Compact tridentate ligands for enhanced aqueous stability of quantum dots and in vivo imaging†
Edmond
Gravel
a,
Chloé
Tanguy
a,
Elsa
Cassette
b,
Thomas
Pons
b,
Fabien
Knittel
a,
Nicholas
Bernards
c,
Anikitos
Garofalakis
c,
Frédéric
Ducongé
c,
Benoît
Dubertret
*b and
Eric
Doris
*a
aCEA, iBiTecS, Service de Chimie Bioorganique et de Marquage, 91191 Gif-sur-Yvette, France. E-mail: eric.doris@cea.fr; Fax: +33 169 08 79 91; Tel: +33 169 08 80 71
bLaboratoire de Physique et d'Etude des Matériaux, UMR8213 du CNRS, ESPCI, 10 rue Vauquelin, 75005 Paris, France. E-mail: benoit.dubertret@espci.fr; Tel: +33 140 79 45 92
cCEA, I2BM, Service Hospitalier Frédéric Joliot, INSERM U1023, Laboratoire d'Imagerie Moléculaire Expérimentale, Université Paris Sud, 4 place du général Leclerc, 91401 Orsay, France
First published on 11th October 2012
Abstract
We describe here a simple and versatile route for the preparation of a tridentate ligand that not only provides stable interactions with colloidal semiconductor nanocrystals (quantum dots, QDs) but also makes them water soluble. The designed ligand incorporates a biocompatible solubilizing polyethylene glycol (PEG) chain connected to a tridentate thiol motif with improved affinity towards the QDs surface. Stability experiments revealed better resistance to various drastic conditions compared to classical dihydrolipoic-based ligands. The enhanced stability of the assembly also enabled in vivo imaging experiments using near infrared-emitting QDs.
Introduction
Since their discovery in the 1980s, semiconductor nanocrystals, also known as quantum dots (QDs), have drawn a great deal of attention for various types of applications, including biomedical imaging. Quantum dots display size-dependent fluorescence emission, high quantum yield, and improved resistance to photobleaching compared to classical organic dyes.1–4 In addition, recent developments led to the synthesis of nanocrystals emitting in the most appropriate optical window for in vivo applications that is in the near infrared (NIR) region.5–13QDs are classically synthesized using the pyrolysis technique which produces nanocrystals coated with hydrophobic ligands.14 However, for biomedical applications hydrophilic QDs are required and various strategies have been developed to convert hydrophobic nanoparticles into hydrophilic QDs. For example, one can either (i) use micellar surfactants to encapsulate the nanocrystals (Fig. 1a),15–20 (ii) perform cap exchange with precursors to grow an inorganic hydrophilic shell around the crystals (e.g. silica coating) (Fig. 1b),21–23 or (iii) exchange the primary lipophilic ligands with hydrophilic moieties (Fig. 1c).24,25 In the latter case, and if biological applications are foreseen, the new ligands must, in addition, provide biocompatibility to the QDs through suitable solubilizing units such as polyethylene glycol chains (PEG).26 For improved stability, anchoring of the ligands to the nanocrystals must be robust and stand various stress conditions that are likely to be encountered in biological medium. Amongst the various ligands developed in the literature, thiols have emerged as the most widespread group of ligands since thiolate groups (–S−) are highly affine toward the surface of the QDs.27–29 However, the interaction can be disrupted under oxidative conditions30 that induce the formation of disulfide bridges and release of the ligands from the QD surface. Simple thiol groups are also readily displaced from the QD's surface by competing sulphur-containing molecules which are abundant species in biological media. However, this stability issue can be partly circumvented using bidentate dihydrolipoic acid (DHLA) ligands that were originally developed by Mattoussi and Bawendi.31–35
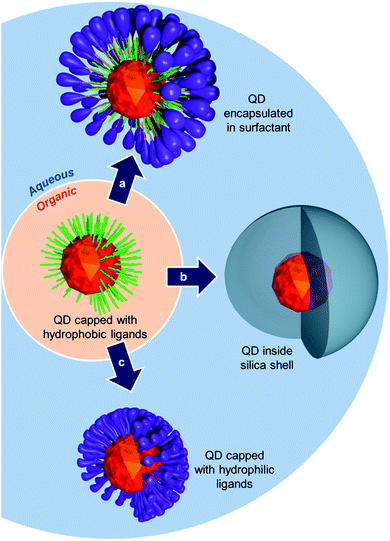 | ||
| Fig. 1 Schematic representation of different possible routes to transfer QDs from organic to aqueous medium. | ||
DHLA-based ligands were shown to exhibit stronger anchoring properties compared to their monothiol equivalents due to the chelating effect of the bidentate group (two thiol units). Recent development also focused on multidentate ligands36–38 including poly-DHLA39–41 and poly-thiols42 whose increased coordination units gave access to more stable nanocrystals.
