Oxidation-driven self-assembly gives access to high-nuclearity molecular copper vanadium oxide clusters†‡
Johannes
Forster
a,
Benedikt
Rösner
b,
Rainer H.
Fink
b,
Leanne C.
Nye
c,
Ivana
Ivanovic-Burmazovic
c,
Katharina
Kastner
a,
Johannes
Tucher
a and
Carsten
Streb
*a
aInorganic Chemistry II, Friedrich-Alexander-University Erlangen-Nuremberg, Egerlandstr. 1, 91058 Erlangen, Germany. E-mail: carsten.streb@chemie.uni-erlangen.de; Fax: +49-9131-8527367; Tel: +49-9131-8529437
bPhysical Chemistry II, Friedrich-Alexander-University Erlangen-Nuremberg, Egerlandstr. 3, 91058 Erlangen, Germany
cBioinorganic Chemistry, Friedrich-Alexander-University Erlangen-Nuremberg, Egerlandstr. 1, 91058 Erlangen, Germany
First published on 15th October 2012
Abstract
We report a general fragmentation-and-re-assembly route which gives access to high-nuclearity, mixed-metal polyoxometalate clusters. Reduced vanadium(IV) precursors are oxidatively dis-assembled into reactive fragments which subsequently re-aggregate under template control in a one-pot reaction. It is shown that the oxidative dis-assembly is required, as the use of vanadium(V)-based precursors results in the formation of smaller clusters. The principle is exemplified by the synthesis of a ca. 1.8 × 1.7 × 1.0 nm3, 36-nuclear copper vanadium oxide cluster, (nBu4N)4[Cu6V30O82(NO3)2(CH3CN)6]. The cluster is characterized in the solid-state and in solution by single-crystal XRD, ESI-MS and other spectroscopic and electrochemical measurements. Several lines of evidence show that the compound is indeed formed exclusively by fully oxidized vanadium(V) centres. In addition, primary fragmentation products of the type [VO(dmso)5]2+ were isolated. The cuprovanadate cluster features pentagonal secondary building units of the type {(V)M5} (M = Cu, V) which show similar structural function as the well-known {(Mo)Mo5} pentagons observed in giant molybdate clusters. The observation suggests that more complex vanadate clusters might be accessible based on these pentagonal units.
Introduction
The development of new bottom-up assembly routes for metal oxide-based nanostructured materials holds great promise for academic and application-driven research alike, as it gives access to new classes of functional materials with a wide range of potential fields of applications.1 In addition, the detailed analysis of these systems allows us to gain insight into the complex mechanisms which control the aggregation of small inorganic precursors into complex nanostructured architectures.2 In particular the controlled bottom-up assembly of molecular metal oxide clusters in the nanometer regime, so-called polyoxometalates (POMs),1 is an attractive route towards functional materials with applications in magnetism,3 catalysis,4 materials chemistry5 and energy conversion and storage.6 Various routes for the assembly of polyoxometalate clusters have been devised and reported in the literature. By far the most prominent synthesis is the one-pot self-assembly of the cluster shell under aqueous conditions, starting from simple oxometalate precursors such as Na2MoO4, Na2WO4 or NaVO3.7 This highly successful and versatile approach has led to the discovery of POM clusters ranging from relatively small, molecular units such as the archetypal Keggin anion [XM12O40]n− (X = B, Si, P, etc.; M = Mo, W) to highly complex, supramolecular architectures containing up to 368 metal centres.8In contrast, cluster assembly in organic media is a less common approach, although it has been shown that cluster synthesis under non-aqueous conditions can be used for the tailored assembly of a large number of small, heterometallic species based on the Lindqvist cluster [(L)nM′M5O19]m− (M′ = various metal centres; M = W, Mo), thereby giving facile access to a range of pre-defined molecular model systems.9 In addition, it has been highlighted how novel vanadium oxide cluster architectures can be accessed using non-aqueous assembly routes.10
Recently, we have started to investigate the assembly of vanadium-based polyoxometalates under non-aqueous conditions as an alternative route towards functional molecular metal oxides for applications in catalysis as well as electron transfer and storage.11 In our initial study, we discovered that controlled fragmentation of labile vanadium oxide cluster precursors such as [V4O12]4− in coordinating organic solvents such as acetonitrile or N,N-dimethyl formamide can be used to initiate cluster re-arrangements in solution.11 It was proposed that these re-arrangements proceed via the formation of reactive vanadate fragments of the type [VO(L)n]z+ (L = solvent ligand, OHx, x = 0–2) which subsequently undergo condensation reactions, yielding new polyoxovanadate clusters, see Scheme 1. Due to the non-aqueous reaction conditions, it was suggested that cluster assembly involves uncommon secondary building units and gives rise to novel cluster architectures.10
