
Synthesis of nitrogen-rich imidazole, 1,2,4-triazole and tetrazole-based compounds†
Abstract
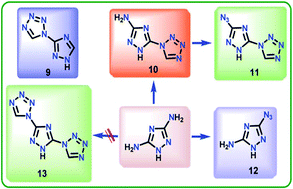
Imidazole, 1,2,4-triazole and tetrazole based molecules were prepared for their possible applications in nitrogen-rich gas generators. The energetic salts of 1-(1H-1,2,4-triazol-3-yl)-1H-tetrazole (9), 5-(1H-tetrazol-1-yl)-1H-1,2,4-triazol-3-amine (10), 1-(3-azido-1H-1,2,4-triazol-5-yl)-1H-tetrazole (11) and 3-azido-1H-1,2,4-triazol-5-amine (12) were prepared with various cationic moieties. Their densities, heats of formation, chemical energy of detonation, detonation velocities and pressures were calculated. All of the compounds possessed high positive heats of formation due to high energy contribution from the molecular backbone of the corresponding compounds. The effect of the azole rings and nitro, amino, and azido groups on their physicochemical properties was examined and discussed.
 Please wait while we load your content...
Please wait while we load your content...

