A ferritin mediated photochemical method to synthesize biocompatible catalytically active gold nanoparticles: size control synthesis for small (∼2 nm), medium (∼7 nm) or large (∼17 nm) nanoparticles†
Oscar D. Petruccia,
David C. Bucka,
Jeff K. Farrerb and
Richard K. Watt*a
aDepartment of Chemistry and Biochemistry, Brigham Young University, C210 Benson Building, Provo, UT 84602, USA. E-mail: rwatt@chem.byu.edu; Fax: +1 801-422-0153; Tel: +1 801-422-1923
bDepartment of Physics and Astronomy, Brigham Young University, N283 ESC building, Provo, UT 84602, USA
First published on 3rd December 2013
Abstract
Ferritin (Ftn) undergoes photo-induced charge separation reactions that oxidize organic substrates. The liberated electrons are transferred through the protein shell to reduce Au ions to gold nanoparticles (AuNPs). We systematically varied the concentrations of citrate (electron donor), Au3+ or Au+ (electron acceptor), and ferritin (photo catalyst) to determine if careful control of these reactant concentrations would: (1) provide size control; (2) alter the morphology of the resulting AuNPs; and (3) alter the catalytic activity of the resulting AuNPs. The size and phosphate content of the ferritin iron core was also evaluated for its influence in this photocatalysis reaction. We report that as the Ftn concentration was increased to an optimal range, the number of AuNPs increased and showed smaller size, more spherical shape, and narrower distribution. Increasing the citrate concentration (electron donor) increased the rate of AuNP formation producing more spherical, uniform sized AuNPs. Increasing the Au3+ concentrations increased the number and sizes of the AuNPs. Since Au3+ reduction requires 3-electrons we proposed that using Au+ would increase the rate of the reaction. The photochemical reaction with Au+ was faster and produced 2.4 ± 1.0 nm diameter AuNPs providing another method of size control. AuNPs were tested as reduction catalysts to convert 4-nitrophenol into 4-aminophenol. The smaller spherical AuNPs were better reduction catalysts than the larger AuNPs. In summary, using a single photochemical synthesis method we can reproducibly control the size, uniformity and catalytic activity of the resulting AuNPs simply by varying the concentrations or oxidation states of the reactants.
Introduction
Gold Nanoparticles (AuNPs) have been the focus of major research efforts over the last ten years.1–3 Nanoparticles (NP) offer large surface to volume ratios and size constraints that leave exposed atoms capable of catalysing reactions otherwise impossible for the same elements as bulk materials.4 The characteristics essential for NP catalytic ability are: small size, narrow size range, and consistent shape.5 Many laboratories have devoted a significant effort to improve their AuNP synthesis in order to reduce the size, control the size distribution, and avoid aggregation of the resulting AuNPs.1,6–8 In this study, our goal was to use a single synthesis method, and by varying the concentrations of the reactants, control the size, morphology, and catalytic activity of the resulting AuNPs.Another important goal was to prepare AuNPs in biocompatible media because of the increasing number of biomedical applications for AuNPs.9,10 Many AuNP synthesis protocols utilize surfactants, polymers, or substituted allylic compounds like tri-n-octylphosphine oxide, polyvinylpyrrolidne, or dialkyl sulfides to prevent AuNPs from clustering and/or precipitating.11–13 These compounds are either toxic or their safety in biological systems have been called into question.14–16 The method described in this study uses all biocompatible reactants to synthesize AuNPs.
AuNPs have been used in a variety of biological applications. Remarkably, AuNPs preferentially accumulate at tumor sites17 so using AuNPs as vehicles to deliver drugs to tumors is an obvious application.17–22 Other applications include imaging,18 biosensing,18 other targeted drug delivery applications, conjugation to drugs, and photo thermal therapy treatments in oncology.18–22 The structure, size, shape and surface ligation of these AuNPs are critical for each application.
Recently ferritin (Ftn) has been used to synthesize AuNPs inside Ftn,23,24 or outside Ftn where the AuNPs assemble on the Ftn exterior.25,26 Ftn is a 24-subunit protein complex (450 kDa) with a spherical structure measuring 12 nm in diameter with an 8 diameter nm hollow interior.27 Ftn has evolved to bind Fe(II), oxidize it to Fe(III) with its catalytic enzyme site known as the ferroxidase center, and sequester the iron in its interior to form ferrihydrite (FeOOH).28,29
In this work our goal was to synthesize AuNPs on the exterior of Ftn. For AuNP synthesis on the Ftn exterior, the protein nano cage appears to have a two-fold purpose. First, Ftn possesses ideal AuNP nucleation sites on the external surface at the 3-fold channels because there are three cysteine residues at the opening of this channel (Fig. 1).25–27 The cysteine residues are ideal as a nucleation site for AuNP deposition because of the thiophilic nature of gold.30,31 Once bound, Au(III) can be reduced to Au(0) by chemical reductants or by light-driven reactions.25,26 Second, Ftn prevents the aggregation and precipitation of the AuNPs by acting as a capping agent.26 Keyes et al. demonstrated that the AuNPs co-migrated with ferritin through a gel filtration column indicating a sufficiently strong interaction that could be used for targeted AuNP delivery based on cellular destinations for serum ferritin.25 Remarkably, tumor cells are often iron-deficient and iron-deficient cells express receptors that endocytose ferritin to satisfy iron needs.32 Preparing Ftn–AuNP conjugates may be an additional mechanism to deliver AuNPs to tumor cells.
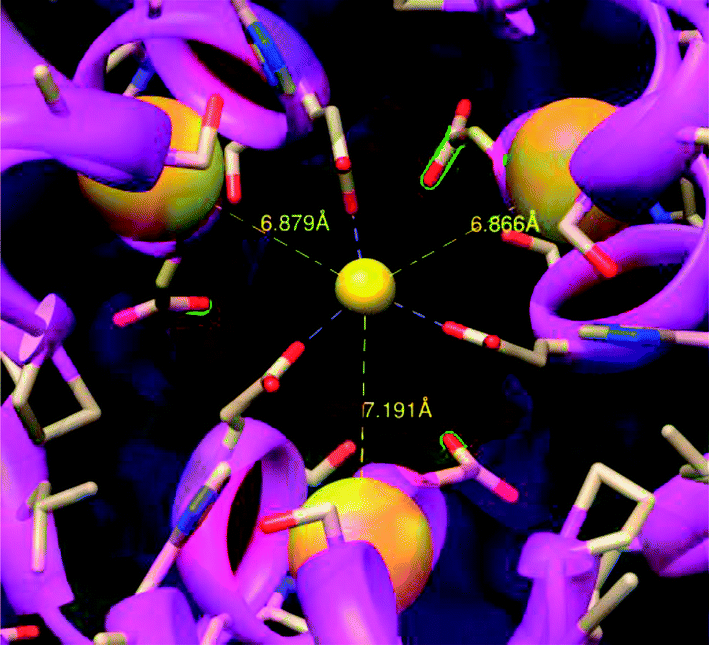 | ||
| Fig. 1 Crystal structure of the 3-fold channel of ferritin – proposed gold nucleation site. This view is from the outside of ferritin looking toward the interior. The conserved negative residues (red atoms) in the 3-fold channel of ferritin attract the cations; the three cysteine residues (the three large yellow spheres) bind the gold atoms facilitating the nucleation of the AuNP. | ||
The photochemical method to synthesize AuNPs with Ftn has additional advantages because it eliminates any contamination from strong chemical reducing agents or their by-products. Ftn is a photo catalyst because the ferrihydrite semi-conductor mineral core can undergo charge separation reactions when exposed to light. Illumination of Ftn promotes an electron from the valence band to the conduction band (2.5–3.5 eV)33 and the excited electrons are ultimately transferred to Au(III) ions bound on the exterior surface of Ftn.25 The electron donors in this system are sacrificial bio-organic molecules that donate electrons to fill the hole created by the photochemical reactions and are typically citrate, tartrate, or oxalate. Scheme 1 represents this process and shows the formation of AuNPs on Ftn.