![Proposed cluster assembly scheme under non-aqueous conditions in coordinating organic solvents, e.g. acetonitrile: fragmentation of the precursor [V4O12]4− leads to the formation of reactive fragments [VO(L)n]z+ which subsequently self-assemble into stable cluster species.](/image/article/2013/SC/c2sc20942j/c2sc20942j-s1.gif) | ||
| Scheme 1 Proposed cluster assembly scheme under non-aqueous conditions in coordinating organic solvents, e.g. acetonitrile: fragmentation of the precursor [V4O12]4− leads to the formation of reactive fragments [VO(L)n]z+ which subsequently self-assemble into stable cluster species. | ||
In the present study, this assembly approach was modified using the controlled oxidative fragmentation14 of a reduced, vanadium(IV) based cluster in organic solvents in order to gain access to novel, reactive vanadium oxide fragments. In a proof-of-principle study it was shown that under oxidative conditions, the prototype VIV cluster12 [H6V18O42]6− can be fragmented and re-assembled, resulting in the formation of the well-known mixed-valence [VIV2VV8O26]4− cluster,13 see Fig. 1 (for details see ESI‡). This observation supports earlier findings where it was shown that similar oxidative fragmentation routes can be used for the assembly of molybdate clusters under aqueous conditions14a and for pure vanadate clusters under non-aqueous conditions.14b
![Oxidative fragmentation of the fully VIV-based cluster [H6V18O42]6− giving access to the mixed-valent species [VIV2VV8O26]4−. Oxidation can be achieved using oxygen or other oxidants. Colour scheme: VIV-based polyhedra: purple; VV-based polyhedra: blue.](/image/article/2013/SC/c2sc20942j/c2sc20942j-f1.gif) | ||
| Fig. 1 Oxidative fragmentation of the fully VIV-based cluster [H6V18O42]6− giving access to the mixed-valent species [VIV2VV8O26]4−. Oxidation can be achieved using oxygen or other oxidants. Colour scheme: VIV-based polyhedra: purple; VV-based polyhedra: blue. | ||
In order to access larger cluster architectures it was suggested that transition metal centres are employed to link vanadium oxide units. In addition, the incorporation of transition metals can be a facile means to fine-tune the electronic, catalytic and magnetic properties of the cluster. In the present study, we used CuII as a model to demonstrate the incorporation of heterometals into the cluster shell. CuII was chosen as it is redox-inert and does not interfere with the oxidative cluster fragmentation under the given reaction conditions; further, it adopts coordination geometries comparable to vanadium centres in polyoxovanadates.15
Results and discussion
Synthesis of {Cu666V303030}
Initial synthetic studies showed that the reaction of (nBu4N)6[H6V18O42] with Cu(NO3)2 × 3H2O in acetonitrile under aerated, oxidative conditions results in a characteristic colour change from deep green to intense brown. This is distinctly different from the oxidative reaction observed in the absence of Cu(NO3)2 × H2O where the solution changes colour to deep purple (indicative of the formation of [VIV2VV8O26]4−, see Fig. 1). Crystallization of the reaction solution by diffusion of diethyl ether allowed the isolation of compound 1 as a single-crystalline product in yields of ca. 37%. 1 was analyzed using single-crystal X-ray diffraction and gave (nBu4N)4[Cu6V30O82(NO3)2(CH3CN)6] × solvent§ (= (nBu4N)4{Cu666V303030} × solvent), making 1 the largest cuprovanadate characterized to-date. The compound crystallizes in the trigonal space group P3221 with unit cell parameters a = b = 20.411(3), c = 40.340(5).§ Structural analysis shows that the crystal lattice contains a 36-nuclear, heterometallic copper vanadium oxide cluster with dimensions of ca. 1.8 × 1.7 × 1.0 nm3. The anionic {Cu666V303030} cluster can be described as a dimer based on two nitrate-templated [CuV15O41(NO3)]6− (= {CuV15}) sub-shells which are linked by two peripheral, dinuclear [Cu2(CH3CN)3]4+ (= {Cu2}) units so that the complete cluster can be rationalized as {Cu666V303030} = {CuV15}2{Cu2}2 (see Fig. 2).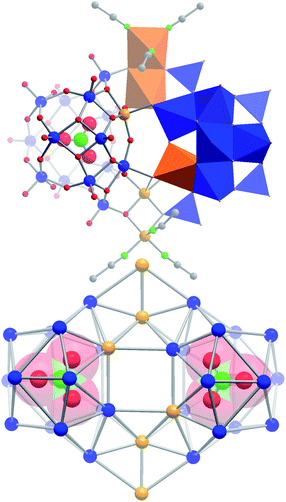 | ||