We describe here the synthesis and evaluation of a novel family of QD ligands made of a compact tris(mercaptomethyl) head group (three thiol units) and a PEG chain. The tris(mercaptomethyl) chelating group (TMM) has already been shown to bind strongly to metal surfaces while maintaining dense coverage. Previous studies on gold surfaces43 and nanoparticles44 demonstrated that tris(mercaptomethyl) groups are stronger chelates than their dithiol counterparts while providing comparable ligand density. A TMM-based ligand was therefore synthesized and used in the capping of QDs before the nanoparticles were evaluated as regards their overall colloidal stability and in vivo behavior.
Results and discussion
Design and synthesis of the tridentate ligands
For in vivo applications, the ideal QD ligands should, (i) be robustly attached to the nanocrystal surface, (ii) provide stable colloidal dispersion, and (iii) confer high biocompatibility to the nanoparticle. These properties could be respectively attained by combining multiple coordinating groups in the same structure to reinforce surface interactions, attaching a highly solubilizing group, and using a non-toxic stealth ligand. With these prerequisites in mind, the compact tris(mercaptomethyl) head was selected as an anchoring group because of its improved chelating properties.41,42 The latter was connected to a solubilizing PEG chain that classically promotes extended blood residence time while being non-toxic.26 A simple and efficient route to the pegylated tridentate ligand was thus developed.We conceived that connection of the TMM chelating unit to the polyethylene glycol solubilizing chain using an ether linkage could improve the overall robustness of the ligand as the bridging C–O–C bond is non-hydrolysable. To this end, the hydroxyl group of PEG2000–monomethyl ether was first deprotonated by treatment with sodium hydride in refluxing THF before tetrakis(bromomethyl) methane in excess was added. Nucleophilic substitution of one of the bromine atoms permitted to anchor the PEG chain to the trithiol precursor unit. In this step, the addition of over-stoichiometric amounts of the bromomethyl electrophile enabled us to substantially minimize the formation of poly-substituted adducts (multiple addition of PEG chains to the same tetrakis(bromomethyl) methane molecule) and recover, as the main product, the expected mono-pegylated tribromide compound 2. The latter was thereafter converted into the corresponding trithioacetate 3 by nucleophilic displacement of the three remaining bromine atoms by potassium thioacetate. Compound 3, which corresponds to the masked form of our target ligand, was finally converted to the corresponding free trithiol 1 by lithium aluminum hydride deprotection of the thiol groups (Scheme 1). This simple route thus permitted the multigram scale synthesis of the tridentate ligand. It is to be noted that the resulting trithiol 1 was stable over time and could be stored for more than a year at −20 °C with neither noticeable oxidation of the SH groups nor degradation.