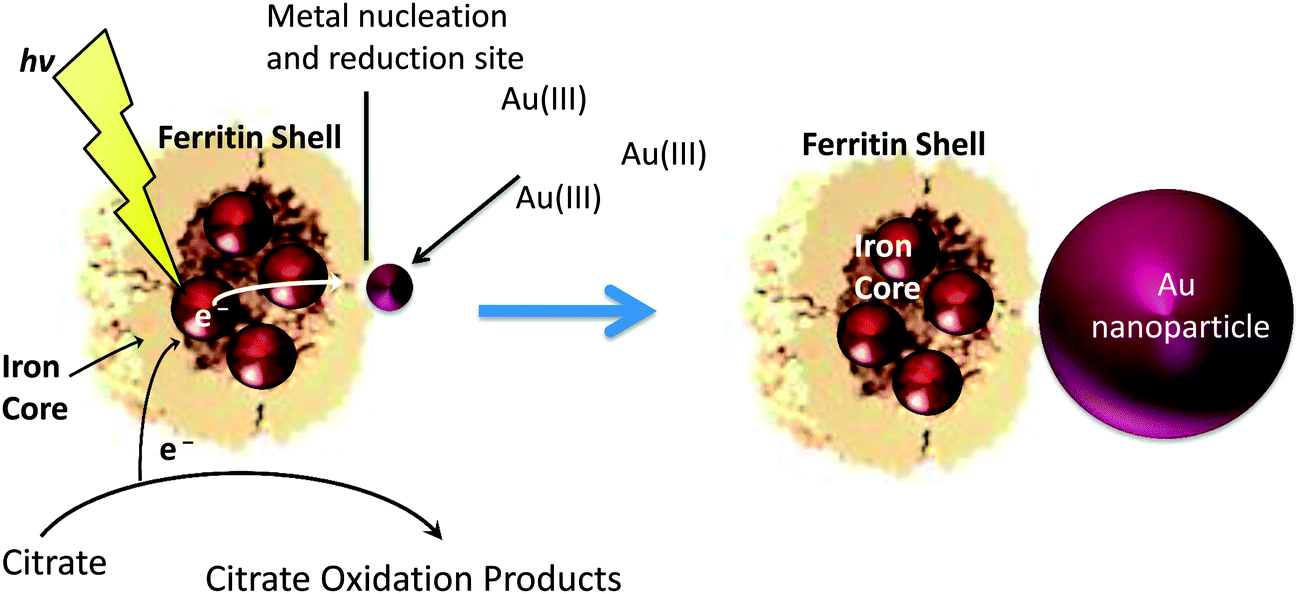 | ||
| Scheme 1 Ferritin containing an iron mineral absorbs light and transfers the excited electrons to Au(III) ions bound to the nucleation sites on the exterior of the ferritin protein shell. This process oxidizes citrate to replenish the electrons for further reactions. Continued light exposure allows the AuNPs to grow to larger sizes. | ||
In this study, reaction conditions were varied to allow control over the morphology, size, and catalytic properties of the resulting AuNPs synthesized by the photochemical method. We hypothesized that changing the concentrations of Ftn, citrate, and AuCl4− could influence the size, shape, quantity, and aggregation state of the AuNPs. Based on the proposal that the cysteine residues in the 3-fold channels of Ftn act as AuNP nucleation sites (Fig. 1), we predicted that higher Ftn concentrations would produce more AuNPs with smaller sizes using the same AuCl4− concentration, whereas lower Ftn concentrations would produce fewer but larger AuNPs (Scheme 2). Because AuNPs can also act as nucleation sites for Au3+, we also predicted that the AuNPs formed with low Ftn concentrations would be more irregularly shaped due to the non-specific nucleation of Au3+ on the growing AuNPs (gold seed effect).26
 | ||
| Scheme 2 AuNPs formation model. The NP size is affected by the concentration of ferritin (Ftn) in solution and by the number of nucleation sites. At low concentrations of Ftn the gold concentrates on fewer nucleation sites, thus forming larger particles (top figure). If more nucleation sites are present the Au3+ will be distributed on a larger number of AuNPs, which will remain smaller (center and bottom figure). | ||
We also hypothesized that high citrate concentrations should favour the specific Ftn-mediated photo-catalysis reactions and minimize the non-specific reactions. Additionally, citrate will assist in the passivation of the AuNPs, whereas low citrate concentrations would increase non-photo catalysed nucleation and synthesis events and gold seed growth. Also, in low citrate conditions Ftn can act as an electron donor; thus under limited electron donor conditions Ftn is damaged by photo-oxidation.34 Therefore, low citrate concentrations could lead to enhanced Ftn oxidation and decreased AuNP passivation. The resulting AuNPs would show increased clustering and precipitation and reduced catalysis. Hence, the results of the lower citrate concentrations should somewhat resemble those of low Ftn concentrations. Finally, increasing the AuCl4− concentration should favour self-nucleating (gold seeds) and auto catalytic events, whereas lower AuCl4− concentrations should slow down the formation of AuNPs. The results presented in this paper are consistent with the model explained above and summarized in Scheme 2.
Materials and methods
Horse spleen Ftn (also referred to as native Ftn) was purchased from Sigma-Aldrich (Sigma F4503) and treated by the thioglycolic method to prepare iron-free or apo Ftn (Apo) as described previously.35 Ftn protein concentrations were determined by the Lowry method.36 After the preparation of apo Ftn, the buffer was exchanged by dialysis to 25 mM 3-(N-morpholino)propanesulfonic acid (MOPS) pH 7.4 with 100 mM NaCl. The concentration of the Apo after the dialysis steps was 28 mg mL−1.Two, 2 mL samples of the Apo were reconstituted with 600 or 1800 Fe/Ftn, by adding the appropriate volumes of ferrous ammonium sulfate prepared in 40 mM HCl and maintaining the pH of the Ftn sample above 7.0 by adding NaOH. The Fe2+–HCl and the NaOH solutions were injected at 6 μL min−1 into the Apo solution using a micro-syringe device (Harvard Instruments) while aerobically stirring the solution throughout the titration. The re-mineralized or reconstituted Ftn was dialyzed against 100 mM NaCl and 10 mM TRIS–HCl pH 7.4 to exchange the buffer.
After the dialysis, 1 mL of the 600 Fe/Ftn and 1 mL of the 1800 Fe/Ftn were removed from the dialysis bags, transferred into clean tubes, and stored at 4 °C, while 1 mL of both the 600 Fe/Ftn and the 1800 Fe/Ftn samples were left in the dialysis bag and further dialyzed against 100 mM NaCl and 10 mM TRIS–HCl pH 7.4 containing 5 mM phosphate. After 48 hours both samples were dialyzed again (three times) against 100 mM NaCl and 10 mM TRIS–HCl pH 7.4 to remove the unbound phosphate. The protein, phosphate, and iron contents in all Ftn samples were quantified using the Lowry, the phospho-molybdate, and the bipyridyl methods described previously.37
Samples (1.0 mL in quartz cuvettes) were prepared for the photochemistry reactions in an Agilent 8453 UV/Vis spectrophotometer with a temperature controlled water-jacketed cuvette holder maintained at 25 °C by a circulating water bath. Each sample was exposed to a UV-Vis floodlight (Integrated Dispensing Solutions, Inc.) equipped with a 400 W metal halide bulb and the reaction was monitored in the 200–1100 nm range in kinetic mode for the length of the reaction.
We took advantage of AuNP surface plasmon resonance (SPR) to monitor the progress of the photo catalytic AuNP synthesis in real time. When AuNPs reach 2 nm in diameter they start producing a SPR peak between 520 and 530 nm.38 The peak of absorbance shifts to wavelengths longer than 530 nm (red-shift) when the AuNPs grow larger than 20 nm, or if the AuNPs cluster together.
Our synthesis utilized Ftn both as photo-catalyst and capping agent, citrate as sacrificial electron donor and additional capping agent, and AuCl4− or AuCl as sources of Au(III) and Au(I), respectively. A UV-Vis floodlight (Integrated Dispensing Solutions, Inc.) provided the energy necessary to transfer the electrons from the citric acid to the Au3+ ions. A similar photochemical system to form nanoparticles has been used by others.25,39–41
We were interested in discovering how varying the concentrations of the reagents used in the photochemical synthesis of AuNPs affect the rate of the reaction and the characteristics of the final AuNPs produced. The conditions reported by Keyes et al. were used as our starting (basal) conditions to create the AuNP25 and are: 50 mM NaCl, 20 mM TRIS–HCl pH 7.4, 400 μM KAuCl4, 30 mM Na-citrate, 150 μg mL−1 Ftn (0.34 μM) (all from Sigma-Aldrich).