| Fig. 2 Top: polyhedral and ball-and-stick representation of the dimeric structure of {Cu666V303030} showing the assembly from two {CuV15} sub-shells which are cross-linked by two dinuclear {Cu2} linkers. Bottom: skeletal representation of the metal framework, showing the topology of the cluster anion and the nitrate templates as space-filling models. Colour scheme: Cu: orange; V: blue; N: green, O: red, C: grey. | ||
Structural analysis
The {CuV15} unit is templated by a nitrate anion in the centre of the sub-shell. The metal oxide framework of each {CuV15} unit is formed by 13 [VO5] square pyramids, one [CuO5] square pyramid and two peripheral [VO4] tetrahedra which link the {CuV15} sub-shell to the {Cu2} bridging units (see Fig. 2). {CuV15} can be described as a derivative of the bi-capped Keggin cluster [(PO4)V14O38]9−,16 where the phosphate template is replaced by a nitrate and a {V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O} unit is replaced by a {Cu} group, giving [(NO3)CuV13O37]8−. This unit is capped with two additional [VO2]2+ groups, giving the full {CuV15} sub-shell [(NO3)CuV15O41]6−. The {CuV15} sub-shells in {Cu666V303030} are bridged by two {Cu2} units which are based on two square pyramidal CuII centres: the central CuII ion links the {CuV15} sub-shells via five μ3-oxo ligands; in contrast, the peripheral Cu centre is connected to the cluster shell via two μ3-oxo ligands and is further stabilized by three terminal acetonitrile ligands.
O} unit is replaced by a {Cu} group, giving [(NO3)CuV13O37]8−. This unit is capped with two additional [VO2]2+ groups, giving the full {CuV15} sub-shell [(NO3)CuV15O41]6−. The {CuV15} sub-shells in {Cu666V303030} are bridged by two {Cu2} units which are based on two square pyramidal CuII centres: the central CuII ion links the {CuV15} sub-shells via five μ3-oxo ligands; in contrast, the peripheral Cu centre is connected to the cluster shell via two μ3-oxo ligands and is further stabilized by three terminal acetonitrile ligands.
It is interesting to note that the CuII centres within the {CuV15} sub-shells are facing each other in opposite, central positions, and are disordered crystallographically over two adjacent positions (see ESI‡). The location of the CuII centres is further substantiated as these positions are the only ones which do not feature terminal oxo ligands (see Fig. 2). The remaining vanadium centres in {Cu666V303030} feature one terminal VO bond each with characteristic bond lengths of dV–Oca. 1.6 Å. Further evidence of the cluster composition and metal ratio is given by elemental analysis, flame atomic absorption spectroscopy and electrospray ionization (ESI) mass spectrometry, all of which support the above formula assignment and the structural description of {Cu666V303030}.
Vanadium oxidation state
Based on the synthetic procedure it was initially assumed that the cluster shell might contain one or several reduced vanadium(IV) centres together with a larger number of VV ions. However, crystallographic analysis in combination with empirical bond-valence sum (BVS) analysis17 gave first indication that all vanadium centres are fully oxidized. In order to further evaluate the oxidation state of the vanadium centres, the near-edge X-ray absorption fine structure (NEXAFS) of the native {Cu666V303030} cluster compound was investigated using scanning transmission X-ray microscopy (STXM) conducted at the Swiss Light Source.18 The L2,3-edge absorption bands of vanadium show a linear shift with the oxidation state, e.g. from 515.5 eV for metallic vanadium to 519.0 eV (2p3/2) for the fully oxidized vanadium(V) species (see ESI‡).19Fig. 3 shows the L2,3-edge absorption for {Cu666V303030}. The two most prominent features correspond to the spin-orbit split 2p3/2 and 2p1/2 initial states with smaller intensities on the low-energy shoulders. The spectral features above 530 eV are due to oxygen K-edge absorption. The results allow the conclusion that {Cu666V303030} is completely based on vanadium(V) ions, which is in good agreement with the literature.19 The copper centres were identified as CuII based on geometric considerations, NEXAFS spectroscopy and BVS calculations, see ESI.‡ In addition, chemical redox titration of {Cu666V303030} using cerium ammonium nitrate as the oxidant shows that no reduced vanadium(IV) is present.