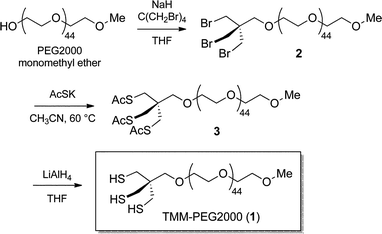 | ||
| Scheme 1 Synthesis of the TMM–PEG2000 ligand (1). | ||
Preparation of PEG-capped hydrophilic CdSe/CdS/ZnS core–shell QDs
To make fluorescent nanocrystals water soluble, the native capping oleic acid and oleylamine ligands were replaced by our newly designed TMM–PEG ligand (Fig. 2). Evaluation of the performances of the tridentate TMM–PEG as a colloidal stabilizer of QDs was then assessed by comparison with those of the thoroughly investigated bidentate DHLA–PEG ligands.32 Toward this goal, multilayered CdSe/CdS/ZnS QDs were synthesized using the high temperature pyrolysis process, as described elsewhere.45 The resulting oleic acid/oleylamine capped nanocrystals were then precipitated in ethanol and transferred to chloroform to perform the ligand exchange reaction.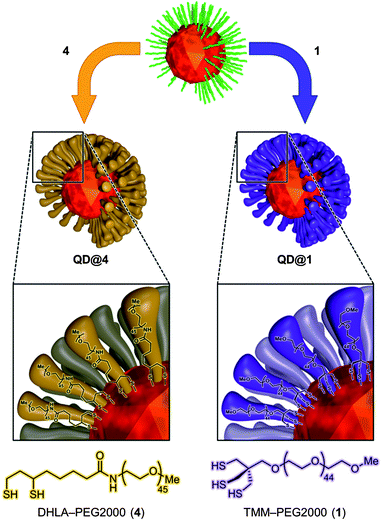 | ||
| Fig. 2 Schematic representation of ligand exchange and interaction with the crystal surface. | ||
The native organic layer was replaced by thiol-based ligands by overnight stirring of the QD suspension in refluxing chloroform and in the presence of an excess of either TMM–PEG2000 (1) or DHLA–PEG2000 (4). The ligand excess was then removed by precipitation of the QDs in a mixture of chloroform–ethanol–hexane, followed by centrifugation. The supernatant was discarded and the precipitate (containing the QDs) was suspended in water. Further purification by ultracentrifugation finally afforded a clear and homogeneous QD colloidal suspension. Both TMM and DHLA hence provided access to aggregate-free aqueous dispersions of the fluorescent nanoparticles that were stable for several months at 4 °C. The hydrodynamic radii of QDs encapsulated with TMM–PEG2000 or DHLA–PEG2000 were measured by dynamic light scattering (DLS) and values of 11.2 ± 0.5 nm and 9.3 ± 0.5 nm were obtained, respectively (see Fig. S1 in the ESI†). These values are consistent with those previously reported in the literature for shorter PEG-chain ligands.13,46
Optical characterization of the QDs capped with multidentate ligands
Absorption and fluorescence spectra were recorded in hexane for QDs capped with the original oleic acid/oleylamine ligands and in pure water for the two hydrophilic QDs (Fig. 3). The aqueous QD dispersions that were produced by ligand exchange with TMM–PEG (QD@1) or DHLA–PEG (QD@4) exhibited absorption and emission profiles that were nearly identical to those of the starting QDs. As the optical properties of the QD are retained after the exchange step, one can speculate that the replacement of the original organic layer by the pegylated multidentate ligands did not alter the inorganic core of the nanocrystals. It is noteworthy, that while the photoluminescence quantum yield of the organic grown QDs was 44%, those of the aqueous colloidal QDs dropped to ca. 25%. This feature of QDs that were transferred into aqueous medium has already been observed and commented on by others.31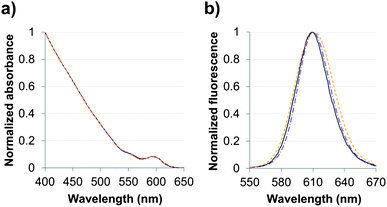 | ||
Fig. 3 (a) Absorbance and (b) fluorescence spectra of CdSe/CdS/ZnS QDs capped with organic ligands ( ) in hexane or with either DHLA–PEG2000 ( ) in hexane or with either DHLA–PEG2000 ( ) or TMM–PEG2000 ( ) or TMM–PEG2000 ( ) in water. ) in water. | ||
Evaluation of the stability of the QDs capped with multidentate ligands
Colloidal stability of the aqueous dispersions of TMM–PEG2000-capped nanocrystals was evaluated under various conditions including a broad pH range, oxidizing conditions, and in the presence of the competing ligand dithiothreitol (DTT). The performances of our newly designed ligand under diverse conditions were compared to those of the well-established DHLA–PEG ligands.First, colloidal stability of the two coated QDs was assessed by measuring their fluorescence intensities (excitation at 400 nm) after incubation in buffered solutions of pH values ranging from 1.5 to 12. Fig. 4 shows the fluorescence intensities of TMM–PEG- and DHLA–PEG-coated QDs at various pH values. The first observation was that under acidic conditions and the fluorescence of QD@1 remained steady until pH 3 was reached. Conversely, we observed a drop in the fluorescence intensity of QD@4 which started from pH 6 as a 20% loss of the initial fluorescence was detected at this value. This effect was even more accentuated at lower pH values with a decrease of fluorescence of ca. 40% and 50% at pH 4 and pH 3, respectively. These results underline the higher stability of the TMM ligand over DHLA under mildly acidic conditions. However, it should be noted that fluorescence of both samples was nearly extinguished under extreme acidic conditions (i.e. pH 1.5). The same trend, that is higher stability of TMM–PEG ligand, was observed under basic conditions, although fewer differences were evidenced at mildly basic pH 8 where both samples lost approximately 45% of their initial fluorescence. At higher pH values (e.g. pH 10), a more significant benefit of the TMM–PEG coating was detected with 45% of the initial fluorescence remaining. Under the same conditions, DHLA–PEG QDs lost 90% of their photoluminescence. At extreme basic pH values (above 12), virtually all fluorescence had vanished from the two QD samples.