We systematically altered one variable in each experiment as follows: Ftn (300 μg mL−1, 150 μg mL−1, 50 μg mL−1, 10 μg mL−1, and zero Ftn as the no Ftn control), citrate (100 mM, 30 mM, 10 mM, 2 mM, and zero citrate), and Au3+ (500 μM, 400 μM, 300 μM, 200 μM, and 40 μM). For AuNPs synthesis from Au+ the following reagent concentrations were used: 50 mM NaCl, 20 mM TRIS–HCl pH 7.4, 400 μM AuCl, 100 mM Na-citrate, 200 μg mL−1 Ftn (0.45 μM).
Transmission electron microscopy (TEM) and high-resolution electron microscopy (HRTEM) experiments were performed on a Tecnai F30 operating at 300 kV. AuNP samples for TEM analysis were collected at the peak of absorbance measured at 530 nm.
Catalytic properties of the AuNPs
As a model to test the catalytic properties of the AuNPs produced by our method we followed the protocol described by Zhang et al.26 modified as follows: 1.25 mL of deionized water, 500 μL of 0.2 mM 4-nitrophenol, and 50 μL of freshly prepared 0.1 M NaBH4 solutions were placed in a standard quartz cuvette, which was then placed in the spectrophotometer cuvette holder set at 25 °C under constant stirring. The spectrophotometric recording was started in kinetic mode at 10 seconds intervals upon addition of 200 μL of freshly prepared AuNPs. Reaction controls contained buffer and citrate alone or buffer, citrate, and ferritin, but without AuNPs.Results
Spectrophotometric methods have been used previously to monitor the photochemical formation of AuNPs.25,26,42–44 In this report, a series of experiments were performed varying the concentrations of the reactants to determine if changes in reactant concentrations could be used to control the size, morphology or catalytic reactivity of the resulting AuNPs. The rate of the reactions was monitored and the resulting products were characterized.Varying Ftn concentrations
Fig. 2A monitors kinetically the formation of the AuNPs by following the appearance of the AuNPs surface plasmon resonance (SPR) at 530 nm.45 It is important to note that each reaction has a lag phase prior to the initiation of AuNP absorbance. The plasmon resonance of AuNPs is not observable until the AuNPs are ∼2 nm in diameter so the AuNPs must reach this size before they are detected by the spectrophotometer. No formation of AuNPs occurs in the absence of Ftn or the absence of light, demonstrating the absolute requirement of Ftn as the photo catalyst.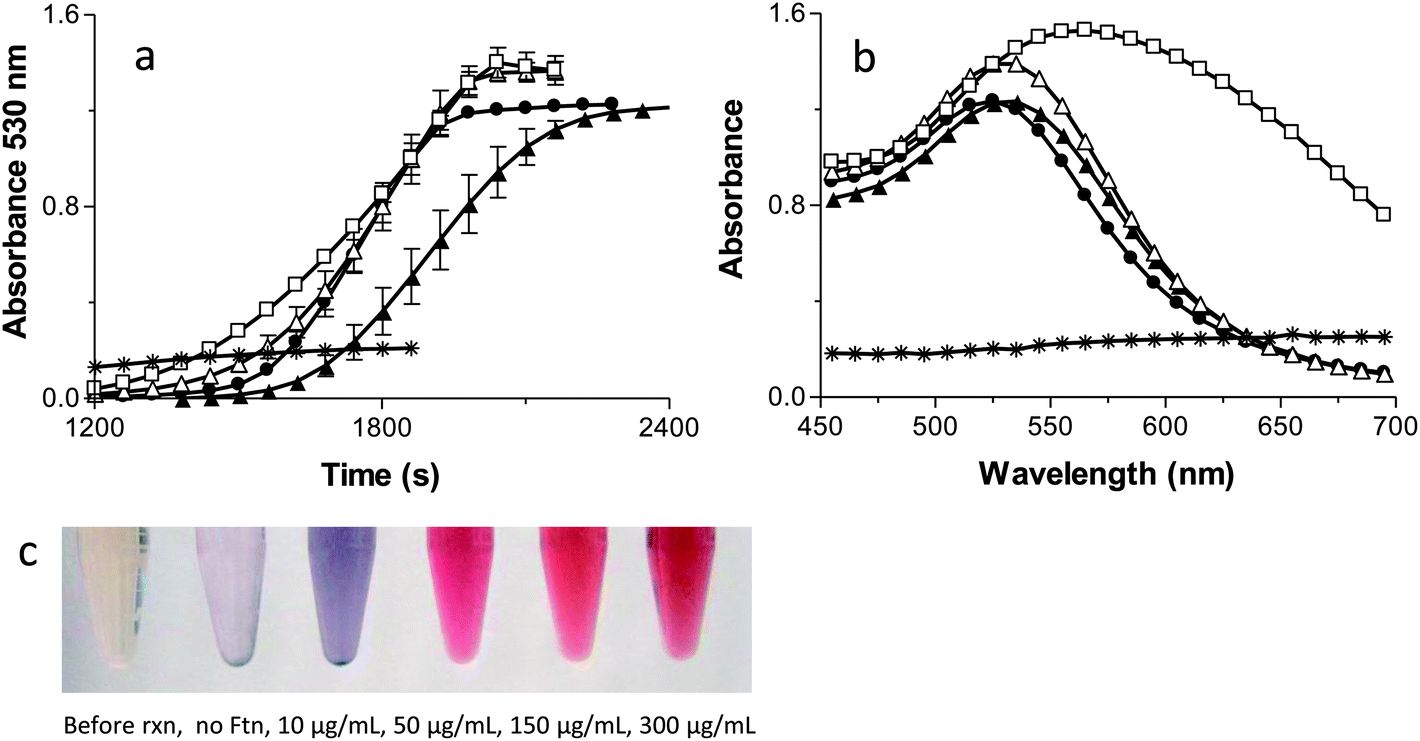 | ||
| Fig. 2 Kinetic traces (a) and absorption spectra (b) of AuNPs formed in the presence of no ferritin (Ftn) (*), 10 μg mL−1 Ftn (□), 50 μg mL−1 Ftn (△), 150 μg mL−1 Ftn (●), or 300 μg mL−1 Ftn (▲). The results are the means ± the S.D. of at least two independent experiments. (c) Tubes containing the AuNP reaction solutions before the reaction (far left), with no Ftn (reaction control, second from the left), and with increasing concentrations of Ftn corresponding to the samples shown in (a) and (b) (10 μg mL−1 Ftn (□), 50 μg mL−1 Ftn (△), 150 μg mL−1 Ftn (●), or 300 μg mL−1 Ftn (▲)). No reactions occurred in the absence of light. | ||
The first point of interest in Fig. 2A is that the highest Ftn concentration has the longest lag phase before AuNP detection occurs. The model in Scheme 2 shows that as Ftn increases, the number of AuNPs increases but the size of the AuNPs decreases. The large number of seed AuNPs bound to the higher concentrations of Ftn all compete for Au3+ ions in solution and it requires a longer time for each of these seed AuNPs to reach 2 nm where the SPR absorbance occurs. When the Ftn concentration is low, there are fewer nucleation sites allowing the few AuNPs that form to grow more rapidly because there are fewer AuNPs competing for free Au3+ ions in solution. These particles reach 2 nm more rapidly and have a shorter lag phase.
The slope in Fig. 2A monitors the rate of formation of the AuNPs once the AuNPs reaches the 2 nm diameter threshold where the SPR can be observed. As the Ftn concentration increases, the rate of AuNPs formed will also increase because there will be more seed AuNPs for growth. Consistent with this hypothesis, Fig. 2A shows the slope increasing with increasing Ftn concentrations, up to 150 μg Ftn mL−1. It is interesting to note that the slope decreases in the 300 μg Ftn mL−1 sample (Fig. 2A). The decrease in slope in the 300 μg Ftn mL−1 sample occurs because the formation of the large number of AuNP seed particles has depleted the Au3+ ions in solution, and the Au3+ ions are limiting in the reaction. In fact, using more gold (800 μM) in the presence of 300 μg Ftn mL−1 increases the steepness of the slope as expected if the gold ions were limiting in the previous reaction (data not shown).