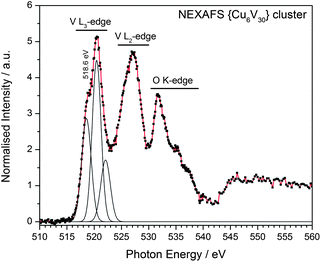 | ||
| Fig. 3 NEXAFS spectrum and Gaussian peak fit of the {Cu666V303030} cluster in the vanadium L-edge region (partly overlapping with the O K-edge absorption). The V L3-edge resonance indicates a fully oxidized vanadium(V) species. For detailed analysis, see ESI.‡ | ||
Electrospray ionization mass spectrometry
In order to determine the cluster stability and potential dynamic behaviour of {Cu666V303030}, electrospray ionization mass spectrometry (ESI-MS) was performed on a sample of {Cu666V303030} ([{Cu666V303030}] ca. 9 × 10−5 M) in acetonitrile using the negative ion mode (−ve). Data analysis indicated the presence of the full cluster anion at m/z = 836.34 assigned as [Cu6V30O82(NO3)2]4−, indicating the loss of all coordinated acetonitrile ligands and implying an all-VV oxidation state (see Fig. 4). In addition, several fragment peaks were observed which feature the {CuV15} sub-shell only, e.g. m/z = 1016.24, assigned to (nBu4N)2[H2CuV15O41(NO3)]2−. This observation suggests that under optimized reaction conditions, it might be possible to obtain the {CuV15} sub-shells as isolated cluster species; alternatively, it might suggest that in solution, {Cu666V303030} exists in an equilibrium with the monomeric {CuV15} species and further in situ analyses are required to fully understand the underlying solution-behaviour of {Cu666V303030}. Other signals where observed which also correspond to the complete metal-oxo shell of {Cu666V303030} with varying amounts of organic counterions, for complete analysis see ESI.‡![Negative-mode high resolution ESI mass spectra, showing the full {Cu666V303030} cluster shell [Cu6V30O82(NO3)2]4− at m/z = 836.34 (left) and the {CuV15} cluster sub-shell (nBu4N)2[H2CuV15O41(NO3)]2− at m/z = 1016.24 (right).](/image/article/2013/SC/c2sc20942j/c2sc20942j-f4.gif) | ||
| Fig. 4 Negative-mode high resolution ESI mass spectra, showing the full {Cu666V303030} cluster shell [Cu6V30O82(NO3)2]4− at m/z = 836.34 (left) and the {CuV15} cluster sub-shell (nBu4N)2[H2CuV15O41(NO3)]2− at m/z = 1016.24 (right). | ||
Non-oxidative synthesis attempts
In order to better understand the minimum synthetic conditions required for the formation of {Cu666V303030}, a series of aggregation reactions were conducted. Fully oxidized, mixed-valent and fully reduced vanadates were reacted with Cu(NO3)2 × 3H2O with variation of the reagent ratio and concentration. The results of this study suggest that the formation of {Cu666V303030} requires the presence of reduced VIV-precursors (see Scheme 2). When the fully reduced [H6V18O42]6− cluster or the mixed-valence [VIV2VV8O26]4− cluster were employed, {Cu666V303030} was obtained as the only crystalline product. However, using fully oxidized species such as [V4O12]4− or [H3V10O28]3− resulted in the formation of the smaller, VV-based cuprovanadate cluster, [Cu2V8O24]4− which has been observed previously.15 In addition we could show that under non-oxidative conditions in de-aerated, N2-saturated solvents, the reduced cluster species do not undergo the {Cu666V303030} assembly and only the starting materials are recovered.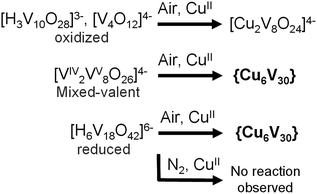 | ||
| Scheme 2 Synthetic conditions required for the formation of {Cu666V303030} and alternative product formation pathways observed. | ||
Observation of pentagonal units in vanadates
A major question in polyoxometalate chemistry is which building units are involved in the formation of complex cluster architectures starting from simple precursors. Seminal work by Müller and Roy on giant molybdate clusters20 and more recent reports on structurally comparable tungstate21 and niobate22 clusters indicate that pentagonal secondary building units (SBUs) of the type {(M)M5} (M = Mo, W, Nb)20–22 play a vital role in the assembly of these complex molecular architectures. Cronin et al. have recently substantiated this by identifying similar cluster building blocks involved in the templating processes which lead to the formation of Müller's giant Bielefeld wheel structures.2In contrast to tungstate and molybdate chemistry, vanadates do not form equally complex, molecular aggregates, as cluster growth is limited by the lower oxidation state accessible by vanadium (VVvs. MoVI/WVI). In addition, the formation of pentagonal units such as {(Mo)Mo5} is difficult as the smaller VV ions cannot adopt the pentagonal bipyramidal central structure observed in the molybdate and tungstate pentagons. However, structural analysis of {Cu666V303030} and of other larger vanadates23 show the presence of a pentagonal unit {(V)M5} (M = Cu, V) which might fulfill an analogous structural function as the molybdate and tungstate pentagons in their respective fields.