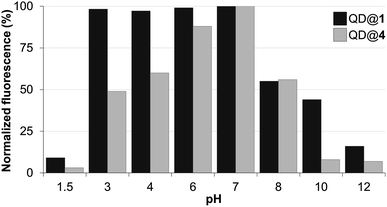 | ||
| Fig. 4 Fluorescence intensity of QDs capped with DHLA–PEG2000 (QD@4) or TMM–PEG2000 (QD@1) at different pH values. | ||
The stability of the colloidal QDs was also assessed under oxidizing conditions by monitoring fluorescence intensity variations in the presence of molecular iodine. In fact, iodine is a well-established reagent for the mild oxidation of thiols into the corresponding disulfides.47,48 If this reaction were to occur with the thiolated QD-capping ligands (TMM–PEG or DHLA–PEG), they would detach from the QD surface and lead to aggregation of the nanocrystals and loss of the fluorescence properties. Accordingly, a 0.25 μM colloidal solution of QD capped with either TMM–PEG or DHLA–PEG was stirred with increasing concentrations of molecular iodine ranging from 33 to 300 μM before fluorescence emission (excitation at 400 nm) was recorded. The performances of the TMM–PEG QDs under the oxidizing conditions were obviously superior to those of the DHLA–PEG QDs since lower fluorescence extinction was observed at all of the studied iodine concentrations. The emission of the DHLA QDs sample decreased rapidly and steadily at the same time as the local concentration of iodine increased and less than 15% of the initial fluorescence was detected at 300 μM of I2. In contrast, under the highest iodine concentrations (i.e. 300 μM), TMM–PEG QDs retained nearly 60% of their fluorescence (Fig. 5). These observations demonstrate the higher resistance of the newly synthesized TMM-based ligands (compared to DHLA ligands) regarding their susceptibility to oxidation which is often challenging when working with thiol derivatives.
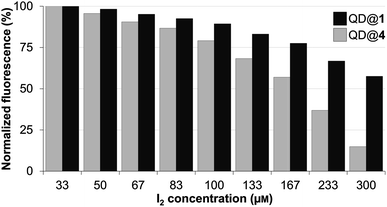 | ||
| Fig. 5 Fluorescence intensity of QDs capped with DHLA–PEG2000 (QD@4) or TMM–PEG2000 (QD@1) when exposed to increasing concentrations of molecular iodine. | ||
Finally, stability of the colloidal QDs was evaluated in the presence of a competing ligand.40 Considering that QDs coated with TMM–PEG ligands are intended to be used in vivo, it appeared important to investigate their stability upon exposure to other thiol groups which are abundant species in biological environments. Dithiothreitol (DTT), a dithiol derivative of threose, was chosen as a test competitor. DTT is made of two thiol units that are able to bind to the surface of the QDs. However, DTT cannot confer aqueous solubility to the QD which will therefore precipitate. QDs capped with either DHLA–PEG or TMM–PEG were diluted in 1.5 M aqueous DTT and aggregation kinetics were evaluated by measuring the absorption spectra of each sample at different time frames, after mild centrifugation to eliminate QD aggregates. Concentration of the non-aggregated QDs (those which were unaffected by DTT) was evaluated by optical density (OD) measurement at 400 nm. While we observed no changes for the TMM–PEG QDs throughout the duration of the experiment (720 h), DHLA–PEG QDs precipitated after 200 hours of exposure to DTT to reach a plateau at ca. 10% of the original absorbance (Fig. 6). These experiments highlight the higher stability of QDs capped with TMM ligands when exposed to competing species.