Fig. 2B shows the final spectrum of each of the samples prepared at varying Ftn concentrations. The no Ftn control shows very little production of AuNPs (no absorbance peak at 530 nm). The 10 μg Ftn mL−1 sample has a broad absorption peak around 560 nm but as the Ftn concentration increases the AuNP peak wavelength moves to shorter wavelengths and becomes sharper. These data suggest that the AuNPs forming at higher concentrations of Ftn are smaller and do not aggregate.45 Fig. 2C shows photos of the samples at the end of the reactions demonstrating the effect of altered Ftn concentrations on the colour of the final product.
The TEM images shown in Fig. 3 confirm that the AuNPs formed in the presence of 10 and 50 μg Ftn mL−1 are larger, more irregularly shaped particles, and tend to aggregate more than the samples that form at higher Ftn concentrations (150 and 300 μg Ftn mL−1). Histograms in Fig. 3 (sizes measured from multiple TEM images, some exemplified in supplemental Fig. 1†) demonstrate that as the Ftn concentration increases, the number of AuNPs increases and the resulting AuNPs are smaller due to the larger number of nucleation sites and the capping of the AuNPs by Ftn. Additionally, as the Ftn concentration increases, the distribution in size of the particles decreases (compare the distribution of the 50 μg mL−1 sample with the 150 μg mL−1 sample). TEM images confirm that when Ftn concentrations are 150–300 μg Ftn mL−1, the formation of AuNPs is more consistent in size and shape with an average diameter around 7 nm.
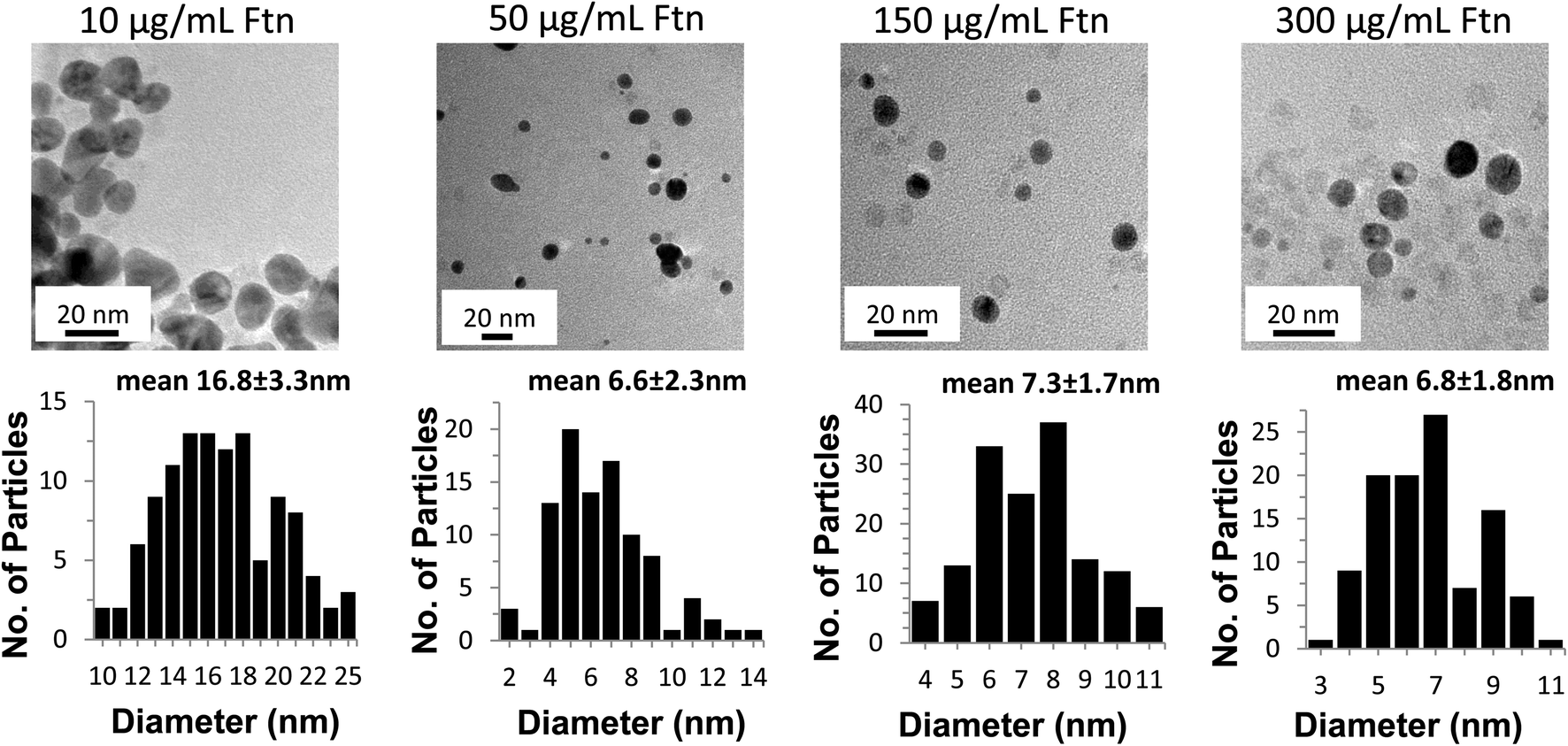 | ||
| Fig. 3 TEM images of the AuNPs at increasing concentrations of ferritin (Ftn) (top row, left to right) and histograms of the corresponding particle size distributions (bottom row). The size distribution of the particles is the result of data collected from several micrographs. The light grey dots visible in the micrographs are the 7–8 nm iron cores of ferritin. | ||
Varying citrate concentrations
Fig. 4A and B show the kinetics and spectra for the formation of AuNPs as the citrate concentration is varied. The standard citrate concentration used by Keyes et al. (30 mM) yields a sigmoidal shape for the kinetic curve of AuNP formation. Increasing the concentration of citrate to 100 mM reduces the lag phase and makes the slope steeper, consistent with a more abundant supply of electrons (Fig 4A). Additionally, the higher concentration of citrate shifts the absorbance maximum peak from 530 nm to 520 nm (Fig. 4B), producing a lighter red solution (Fig. 4C), and forms spherical AuNPs of smaller average size (Fig 5). The lower overall absorbance of the AuNPs at 100 mM citrate is due to the lower extinction coefficient of smaller AuNP.45 Taken together, these data suggest that the higher citrate concentration enhances the photo catalysed AuNP formation reactions by providing an abundant supply of electrons.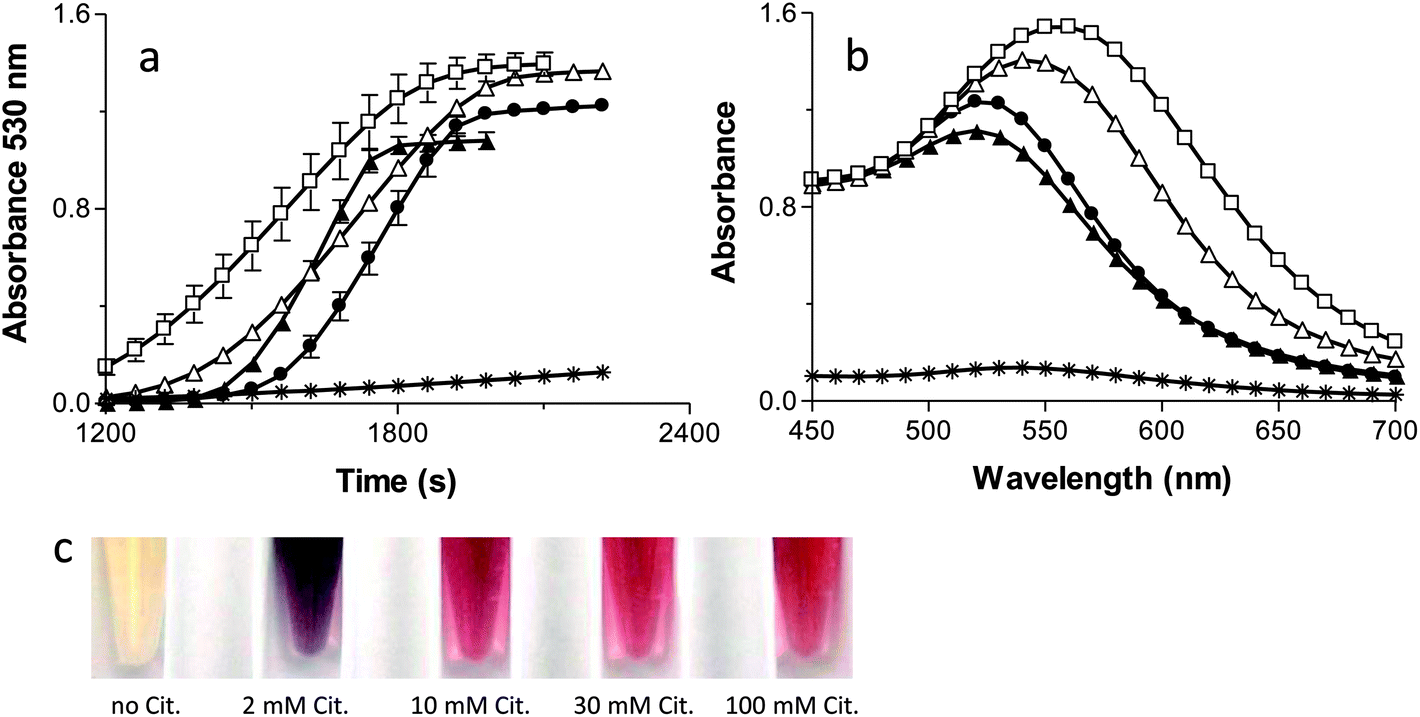 | ||
