB cells were seeded in a 96-well plate at a density of 5 × 103 cells per well in 200 μL of DMEM containing 10% FBS and 1% PS and incubated in a humidified 5% CO2 atmosphere at 37 °C for 24 h. The cell culture medium was replaced with 200 μL of a fresh one containing 20 μL of PBS or 20 μL of a PBS dispersion of 4 or 6 (0.1–10 μM) containing 0.5 wt% of pluronic F127 surfactant. The cells were incubated for 1 day at 37 °C. Subsequently, the cells were incubated with 200 μL of a fresh medium containing 20 μL of methylthiazolyldiphenyl-tetrazolium bromide (MTT) (5 mg mL−1) for an additional 4 h at 37 °C, and then the medium was gently removed. The purple, water insoluble crystals formed by live cells remaining at the bottom of the wells were dissolved with 200 μL of DMSO and the solution was gently shaken for 10 min. UV absorption of the solution at 590 nm was measured by a multi-well plate reader. The experiment was performed 3 times to obtain a standard deviation.In vitro phototoxicity of 4 and 6 to KB cells
KB cells were seeded in a 96-well plate at a density of 5 × 103 cells per well in 200 μL of DMEM containing 10% FBS and 1% PS and incubated in a humidified 5% CO2 atmosphere at 37 °C for 1 day. The cell culture medium was replaced with 200 μL of a fresh one containing 20 μL of PBS or 20 μL of a PBS dispersion of 4 or 6 (0.1–10 μM) containing 0.5 wt% of pluronic F127 surfactant. The cells were incubated for 1 day at 37 °C. A 660 nm laser (500 mW cm−2) was irradiated to the cells for 2.5 min. After irradiation, the cells were incubated for 1 day at 37 °C. Subsequently, the cells were incubated with 200 μL of a fresh medium containing 20 μL of MTT (5 mg mL−1) for an additional 4 h at 37 °C, and then the medium was gently removed. The purple, water insoluble crystals formed by live cells remaining at the bottom of the wells were dissolved with 200 μL of DMSO and the solution was gently shaken for 10 min. UV absorption of the solution at 590 nm was measured by a multi-well plate reader. The experiment was performed 3 times to obtain standard deviation.Notes and references
- N. B. McKeown, Phthalocyanine Materials: Synthesis, Structure and Function, Cambridge University Press, Cambridge, 1998 Search PubMed.
- G. de la Torre, M. Nicolau, T. Torres, in Supramolecular Photosensitive and Electroactive Materials, Academic Press, New York, 2001 Search PubMed.
- G. de la Torre, P. Vazquez, F. Agullo-Lopez and T. Torres, J. Mater. Chem., 1998, 8, 1671 RSC.
- G. de la Torre, P. Vazquez, F. Agullo-Lopez and T. Torres, Chem. Rev., 2004, 104, 3723 CrossRef CAS.
- J. A. A. W. Elemans, R. Van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251 CrossRef CAS.
- G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000 RSC.
- G. Torre, G. Bottari, U. Hahn and T. Torres, Struct. Bonding, 2010, 135, 1 CrossRef.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19 RSC.
- E. A. Lukyanets, J. Porphyrins Phthalocyanines, 1999, 3, 424 CrossRef CAS.
- M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897 CrossRef CAS.
- M. Zhang, T. Murakami, K. Ajima, K. Tsuchida, A. S. D. Sandanayaka, O. Ito, S. Iijima and M. Yudasaka, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14773 CrossRef CAS.
- W.-D. Jang, Y. Nakagishi, N. Nishiyama, S. Kawauchi, Y. Morimoto, M. Kikuchi and K. Kataoka, J. Controlled Release, 2006, 113, 73 CrossRef CAS.
- N. Nishiyama, Y. Nakagishi, Y. Morimoto, P.-S. Lai, K. Miyazaki, K. Urano, S. Horie, M. Kumagai, S. Fukushima, Y. Cheng, W.-D. Jang, M. Kikuchi and K. Kataoka, J. Controlled Release, 2009, 133, 245 CrossRef CAS.
- A. M. Master, M. E. Rodriguez, M. E. Kenney, N. L. Oleinik and A. S. Gupta, J. Pharm. Sci., 2010, 99, 2386 CAS.
- H. L. Lu, W. J. Syu, N. Nishiyama, K. Kataoka and P. S. Lai, J. Controlled Release, 2011, 155, 458 CrossRef CAS.
- P. Ngweniform, G. Abbineni, B. Cao and C. Mao, Small, 2009, 5, 1963 CrossRef CAS.