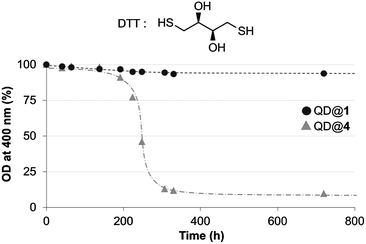 | ||
| Fig. 6 Time dependant evolution of the optical density at 400 nm measured for QDs capped with DHLA–PEG2000 (QD@4) or TMM–PEG2000 (QD@1) incubated with DTT (structure shown). | ||
In vivo imaging studies
With encouraging results from the in vitro stability tests, the next step was to evaluate the in vivo behavior of TMM–PEG-capped QDs. For optimal in vivo imaging applications, the QD emission wavelength should ideally be set in a region of the spectrum where blood and other tissues have low absorption which corresponds to the near-infrared domain (NIR). Thus, cadmium-free near infrared-emitting CuInS2/ZnS quantum dots (NIR-QDs) were synthesized and subsequently capped with our newly developed tridentate–PEG ligand 1. Absorption and emission spectra of the TMM–PEG coated nanoparticles are depicted in Fig. 7 which shows that the absorption spectrum of the TMM–PEG NIR-QDs is continuous up to ca. 700 nm and that the maximum emission is detected at 760 nm. It is to be noted that the hydrodynamic diameter of the NIR-emitting QDs could not be measured because of the strong absorption of the quantum dots at 633 nm, which is the operating wavelength of the DLS laser beam. Since the inorganic core–shell diameter of NIR-QDs is only slightly smaller (typically 3–4 nm) than that of visible-emitting CdSe/CdS/ZnS QDs (typically 6–7 nm), one can assume that their final (PEG-coated) hydrodynamic diameter is in the same range, that is approximately 20 nm.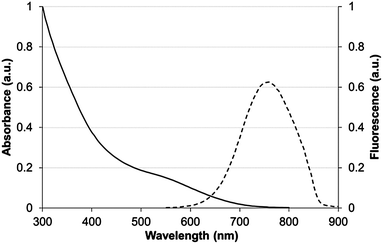 | ||
| Fig. 7 Absorbance (solid line) and fluorescence (dashed line) spectra of CuInS2/ZnS QDs capped with TMM–PEG2000 in water. | ||
Prior to in vivo experiments, preliminary toxicity studies were undertaken. No harmful effect of the TMM–PEG NIR-QDs was detected at the QD-concentration (ca. 100 nM) used for the in vivo experiments and cell proliferation assays on MDA-MB-231 cells indicated an IC50 value of 12.2 ± 1.1 μM (see Fig. S2 in the ESI†). The water soluble TMM–PEG NIR-QDs were thereafter injected intravenously to female nude mice (n = 3) at a dose of 10 nmol kg−1 (ca. 200 pmol per animal) and their biodistribution was monitored in real-time using whole body planar fluorescence imaging (Fig. 8). The first observation is that the TMM–PEG coated QDs produced a clear in vivo image contrast which demonstrated their effectiveness as NIR optical probes. During the first two hours after injection, a high contrast of the blood vessels was observed, suggesting that most of the injected QDs remained in the bloodstream. As time elapsed, the fluorescence distribution in the mice became more diffuse but the vessels were still clearly visible 4 h after injection. However, nearly all fluorescence had vanished from the bloodstream 24 h after injection. A comparable overall in vivo behavior was observed for DHLA–PEG2000-coated NIR-QDs (see Fig. S3 in the ESI†).