| Fig. 4 Kinetic traces (a) and absorption spectra (b) of AuNPs formation in the presence of no citrate (*), 2 mM (□), 10 mM (△), 30 mM (●), or 100 mM citrate (▲). The results are the means ± the S.D. of at least two independent experiments. (c) Tubes containing the AuNP reaction solutions with no citrate (far left), and with increasing concentrations of citrate (left to right) corresponding with the concentrations given above (2 mM (□), 10 mM (△), 30 mM (●), or 100 mM citrate). | ||
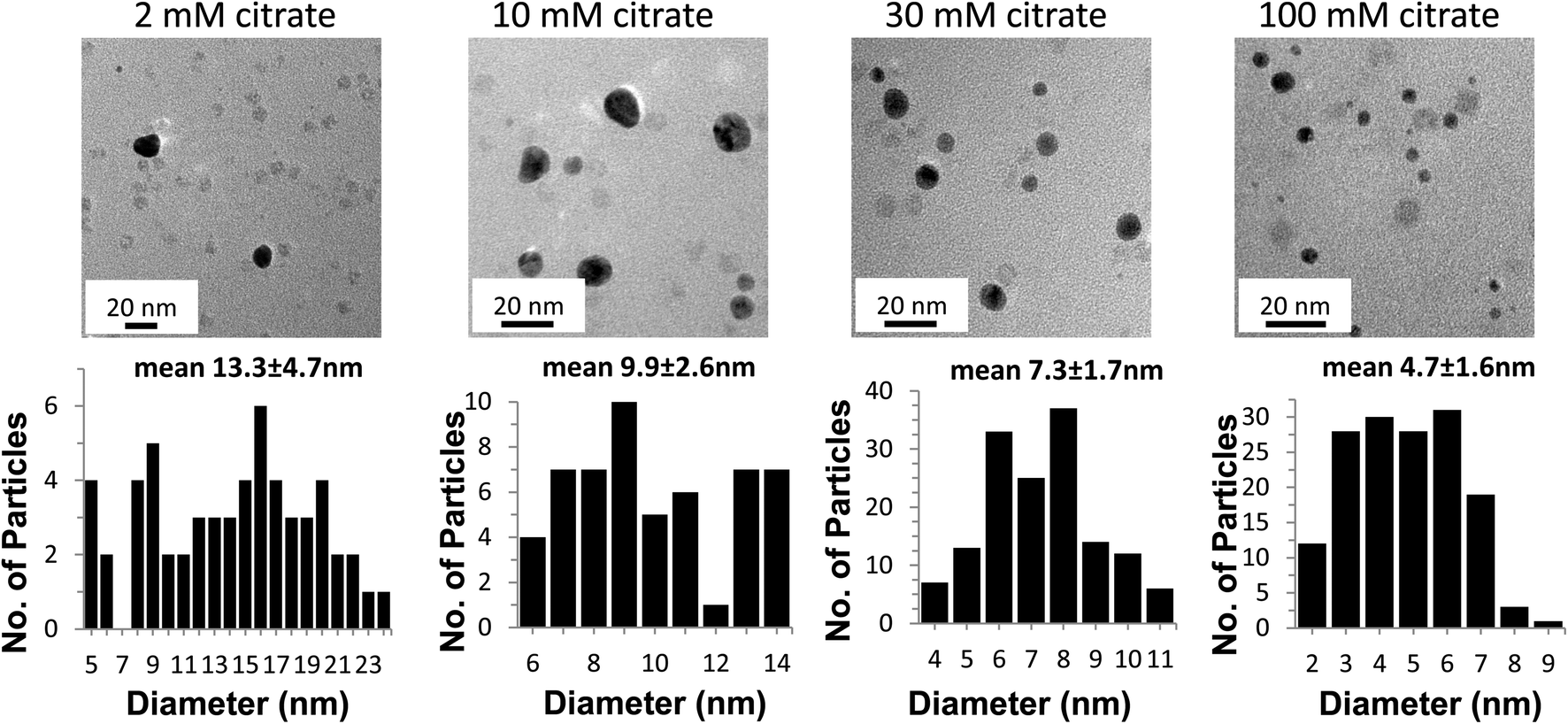 | ||
| Fig. 5 TEM images of the AuNPs at increasing concentrations of citrate (top row, left to right) and histograms of the corresponding particle size distributions (bottom row). The size distribution of the particles is the result of data collected from several micrographs. The light grey dots visible in the micrographs are the 7–8 nm iron cores of ferritin. | ||
When the citrate concentration is decreased below 30 mM, the lag phase shortens, the kinetic slope becomes shallow (Fig. 4A), the spectrum broadens and shifts to longer wavelengths (Fig. 4B), and the particles become fewer, larger, and more irregularly shaped (Fig. 5). These observations are all consistent with slower reactions that are associated with a decreased source of electrons and less capping of the AuNPs as they form, resulting in non-specific AuNP production.25 Additionally, the lack of passivation by citrate contributes to the formation of larger NP due to aggregation.46 One piece of data that seems inconsistent with this interpretation is the fact that the lag phase is shorter with lower citrate concentrations. Nikandrov also reported this phenomenon and demonstrated that in the absence of a suitable electron donor, the Ftn protein shell became the electron donor and was degraded.34 The protein-mediated reduction of AuNPs and the resulting damage to the protein causes an increase in the absorbance throughout the entire spectrum of the sample and caused a shortened lag phase. Also, all of the gold ions available will assemble onto the few particles formed, allowing them to quickly grow above 2 nm, thus recapitulating the results seen at low Ftn concentrations.
Fig. 5 shows the TEM micrographs and size histograms (sizes measured from multiple TEM images, some exemplified in supplemental Fig. 2†) of AuNPs in the presence of 150 μg mL−1 of Ftn with 2 mM, 10 mM, 30 mM, and 100 mM citrate, respectively. At 2 mM citrate the NP tend to grow larger than 20 nm, fuse to each other and take an amorphous shape, whereas the AuNPs become smaller and more spherical as the citrate concentration increases from 10 mM to 100 mM. Other evidence of the increase in size can be seen visually (Fig. 4C) as the samples change from red to purple with the decrease of the citrate concentration. It is clearly visible that the 10 mM and 2 mM citrate samples appear a blue-violet colour instead of the red colour observed in the other samples. The aggregated AuNPs in the 2 mM sample partially precipitated in a few hours.