- N. Nombona, K. Maduray, E. Antunes, A. Karsten and T. Nyokong, J. Photochem. Photobiol., B, 2012, 107, 35 CrossRef CAS.
- P. P. S. Lee, T. Ngai, J.-D. Huang, C. Wu, W.-P. Fong and D. K. P. Ng, Macromolecules, 2003, 36, 7527 CrossRef CAS.
- C. Liu, J. Guo, W. Yang, J. Hu, C. Wang and S. Fu, J. Mater. Chem., 2009, 19, 4764 RSC.
- H. S. Qian, H. C. Guo, P. C.-L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285 CrossRef CAS.
- M. K. K. Oo, Y. Yang, Y. Hu, M. Gomez, H. Du and H. Wang, ACS Nano, 2012, 6, 1939 CrossRef.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Angew. Chem., Int. Ed., 2012, 51, 6158 CrossRef CAS.
- R. Solaro, F. Chiellini and A. Battisti, Materials, 2010, 3, 1928 CrossRef CAS.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. yang, P. L. Rachel Ee, P. T. Hammond and Y. Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228 CrossRef CAS.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840 CrossRef CAS.
- H. Maeda, J. Controlled Release, 1992, 19, 315 CrossRef CAS.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189 CrossRef CAS.
- H. Maeda, J. Fang, T. Inutsuka and Y. Kitamoto, Int. Immunopharmacol., 2003, 3, 319 CrossRef CAS.
- D. Kim, E. Kim, J. Kim, K. M. Park, K. Baek, M. Jung, Y. H. Ko, W. Sung, H. S. Kim, J. H. Suh, C. G. Park, O. S. Na, D.-k. Lee, K. E. Lee, S. S. Han and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 3471 CrossRef CAS.
- W. Kuykendall and S. C. Zimmerman, Nat. Nanotechnol., 2007, 2, 201 CrossRef.
- E. Kim, J. Lee, D. Kim, K. E. Lee, S. S. Han, N. Lim, J. Kang, C. G. Park and K. Kim, Chem. Commun., 2009, 1472 RSC.
- E. Kim, D. Kim, H. Jung, J. Lee, S. Paul, N. Selvapalam, Y. Yang, N. Lim, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2010, 49, 4405 CrossRef CAS.
- D. Kim, E. Kim, J. Lee, S. Hong, W. Sung, N. Lim, C. G. Park and K. Kim, J. Am. Chem. Soc., 2010, 132, 9908 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS.
- X.-F. Zhang, Q. Xi and J. Zhao, J. Mater. Chem., 2010, 20, 6726 RSC.
- D. V. O'Connor and D. Phillips, Time Correlated Single Photon Counting, Academic Press, London, 1984 Search PubMed.
- K. Wynne and R. M. Hochstrasser, Chem. Phys., 1993, 171, 179 CrossRef CAS.
- G. R. Fleming, Chemical Application of Ultrafast Spectroscopy, Oxford University Press, New York, 1986 Search PubMed.
- W. Spiller, H. Kliesch, D. Wohrle, S. Hackbarth, B. Roder and G. Schnurpfeil, J. Porphyrins Phthalocyanines, 1998, 2, 145 CrossRef CAS.
- Z. Zhu, Z. Tang, J. A. Phillips, R. Yang, H. Wang and W. Tan, J. Am. Chem. Soc., 2008, 130, 10856 CrossRef CAS.
- R. W. Bost, J. E. Everett and J. Zaia, J. Am. Chem. Soc., 1940, 62, 1752 CrossRef CAS.
- L. Howe and J. Z. Zhang, J. Phys. Chem. A, 1997, 101, 3207 CrossRef CAS.
- H. K. Moon, M. Son, J. E. Park, S. M. Yoon, S. H. Lee and H. C. Choi, NPG Asia Mater., 2012, 4, 1 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis and characterization of 1 and 3b, in vitro cellular uptake. See DOI: 10.1039/c2sc21254d |
| ‡ These authors contributed equally to this work. |
| § Encapsulation of fluorescent probe 5 was performed to facilitate the visualization of the intracellular localization of the nanospheres, to confirm the encapsulation ability of the nanospheres, and to demonstrate their potential as a dual mode nanocarrier for a combination of PDT and chemotherapy. |
| ¶ Although the post-synthetic modification of ZnPc nanospheres significantly improved their dispersibility, utilization of the pluronic F127 was necessary to achieve long-term dispersibility. |
| This journal is © The Royal Society of Chemistry 2013 |