The pentagonal vanadate SBU is observed four times in {Cu666V303030} and forms the main structural part of the {CuV15} sub-shells, where two {(V)M5} units are held together by two [VO4] tetrahedra (= {V1Tet}) and two [VO5] square pyramids (= {V1Pyr}) so that the sub-shells can be rationalized as {CuV15} = {(Cu)V5}{(V)V5}{V1Tet}2{V1Pyr}2. Each {(V)M5} pentagon is formed by six [MO5] square pyramids where a backbone of three edge-sharing [MO5] units are further connected with one central edge-sharing [MO5] moiety on one side and two peripheral [MO5] groups on the opposite side (see Fig. 5). The latter set is further linked by a corner-sharing motif, resulting in an overall bent architecture which allows the assembly of the convex {CuV15} sub-shells. It should be noted the {(V)M5} pentagons do not feature pentagonally coordinated vanadium centres. As a result, the idealized symmetry of {(V)M5} (Cs) is significantly lower than the C5 symmetry in the Müller pentagons, see Fig. 5.
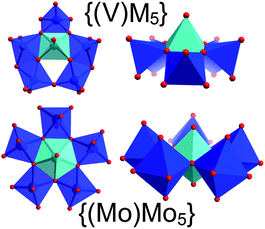 | ||
| Fig. 5 Illustration of the Cs-symmetric {(V)M5} pentagons (M = Cu, V) in {Cu666V303030} and the classical Müller-type {(Mo)Mo5} pentagons observed in Keplerate and Bielefeld wheel-type structures. Note the central square pyramidal V centre in {(V)M5} and the pentagonal-bipyramidal Mo centre in {(Mo)Mo5} highlighted in light blue. | ||
Observation of initial fragments
To better understand the cluster assembly mechanism, the identification of initial fragments of the reduced VIV precursor [H6V18O42]6− was attempted. Based on experimental observations and previous reports24 it was known that the solution acidity controls the fragmentation and re-assembly process and it was observed that the oxidation kinetics under acidic conditions were slow enough to allow the crystallization of an initial fragmentation product, compound 2. The VIV species, which was isolated and analyzed using single-crystal X-ray diffraction is a solvent-stabilized vanadyl unit, [VO(dmso)5](NO3)2 which is structurally closely related to the fragment species proposed in Scheme 1 (see Fig. 6).‡![Illustration of the fragment species 2, [VO(dmso)5]2+, observed during the fragmentation of the reduced cluster species [H6V18O42]6−.](/image/article/2013/SC/c2sc20942j/c2sc20942j-f6.gif) | ||
| Fig. 6 Illustration of the fragment species 2, [VO(dmso)5]2+, observed during the fragmentation of the reduced cluster species [H6V18O42]6−. | ||
It should be noted that similar vanadyl species are observed under highly acidic aqueous conditions,24 but to the best of our knowledge, the species described above has not been reported before. Observation of this species is important as it illustrates the type of fragmentation products principally available for the assembly of vanadate clusters under non-aqueous reaction conditions. It also offers a starting point to gain insight into the formation of larger, stable polyoxovanadates, and might help us to understand the role of {(V)M5} secondary building units.