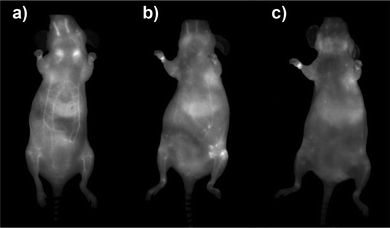 | ||
| Fig. 8 Whole body fluorescence images recorded (a) 2 h, (b) 4 h, and (c) 24 h after intravenous injection of TMM–PEG2000-capped NIR-QDs to nude mice (ventral side of the animal). | ||
The biodistribution of a nanoparticulate system is classically governed by its size and surface coating. In our case, the slow clearance from the bloodstream is not only due to the minimization of the nanoparticles opsonization thanks to the PEGs but also to the stability of the tridentade QD-complexing unit. Consequently, QD uptake by macrophages is delayed and higher blood residence time is observed. Quantification of fluorescence by in vivo planar imaging is usually difficult to achieve because of the absorption and scattering of photons when migrating through tissues. To have a clearer picture of the TMM–PEG-QD tissue distribution, animals were euthanized 24 h after injection and organ resection permitted ex vivo quantification of mean fluorescence (Fig. 9). Analysis of the different mice groups showed distribution of the QDs in the main organs (Fig. 9a) with a slightly higher uptake (Fig. 9b) in the liver, lymph nodes (especially in brachial lymph nodes), and skin. The uptake of the QDs by the skin was unexpected, although such a phenomenon has already been observed with PEG-coated fluorescent gadolinium oxide nanoparticles.49 On the other hand, the preferential accumulation in the liver and lymph nodes can be ascribed to QD-uptake by local phagocytes that act as nanoparticles traps.9 Interestingly, we did not observe any increase of the uptake in the spleen, although this organ is also rich in phagocytes. Besides, only weak fluorescence was detected in the kidneys or bladder, suggesting low renal excretion. In fact, the hydrodynamic diameter of the TMM–PEG2000-encapsulated QDs is likely above the renal filtration threshold.50 This behavior is in line with previous reports where QDs coated with short polyethylene glycol chains (up to PEG400) were mainly excreted through the urines, while QDs with larger PEG coatings were detected in intestines and lymph nodes.13 Our study thus corroborates the influence of the PEG chain length on the biodistribution and clearance of nanoparticulate systems.
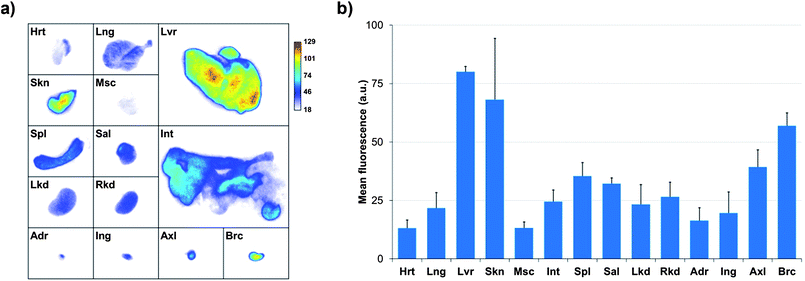 | ||
| Fig. 9 (a) Planar NIR fluorescence images of different organs of nude mice 24 h after injection of TMM–PEG2000-capped NIR-QDs and (b) quantification of the mean fluorescence adjusted by subtracting the autofluorescence of the same organs measured on a non-injected control mouse (Hrt = heart; Lng = lung; Lvr = liver; Skn = skin; Msc = muscle; Int = intestine; Spl = spleen; Sal = salivary gland; Lkd = left kidney; Rkd = right kidney; Adr = adrenal gland; Ing = inguinal lymph node; Axl = axillary lymph node; Brc = brachial lymph node). | ||
Conclusions
A new family of ligands incorporating a tris(mercaptomethyl) chelating head and a polyethylene glycol chain has been synthesized and evaluated for the stabilization of quantum dots in aqueous media. Comparison with well-known DHLA-based ligands demonstrated that colloids obtained from the new ligand were more resistant to pH variations, oxidizing conditions, and against an excess of a competing ligand. Colloids of NIR-emitting QDs capped with TMM–PEG2000 ligands were prepared and injected into nude mice. The acquired images demonstrated that these nanoparticles could be used as NIR optical imaging probes for in vivo imaging and permit the assessment of their in vivo distribution which was confirmed ex vivo, after organ resection. Overall, the TMM–PEG ligands described herein appear well suited for the biocompatibilization of QDs for biomedical applications and compare favorably with previously reported systems in terms of overall stability.Acknowledgements
The “Service de Chimie Bioorganique et de Marquage” belongs to the Laboratory of Excellence in Research on Medication and Innovative Therapeutics (LabEx LERMIT). The “Agence Nationale de la Recherche” (ANR) (Dot Imager and IVF-proteomic projects) is acknowledged for financial support.Notes and references
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602 CrossRef CAS.