Varying gold concentrations
Varying the Au3+ concentration produced some obvious challenges because gold is absolutely essential for product formation. Fig. 6A and B show the kinetics and spectra of the AuNP formation at various Au3+ concentrations. At 40 μM Au3+ there is almost no formation of AuNP as is evident by no change in the kinetic plot (Fig. 6A) and the almost-zero absorbance throughout the whole spectrum (Fig. 6B). The absorbance increases and the lag phase decreases as the Au3+ concentration increases from 200 to 500 μM (Fig. 6A). The spectra peak at 520 shifts to higher wavelengths as the Au3+ concentration increases (it shifts about 2 nm for every 100 μM increase in Au3+ concentration up to 600 μM). Beyond 600 μM the red shift is rapid (data were collected but are not shown here because the absorbance was above the detection limit of the spectrophotometer). As expected, increasing the Au3+ concentration increases the final absorbance due to more AuNP formation. This is evident upon visual inspection as the samples become darker as the Au3+ concentration increases (Fig 6C). The TEM images and histograms (Fig. 7) confirm an increase in the number of the AuNP as well as a slight increase in size of the particles as the gold concentration increases from 200 to 500 μM Au3+ (see supplemental Fig. 3†) for additional TEM images. Note that the 40 μM Au3+ sample did not produce sufficient particles for a proper histogram. | ||
| Fig. 6 Kinetic traces (a) and absorption spectra (b) of AuNPs formation in the presence of 40 μM (□), 200 μM (△), 300 μM (●), 400 μM (▲), or 500 μM Au3+ (♦). The results are the means ± the S.D. of at least two independent experiments. (c) Tubes containing the AuNP solutions with increasing concentrations of Au3+ (left to right). | ||
 | ||
| Fig. 7 TEM images of the AuNPs at increasing concentrations of Au3+ (top row, left to right) and histogram of the corresponding particle size distributions (bottom row). Too few particles were formed at 40 μM gold, hence a statistically significant particle size distribution graph could not be created (bottom left). The size distribution of the particles is the result of data collected from several micrographs. The light grey dots visible in the micrographs are the 7–8 nm iron cores of ferritin. | ||
Effect of ferritin iron core size and phosphate content on photo catalysis
An important part of controlling the size of the AuNPs is gaining a better understanding of the catalyst that is being used. Ensign et al. reported that smaller cores react faster than larger cores during the reduction of Cu(II) to Cu(0).39 However, in their experiments they keep the concentration of the total iron constant but doubled the concentration of the protein. Therefore, as they state in their paper, “it is not certain whether the higher speed of reaction of the smaller core should be attributed to the smaller cores or to the higher concentration of Ftn”.We desired to further evaluate the influence of the core size and, additionally, the effect of phosphate associated with the iron core on the rate of AuNP formation. Samples were prepared by reconstituting Ftn with 100 Fe/Ftn, 600 Fe/Ftn, or 1800 Fe/Ftn. The 600 Fe/Ftn and 1800 Fe/Ftn samples were divided into two fractions and one fraction of each metal concentration was incubated with phosphate to add a phosphate layer onto the iron mineral as previously reported by Treffry et al.35 The samples incubated with phosphate were characterized. The ratios of phosphate/Fe were 1 phosphate for every 16.5 in the 600 Fe/Ftn sample and 1 phosphate for every 19 Fe in the 1800 Fe/Ftn. The rate of AuNP formation by these samples was compared to the native horse spleen Ftn sample that contained a natural core with 1800 Fe/Ftn with 1 phosphate for every 9.5 Fe.
Each of these samples successfully formed AuNPs, however, the length of the lag phase and the steepness of the slope of the reactions differed significantly among these samples (Fig. 8). The trend showed that the smaller cores produced a much shorter lag phase and a steeper slope indicating that they formed AuNP seeds reaching 2 nm faster and that once these AuNP seeds formed, the growth of the AuNPs was faster. Our data indicate that the smaller iron cores catalyse the photochemical reaction faster and that the increased reaction rate is independent of the concentration of the protein.
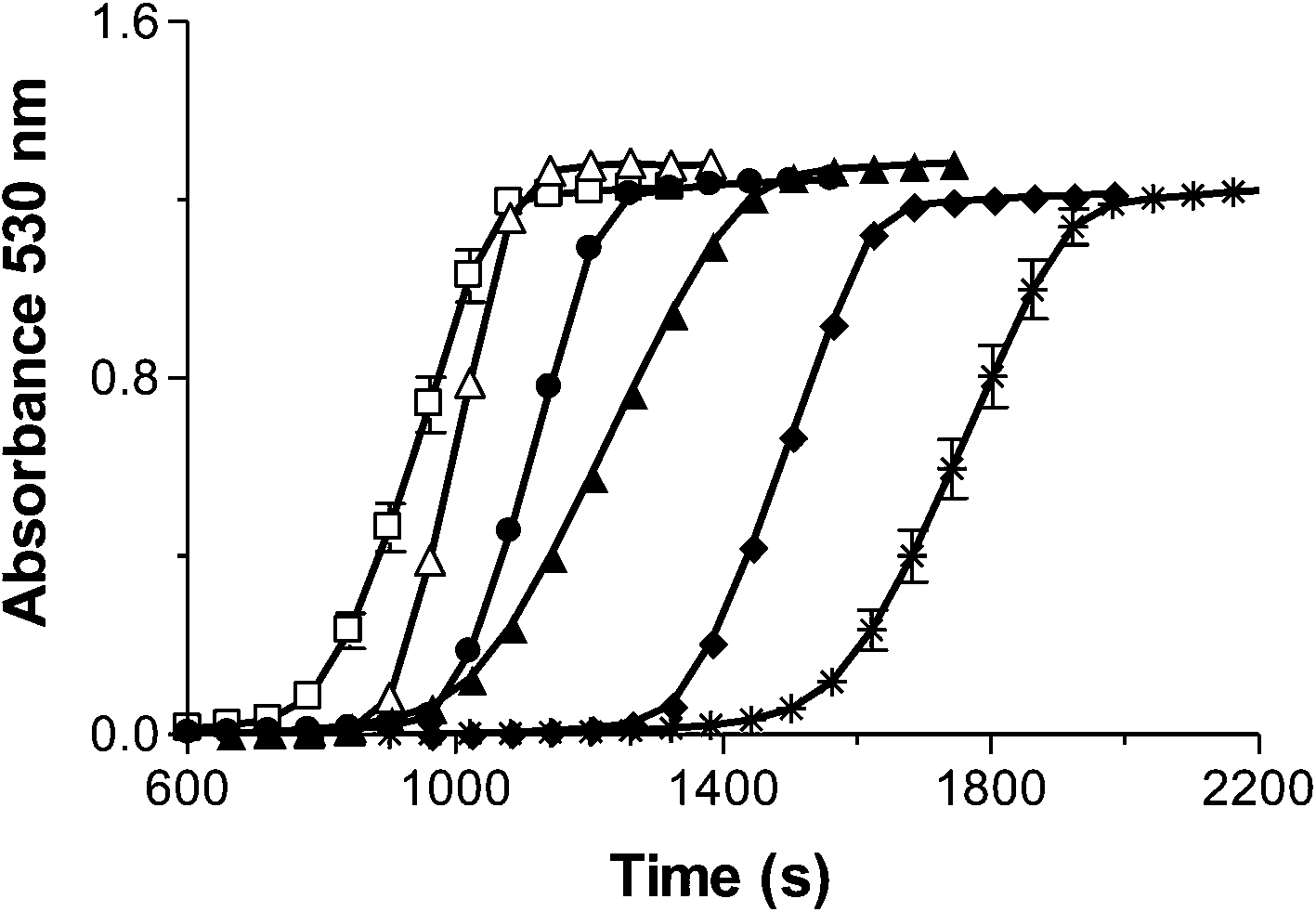 | ||
| Fig. 8 Kinetic traces of AuNP formation dependent on core size and phosphate content: a reconstituted 1800 Fe/ferritin (Ftn) sample (*), reconstituted 1800 Fe/Ftn sample containing phosphate with 1 phosphate for every 19 Fe (Ftn-P) (♦), Native Ftn sample with 1800 Fe/Ftn with 1 phosphate for every 9.5 Fe (▲), reconstituted 600 Fe/Ftn sample (●), 600 Fe/Ftn sample containing phosphate with 1 phosphate for every 16.5 Fe (Ftn-P) (△), reconstituted 100 Fe/Ftn sample (□). The results are the means ± the S.D. of at least two independent experiments. | ||
In addition, for Ftn samples that contained the same number iron atoms, the presence of phosphate decreased the lag phase. Both a smaller core and the presence of phosphate are able to decrease the lag phase and increase the reaction rate (Fig. 8).