Conclusions
In summary, we have presented a new cluster assembly route giving access to high-nuclearity, heterometallic metal oxide clusters. The route is based on the oxidative fragmentation of reduced precursors under non-aqueous conditions. A primary fragmentation product, [VO(dmso)5]2+ was identified and isolated. The new route is used for the assembly of a 36-nuclear copper vanadium oxide cluster, {Cu666V303030}. It is demonstrated that {Cu666V303030} is only accessible when reduced, VIV-based precursors are employed, highlighting that the precursor oxidation state can be a vital secondary reaction parameter which controls cluster aggregation. Intriguingly, {Cu666V303030} features a set of four pentagonal building units, {(V)M5} (M = V, Cu) which show similar features and structural function (but lower symmetry) as the well-known molybdate and tungstate pentagons observed in giant polyoxometalate architectures. As such, these units provide a first glimpse at similar vanadate-based secondary building units which could be employed for the assembly of larger, more complex vanadium oxide architectures.Future work will be focused on the analysis of the complex magnetic and electrochemical properties of {Cu666V303030} so as to understand whether the cluster might be used as a switchable magnetic material. In addition, the role of the {(V)M5} pentagons in the assembly of high-nuclearity polyoxovanadate structures will be further evaluated and attempts will be made to identify this species in solution based on in situ spectroscopic methods. Further, we will investigate whether the reported synthesis is a general route to high-nuclearity mixed-metal polyoxovanadates by replacing CuII with other transition metals. A particular focus will be the embedding of redox-active metals as catalytic sites as well as paramagnetic centres to tune the magnetic cluster properties and to demonstrate the general applicability of the route.
Experimental
Synthesis
Synthesis of 1: (nBu4N)4[Cu6V30O82(NO3)2(CH3CN)6] × solvent: (nBu4N)6[H6V18O42] (200 mg, 66.4 μmol) and Cu(NO3)2 × 3H2O (101 mg, 418 μmol) were dissolved in 15 ml acetonitrile. The resulting solution was vigorously stirred for one hour at ambient temperature. Diffusion of diethyl ether into the reaction mixture over a period of ca. 1 week gave brown needle crystals, which were suitable for X-ray diffraction. Yield: 117 mg (25.7 μmol, 36.8% based on Cu). Elemental analysis for the fully dried compound C64Cu6H144N6O88V30 (all acetonitrile removed) in wt% (calcd): C 18.17 (17.81), H 3.93 (3.36), N 1.53 (1.95), Cu 8.97 (8.83), V 35.63 (35.40).Synthesis of 2: (nBu4N)6[H6V18O42] (298 mg, 97.7 μmol) was dissolved in a DMSO–water mixture (3 ml, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v). Nitric acid (65 wt%, 460 μl, 6.74 mmol) was slowly added. The solution was stirred over night and diffusion of ethyl acetate into the mother liquor gave light blue single crystals. Yield: 450 mg (775 μmol, 44.2% based on V). Elemental analysis for C10H30N2S5VO11 in wt% (calcd): C 20.75 (20.65), H 5.11 (5.19), N 4.79 (4.82), S 27.96 (27.57).
:1, v:v). Nitric acid (65 wt%, 460 μl, 6.74 mmol) was slowly added. The solution was stirred over night and diffusion of ethyl acetate into the mother liquor gave light blue single crystals. Yield: 450 mg (775 μmol, 44.2% based on V). Elemental analysis for C10H30N2S5VO11 in wt% (calcd): C 20.75 (20.65), H 5.11 (5.19), N 4.79 (4.82), S 27.96 (27.57).
Crystallography
Suitable single crystals of {Cu666V303030} were grown and mounted onto the end of a thin glass fiber using Fomblin oil. X-Ray diffraction intensity data were measured at 150 K on a Nonius Kappa CCD diffractometer [λ(Mo-Kα) = 0.71073 Å] equipped with a graphite monochromator. Structure solution and refinement was carried out using the SHELX-97 package via WinGX. Structures were solved by a combination of direct methods and difference Fourier syntheses and refined against F2 by the full-matrix least-squares technique. For details, see ESI.‡ CCDC reference numbers 890307 (1) and 890308 (2). In 1, the carbon atoms of the nBu4N+ tetra-n-butylammonium cation based around N6 were refined isotropically due to heavy structural disorder. One ligand acetonitrile molecule (based on N1) was heavily disordered and was refined isotropically. The SQUEEZE routine was applied to the dataset from within the Platon program suite25 to account for diffuse solvent molecules. In the cluster shell, the Cu centres Cu1A/Cu1B and V centres V4A/V4B are statistically disordered over the two metal positions. The metal oxide cluster atoms in 1 were refined fully anisotropically, giving satisfactory R-values: R1 = 0.0479; wR2 (all data) = 0.1222.ESI-Mass spectrometry
MS measurements were performed on a UHR-TOF Bruker Daltonics maXis using electrospray ionization (ESI). Detection was in negative-ion mode and the source voltage was 4 kV. The flow rates were 500 μl h−1. The drying gas (N2) was held at 180 °C. The machine was calibrated prior to every experiment using the Agilent ESI-TOF low concentration tuning mixture, with an m/z range of singly charged peaks up to 2700 Da.NEXAFS spectroscopy
STXM measurements were conducted at the PolLux beamline at the Swiss Light Source (ref. 16). The {Cu666V303030} cluster was dissolved in acetonitrile, dropped onto a clean silicon nitride membrane and then air-dried. The droplet size was chosen to yield a satisfying optical density. The NEXAFS spectrum was obtained by measuring a stack of 30 × 30 pixel images in the energy range of 506–560 eV. The intensity of the {Cu666V303030} cluster was averaged and normalized to incoming photon intensity. The spectrum was normalized to the edge jump and fitted using Gaussian functions to a chi-square value of 0.006.Acknowledgements
JF and CS acknowledge financial support by the Fonds der Chemischen Industrie (FCI). KK acknowledges financial support by the Deutsche Bundesstiftung Umwelt (DBU) and the Deutsche Forschungsgemeinschaft Graduate School GRK 1626 (Chemical Photocatalysis). RHF and BR acknowledge financial support from the Bundesministerium für Bildung und Forschung (BMBF), project no 05K10WEA.Notes and references
- (a) Chem. Rev., ed. C. Hill, 1998, special issue on polyoxometalates Search PubMed; (b) D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (c) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS.