- A. Rogach, N. Gaponik, J. Lupton, C. Bertoni, D. Gallardo, S. Dunn, N. Li Pira, M. Paderi, P. Repetto, S. G. Romanov, C. O'Dwyer, C. M. Sotomayor Torres and A. Eychmüller, Angew. Chem., Int. Ed., 2008, 47, 6538 CrossRef CAS.
- T. Jamieson, R. Bakhshi, D. Petrova, R. Pocock, M. Imani and A. M. Seifalian, Biomaterials, 2007, 28, 4717 CrossRef CAS.
- Y. He, Y. L. Zhong, Y. Y. Su, Y. M. Lu, Z. Y. Jiang, F. Peng, T. Xu, S. Su, Q. Huang, C. Fan and S. T. Lee, Angew. Chem., Int. Ed., 2011, 50, 5694 Search PubMed.
- P. M. Allen, W. Liu, V. P. Chauhan, J. Lee, A. Y. Ting, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 470 CrossRef CAS.
- L. Li, T. J. Daou, I. Texier, T. K. C. Tran, Q. L. Nguyen and P. Reiss, Chem. Mater., 2009, 21, 2422 CrossRef CAS.
- T. Pons, E. Pic, N. Lequeux, E. Cassette, L. Bezdetnaya, F. Guillemin, F. Marchal and B. Dubertret, ACS Nano, 2010, 4, 2531 CrossRef CAS.
- E. Cassette, T. Pons, C. Bouet, M. Helle, L. Bezdetnaya, F. Marchal and B. Dubertret, Chem. Mater., 2010, 22, 6117 CrossRef CAS.
- Y. Y. Su, F. Peng, Z. Y. Jiang, Y. L. Zhong, Y. M. Lu, X. Jiang, Q. Huang, C. Fan, S.-T. Lee and Y. He, Biomaterials, 2011, 32, 5855 CrossRef CAS.
- M. Praetner, M. Rehberg, P. Bihari, M. Lerchenberger, B. Uhl, M. Holzer, M. E. Eichhorn, R. Fürst, T. Perisic, C. A. Reichel, U. Welsch and F. Krombach, Biomaterials, 2010, 31, 6692 CrossRef CAS.
- H. Yukawa, M. Watanabe, N. Kaji, Y. Okamoto, M. Tokeshi, Y. Miyamoto, H. Noguchi, Y. Baba and S. Hayashi, Biomaterials, 2012, 33, 2177 CrossRef CAS.
- H. S. Choi, B. I. Ipe, P. Misra, J. H. Lee, M. G. Bawendi and J. V. Frangioni, Nano Lett., 2009, 9, 2354 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759 CrossRef CAS.
- X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41 CrossRef CAS.
- T. Pellegrino, L. Manna, S. Kudera, T. Liedl, D. Koktysh, A. L. Rogach, S. Keller, J. Rädler, G. Natile and W. J. Parak, Nano Lett., 2004, 4, 703 CrossRef CAS.
- N. Travert-Branger, F. Dubois, O. Carion, G. Carrot, B. Mahler, B. Dubertret, E. Doris and C. Mioskowski, Langmuir, 2008, 24, 3016 CrossRef CAS.
- N. Travert-Branger, F. Dubois, J. P. Renault, S. Pin, B. Mahler, E. Gravel, B. Dubertret and E. Doris, Langmuir, 2011, 27, 4358 CrossRef CAS.
- E. Genin, O. Carion, B. Mahler, B. Dubertret, N. Arhel, P. Charneau, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2008, 130, 8596 CrossRef CAS.
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861 CrossRef CAS.