For example, among the samples containing 1800 iron atoms, the reconstituted 1800 Fe/Ftn iron core with no phosphate had the longest lag phase and the native ferritin sample with 1800 Fe/Ftn that contained the most phosphate/Fe (1 phosphate for every 9.5 Fe) had the shortest lag phase. The sample reconstituted with 1800 Fe/Ftn incubated with phosphate (1 phosphate for every 19 Fe) had a lag phase intermediate between the other 1800 Fe/Ftn samples. Similar results were observed for the 600 Fe/Ftn samples with and without phosphate. We propose that the phosphate coordinates the iron in the ferrihydrite core of the protein making it more reactive, probably by weakening the crystalline structure of ferrihydrite because phosphate is known to make the mineral core more amorphous46 and/or by increasing the solubility constant (Ksp from 10−36 to 10−22).
Oxidation state of the gold ions
Ensign et al. suggested that the reduction of Cu(II) to Cu(0) was a stepwise 1-electron at a time process.39 Additionally, Zhang et al. reported that chemical reduction of Au3+ to form Au(0) AuNPs on Ftn occurred through a 1-electron at a time pathway.26 If a similar step by step electron transfer mechanism is occurring with Au3+ in our photochemical system, we would predict that using Au+ as the electron acceptor instead of Au3+ should result in a shorter lag phase and a more rapid production of AuNPs.Photochemical reactions with Au+ as the substrate occurred with a much shorter lag phase than Au3+ reactions to produce AuNPs, (Fig. 9A) suggesting that the 1-electron transfer observed by Ensign and Zhang occurs during the photochemical redox reaction as well. The AuNPs formed using Au+ stopped growing at 1–3 nm diameter size (Fig. 9A–E) and are the smallest we have been able to reproducibly synthesize so far. These AuNPs are mono crystalline and spherical with a diameter size of 2.4 ± 1.0 nm (Fig. 9E). Thus, varying the oxidation state of the gold ion substrate has provided another layer of synthetic control over the desired size of the AuNPs produced by this method.
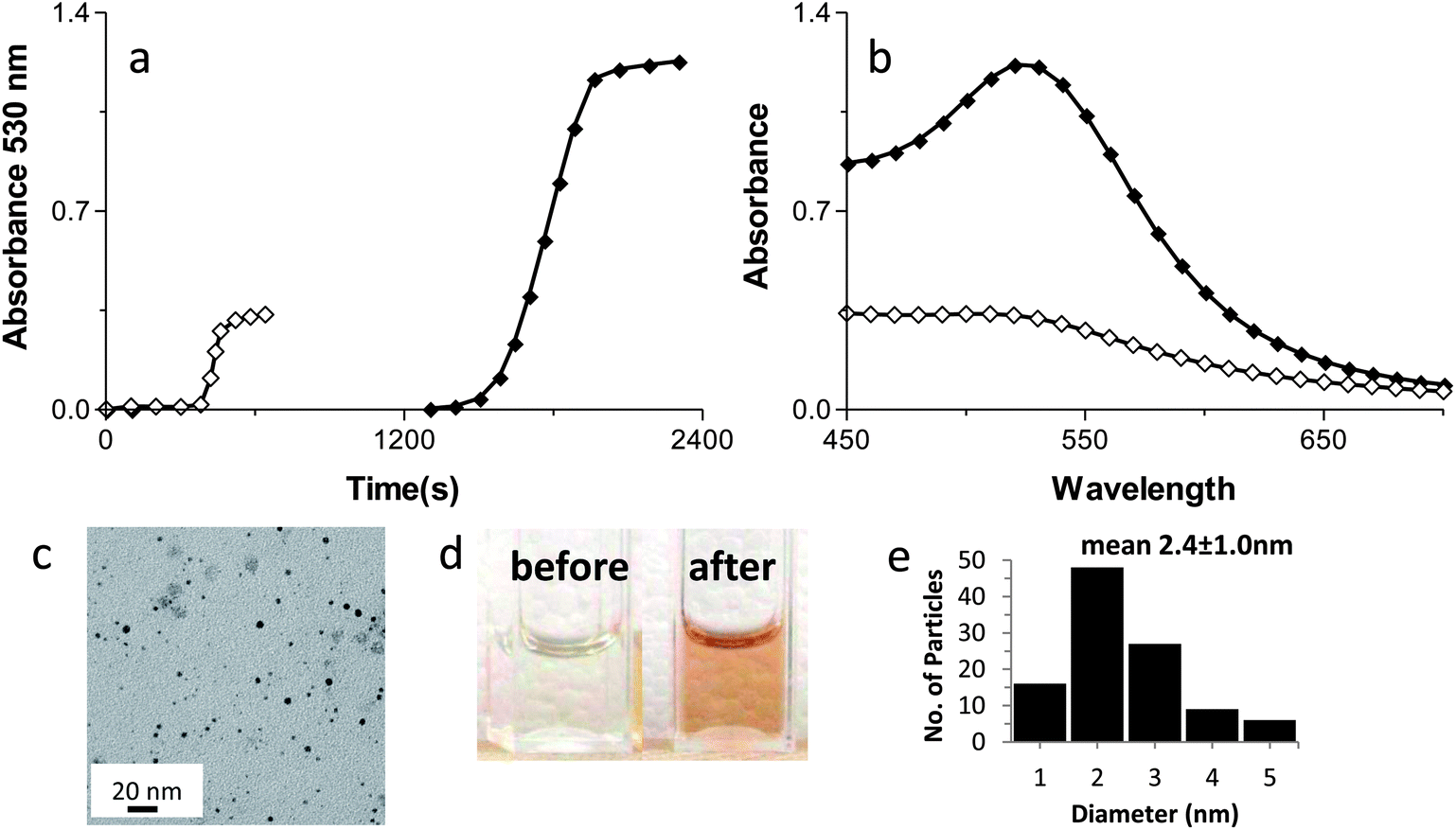 | ||
| Fig. 9 Kinetic traces (a) and absorption spectra (b) of AuNPs formation from AuCl (◊) vs. KAuCl4 (♦). (c) TEM image of AuNPs from Au+. (d) Cuvettes containing the AuNPs solutions before (left) and after (right) the photo reaction. (e) Histogram of the particle size distribution from the Au+ sample (106 particles analyzed). | ||
All of the AuNPs smaller than 10 nm produced by our method are mono crystalline (supplemental Fig. 4†).
Catalytic activity of the AuNPs
An important goal was to measure the catalytic activity of the AuNPs produced by this method. We used the reduction of 4-nitrophenol (PNP) to 4-aminophenol (PAP) with NaBH4 as a model reaction to demonstrate the catalytic reactivity of the AuNPs produced by the Ftn photo catalytic method.
According to the literature, 2–3 nm AuNPs have the highest catalytic properties4,47. We tested AuNPs with diameters of 2.4, 7.3 and 20 nm as they catalysed the reduction of PNP in the presence of sodium borohydride (NaBH4) to produce PAP.26 The rate constants for the reaction (using 10% volume Ftn–AuNPs solution as a catalyst – corresponding to 40 μM gold – or 7.9 ppm), are (linear fitting) k = 5.34 × 10−2 s−1 for the 2.4 nm AuNPs (from Au+), and k = 2.91 × 10−2 s−1 for 7.3 nm AuNPs (from Au3+). AuNPs with a 20 nm diameter did not catalyse the reaction within the time they were allowed to react (800 seconds, data not shown). Consistent with the literature, we observed that the smaller AuNPs were more catalytically active than the larger ones.26 Furthermore, PNP was not reduced by NaBH4 when the AuNPs were not present. These results are shown in Fig. 10.