- H. N. Miras, G. J. T. Cooper, D. L. Long, H. Bögge, A. Müller, C. Streb and L. Cronin, Science, 2010, 327, 72–74 CrossRef CAS.
- (a) C. Ritchie, A. Ferguson, H. Nojiri, H. N. Miras, Y. F. Song, D. L. Long, E. Burkholder, M. Murrie, P. Kögerler, E. K. Brechin and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 5609–5612 CrossRef CAS; (b) U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS; (c) P. Kögerler, B. Tsukerblat and A. Müller, Dalton Trans., 2010, 39, 21–36 RSC.
- (a) C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS; (b) C. L. Hill, J. Mol. Catal. A: Chem., 2007, 262, 2–6 CrossRef CAS; (c) M. Bonchio, M. Carraro, A. Sartorel, G. Scorrano and U. Kortz, J. Mol. Catal. A: Chem., 2006, 251, 93–99 CrossRef CAS.
- (a) C. Streb, R. Tsunashima, D. A. MacLaren, T. McGlone, T. Akutagawa, T. Nakamura, A. Scandurra, B. Pignataro, N. Gadegaard and L. Cronin, Angew. Chem., Int. Ed., 2009, 48, 6490–6493 CrossRef CAS; (b) J. Thiel, C. Ritchie, H. N. Miras, C. Streb, S. G. Mitchell, T. Boyd, M. N. C. Ochoa, M. H. Rosnes, J. McIver, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 6984–6988 CrossRef CAS; (c) C. Streb, T. McGlone, O. Brücher, D. L. Long and L. Cronin, Chem.–Eur. J., 2008, 14, 8861–8868 CrossRef CAS.
- (a) A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5009 CrossRef CAS; (b) Y. V. Geletii, Z. Q. Huang, Y. Hou, D. G. Musaev, T. Q. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522–7524 CrossRef CAS; (c) F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS; (d) Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS; (e) J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J. Am. Chem. Soc., 2011, 133, 188–190 CrossRef CAS; (f) Z. Q. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. S. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Q. Lian, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef CAS; (g) C. Streb, Dalton Trans., 2012, 41, 1651–1658 RSC.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Heidelberg, 1983 Search PubMed.
- A. Müller, E. Beckmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., Int. Ed., 2002, 41, 1162–1165 CrossRef.
- (a) R. J. Errington, in Polyoxometalate Molecular Science, ed. J. J. Borras-Almenar, E. Coronado, A. Müller and M. T. Pope, Kluwer Academic Publishers, Dordrecht, 2003, pp. 55–78 Search PubMed; (b) R. J. Errington, in Comp. Coord. Chem. II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, pp. 759–773 Search PubMed; (c) R. J. Errington, S. S. Petkar, P. S. Middleton and W. McFarlane, J. Am. Chem. Soc., 2007, 129, 12181–12196 CrossRef CAS.
- (a) L. Zhang and W. Schmitt, J. Am. Chem. Soc., 2011, 133, 11240–11248 CrossRef CAS; (b) Y. Hayashi, Coord. Chem. Rev., 2011, 255, 2270–2280 CrossRef CAS; (c) W. G. Klemperer, T. A. Marquart and O. M. Yaghi, Angew. Chem., Int. Ed. Engl., 1992, 31, 49–51 CrossRef; (d) F. Li, D. L. Long, J. Cameron, H. Miras, C. Pradeep, L. Xu and L. Cronin, Dalton Trans., 2012, 41, 9859–9862 RSC.