- S. Selvan, T. Tan and J. Ying, Adv. Mater., 2005, 17, 1620 CrossRef CAS.
- M. Darbandi, G. Urban and M. Krüger, J. Colloid Interface Sci., 2012, 365, 41 CrossRef CAS.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS.
- F. Dubois, B. Mahler, B. Dubertret, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2007, 129, 482 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS.
- W. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. G. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530 CrossRef CAS.
- V. V. Breus, C. D. Heyes, K. Tron and G. U. Nienhaus, ACS Nano, 2009, 3, 2573 CrossRef CAS.
- R. Gill, L. Bahshi, R. Freeman and I. Willner, Angew. Chem., 2008, 120, 1700 CrossRef.
- J. Aldana, N. Lavelle, Y. J. Wang and X. G. Peng, J. Am. Chem. Soc., 2005, 127, 2496 CrossRef CAS.
- H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 12142 CrossRef CAS.
- H. T. Uyeda, I. L. Medintz, J. K. Jaiswal, S. M. Simon and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 3870 CrossRef CAS.
- W. Liu, M. Howarth, A. B. Greytak, Y. Zheng, D. G. Nocera, A. Y. Ting and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 1275 Search PubMed.
- E. Muro, T. Pons, N. Lequeux, A. Fragola, N. Sanson, Z. Lenkei and B. Dubertret, J. Am. Chem. Soc., 2010, 132, 4556 CrossRef CAS.
- K. Susumu, E. Oh, J. B. Delehanty, J. B. Blanco-Canosa, B. J. Johnson, V. Jain, W. J. Hervey, W. R. Algar, K. Boeneman, P. E. Dawson and I. L. Medintz, J. Am. Chem. Soc., 2011, 133, 9480 CrossRef CAS.
- A. M. Smith and S. Nie, J. Am. Chem. Soc., 2008, 130, 11278 CrossRef CAS.
- W. Liu, A. B. Greytak, J. Lee, C. R. Wong, J. Park, L. F. Marshall, W. Jiang, P. N. Curtin, A. Y. Ting, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 472 CrossRef CAS.
- P. F. Zhang, S. H. Liu, D. Y. Gao, D. H. Hu, P. Gong, Z. H. Sheng, J. Z. Deng, Y. F. Ma and L. T. Cai, J. Am. Chem. Soc., 2012, 134, 8388 CrossRef CAS.
- I. Yildiz, E. Deniz, B. McCaughan, S. F. Cruickshank, J. F. Callan and F. M. Raymo, Langmuir, 2010, 26, 11503 CrossRef CAS.
- M. H. Stewart, K. Susumu, B. C. Mei, I. L. Medintz, J. B. Delehanty, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2010, 132, 9804 CrossRef CAS.
- G. Palui, H. B. Na and H. Mattoussi, Langmuir, 2012, 28, 2761 CrossRef CAS.
- M. Thiry, K. Boldt, M. S. Nikolic, F. Schulz, M. Ijeh, A. Panicker, T. Vossmeyer and H. Weller, ACS Nano, 2011, 5, 4965 CrossRef CAS.
- J. S. Park, A. Nguyen Vo, D. Barriet, Y. S. Shon and T. R. Lee, Langmuir, 2005, 21, 2902 CrossRef CAS.
- K. Wojczykowski, D. Meißner, P. Jutzi, I. Ennen, A. Hütten, M. Fricke and D. Volkmer, Chem. Commun., 2006, 3693 RSC.
- L. Qu, Z. A. Peng and X. Peng, Nano Lett., 2001, 1, 333 CrossRef CAS.
- T. Pons, H. T. Uyeda, I. L. Medintz and H. Mattoussi, J. Phys. Chem. B, 2006, 110, 20308 CrossRef CAS.
- H. Fraenkel-Conrat, J. Biol. Chem., 1955, 217, 373 CAS.
- J. P. Danehy and M. Y. Oester, J. Org. Chem., 1967, 32, 1491 CrossRef CAS.
- A. C. Faure, S. Dufort, V. Josserand, P. Perriat, J. L. Coll, S. Roux and O. Tillement, Small, 2009, 5, 2565 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supplementary figures. See DOI: 10.1039/c2sc21113k |
| This journal is © The Royal Society of Chemistry 2013 |