- (a) J. Forster, B. Rösner, M. M. Khusniyarov and C. Streb, Chem. Commun., 2011, 47, 3114–3116 RSC; (b) M. Grabau, K. Heussner, J. Forster and C. Streb, Eur. J. Inorg. Chem., 2011, 1719–1724 CrossRef CAS; (c) J. Tucher, L. C. Nye, I. Ivanovic-Burmazovic, A. Notarnicola and C. Streb, Chem.–Eur. J., 2012, 18, 10949–10953 CrossRef CAS; (d) J. Tucher, Y. Wu, L. C. Nye, I. Ivanovic-Burmazovic, M. M. Khusniyarov and C. Streb, Dalton Trans., 2012, 41, 9938–9943 RSC.
- A. Müller, R. Sessoli, E. Krickemeyer, H. Bögge, J. Meyer, D. Gatteschi, L. Pardi, J. Westphal, K. Hovemeier, R. Rohlfing, J. Döring, F. Hellweg, C. Beugholt and M. Schmidtmann, Inorg. Chem., 1997, 36, 5239–5250 CrossRef.
- Y. Hayashi, N. Miyakoshi, T. Shinguchi and A. Uehara, Chem. Lett., 2001, 170–171 CrossRef CAS.
- (a) A. Müller, J. Meyer, E. Krickemeyer, C. Beugholt, H. Bögge, F. Peters, M. Schmidtmann, P. Kögerler and M. J. Koop, Chem.–Eur. J., 1998, 4, 1000–1006 CrossRef; (b) K. Okaya, T. Kobayashi, Y. Koyama, Y. Hayashi and K. Isobe, Eur. J. Inorg. Chem., 2009, 34, 5156–5163 CrossRef.
- T. Kurata, A. Uehara, Y. Hayashi and K. Isobe, Inorg. Chem., 2005, 44, 2524–2530 CrossRef CAS.
- M. I. Khan, J. Zubieta and P. Toscano, Inorg. Chim. Acta, 1992, 193, 17–20 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- J. Raabe, G. Tzvetkov, U. Flechsig, M. Boge, A. Jaggi, B. Sarafimov, M. G. C. Vernooij, T. Huthwelker, H. Ade, D. Kilcoyne, T. Tyliszczak, R. H. Fink and C. Quitmann, Rev. Sci. Instrum., 2008, 79, 113704–113710 CrossRef CAS.
- (a) B. Gurevich, B. E. Bent, A. V. Teplyakov and J. G. Chen, Surf. Sci., 1999, 442, L971–L976 CrossRef; (b) J. G. Chen, C. M. Kim, B. Frühberger, B. D. Devries and M. S. Touvelle, Surf. Sci., 1994, 321, 145–155 CrossRef CAS.
- A. Müller and S. Roy, Coord. Chem. Rev., 2003, 245, 153–166 CrossRef.
- A. M. Todea, A. Merca, H. Bögge, T. Glaser, L. Engelhardt, R. Prozorov, M. Lubanc and A. Müller, Chem. Commun., 2009, 3351–3353 RSC.
- R. Tsunashima, D. L. Long, H. N. Miras, D. Gabb, C. P. Pradeep and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 113–116 CrossRef CAS.
- (a) A. Wutkowski, F. Niefind, C. Näther and W. Bensch, Z. Anorg. Allg. Chem., 2011, 637, 2198–2204 CrossRef CAS; (b) A. Müller, R. Rohlfing, J. Döring and M. Penk, Angew. Chem., Int. Ed. Engl., 1991, 30, 588–590 CrossRef.
- J. Livage, Chem. Mater., 1991, 3, 578–593 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef.
Footnotes |
| † Dedicated to Professor Michael T. Pope on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic, analytical and crystallographic details are given in the ESI. CCDC numbers 890307 and 890308. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc20942j |
| § During crystal preparation, solvent evaporation from single crystals of 1 was noticed. Consequently, diffuse solvent molecules were detected during crystallographic refinement but their position could not be resolved, although several crystallographic datasets of 1 were recorded. The SQUEEZE routine was therefore applied to the dataset from within the Platon program suite24 to account for diffuse solvent molecules. Based on solvent accessible void calculations, the number of solvent molecules in 1 can be approximated to ca. 70 acetonitrile molecules per unit cell. Thermogravimetric analyses were performed but due to the high volatility of the solvent molecules in 1, varying amounts of solvent were observed. However, thermogravimetry confirmed the amount of nBu4N+ counterions in 1, see ESI for details.‡ |
| This journal is © The Royal Society of Chemistry 2013 |