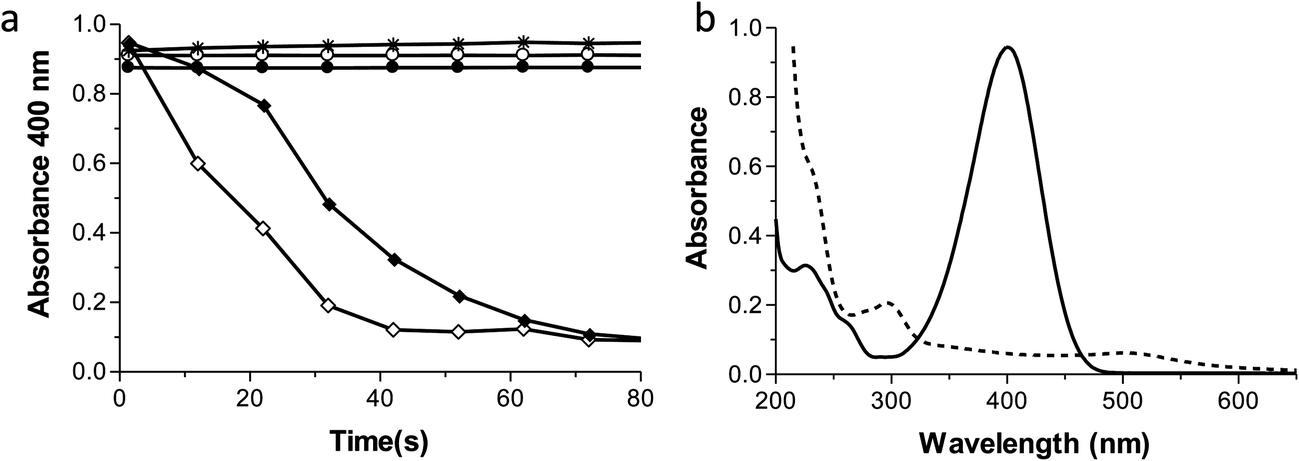 | ||
| Fig. 10 Kinetic traces (a) of the catalytic reduction of 4-nitrophenol (PNP, 50 μM) to 4-aminophenol (PAP) by NaBH4 (2.5 mM) using 20 nm (*), 7.3 nm (♦), or 2.4 nm (◊) Ftn–AuNPs as catalyst. Buffer/citrate alone (●) or buffer/citrate + ferritin (○) reaction controls, all in the presence of 2.5 mM NaBH4. Absorption spectra (b) of 4-nitrophenol before (solid line) and after reduction (dashed line). | ||
Conclusions
The synthesis and assembly of nanoparticles has become central to modern nanotechnology. Medicine, material science, energy research, catalysis, and semiconductor engineering all exploit the unique capabilities of the nanoparticles. The unique properties of nanoparticles are dependent on their size, shape, crystallinity, passivation, and biocompatibility. Synthesis methods developed so far have provided many structures that control these characteristics. Our goal was to develop a single method that could be used to synthesize a variety of sizes of AuNPs by simply altering the concentrations of the reactants.Our work demonstrated that the Ftn-based nanoparticle photosynthesis method could be used to form a broad range of mono disperse AuNPs. Optimizing the reaction conditions allowed us to control the nanoparticle size and catalytic activity. Other than the Au ions, we utilized reagents that are already present in the human body and are therefore biocompatible by definition. Ftn-based photochemical reactions are quick, simple, and inexpensive in that it does not require complex and costly equipment (e.g.: lasers). This method will allow all laboratories to create their own nanoparticles freshly “in house” at the time they are needed. Our work demonstrates a method that allows us to reproducibly prepare biocompatible, quasi-mono disperse ∼2, ∼7 or ∼17 nm diameter AuNPs without the need of additional separations steps. Additionally, the Ftn-citrate-capped AuNPs remain catalytically active and the catalytic activity of the desired particles correlates with the different sized AuNPs.
Notes and references
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- B. Hvolbaek, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Norskov, Nano Today, 2007, 2, 14–18 CrossRef.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef PubMed.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- I. Hussain, S. Graham, Z. Wang, B. Tan, D. C. Sherrington, S. P. Rannard, A. I. Cooper and M. Brust, J. Am. Chem. Soc., 2005, 127, 16398–16399 CrossRef CAS PubMed.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222–234 CrossRef CAS PubMed.
- P. C. Chen, S. C. Mwakwari and A. K. Oyelere, Nanotechnol., Sci. Appl., 2008, 1, 67–78 Search PubMed.
- G. L. Prasad, in Safety of Nanoparticles, ed. T. J. Webster, Springer New York, 2009, pp. 89–109 Search PubMed.
- D. Prime, S. Paul, C. Pearson, M. Green and M. C. Petty, Mater. Sci. Eng., C, 2005, 25, 33–38 CrossRef PubMed.
- D. P. Stankus, S. E. Lohse, J. E. Hutchison and J. A. Nason, Environ. Sci. Technol., 2011, 45, 3238–3244 CrossRef CAS PubMed.
- A. E. Lanterna, E. A. Coronado and A. M. Granados, J. Phys. Chem. C, 2012, 116, 6520–6529 CAS.
- L. A. McConnachie, D. Botta, C. C. White, C. S. Weldy, H. W. Wilkerson, J. Yu, R. Dills, X. Yu, W. C. Griffith, E. M. Faustman, F. M. Farin, S. E. Gill, W. C. Parks, X. Hu, X. Gao, D. L. Eaton and T. J. Kavanagh, PLoS One, 2013, 8, e64165 CAS.
- K. Yoshida, Y. Sakurai, S. Kawahara, T. Takeda, T. Ishikawa, T. Murakami and A. Yoshioka, Int. Arch. Allergy Immunol., 2008, 146, 169–173 CrossRef PubMed.
- R. Munday, E. Manns and E. A. Fowke, Food Chem. Toxicol., 1990, 28, 561–566 CrossRef CAS.
- X. Nie and C. Chen, Sci. China: Life Sci., 2012, 55, 872–883 CrossRef CAS PubMed.
- N. Khlebtsov, V. Bogatyrev, L. Dykman, B. Khlebtsov, S. Staroverov, A. Shirokov, L. Matora, V. Khanadeev, T. Pylaev, N. Tsyganova and G. Terentyuk, Theranostics, 2013, 3, 167–180 CrossRef CAS PubMed.
- S. Akhter, M. Z. Ahmad, F. J. Ahmad, G. Storm and R. J. Kok, Expert Opin. Drug Delivery, 2012, 9, 1225–1243 CrossRef CAS PubMed.
- M. Z. Ahmad, S. Akhter, Z. Rahman, M. Anwar, N. Mallik and F. J. Ahmad, J. Pharm. Pharmacol., 2013, 65, 634–651 CrossRef CAS PubMed.
- E. C. Dreaden, L. A. Austin, M. A. Mackey and M. A. El-Sayed, Ther. Delivery, 2012, 3, 457–478 CrossRef CAS.
- A. Kumar, X. Zhang and X. J. Liang, Biotechnol. Adv., 2013, 31, 593–606 CrossRef CAS PubMed.
- R. L. Fan, S. W. Chew, V. V. Cheong and B. P. Orner, Small, 2010, 6, 1483–1487 CrossRef CAS PubMed.
- C. A. Butts, J. Swift, S. G. Kang, L. Di Costanzo, D. W. Christianson, J. G. Saven and I. J. Dmochowski, Biochemistry, 2008, 47, 12729–12739 CrossRef CAS PubMed.
- J. D. Keyes, R. J. Hilton, J. Farrer and R. K. Watt, J. Nanopart. Res., 2011, 13, 2563–2575 CrossRef CAS.
- L. Zhang, J. Swift, C. A. Butts, V. Yerubandi and I. J. Dmochowski, J. Inorg. Biochem., 2007, 101, 1719–1729 CrossRef CAS PubMed.
- R. R. Crichton and J.-P. Declercq, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 706–718 CrossRef CAS PubMed.
- F. Bou-Abdallah, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 719–731 CrossRef CAS PubMed.
- R. K. Watt, ChemBioChem, 2013, 14, 415–419 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- L. Li, C. J. Fang, J. C. Ryan, E. C. Niemi, J. A. Lebron, P. J. Bjorkman, H. Arase, F. M. Torti, S. V. Torti, M. C. Nakamura and W. E. Seaman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3505–3510 CrossRef CAS PubMed.
- I. Kim, H. A. Hosein, D. R. Strongin and T. Douglas, Chem. Mater., 2002, 14, 4874–4879 CrossRef CAS.
- V. V. Nikandrov, C. K. Gratzel, J. E. Moser and M. Gratzel, J. Photochem. Photobiol., B, 1997, 41, 83–89 CrossRef CAS.
- A. Treffry and P. M. Harrison, Biochem. J., 1978, 171, 313–320 CAS.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- R. J. Hilton, M. C. Seare, N. D. Andros, Z. Kenealey, C. M. Orozco, M. Webb and R. K. Watt, J. Inorg. Biochem., 2012, 110, 1–7 CrossRef CAS PubMed.
- M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shafigullin, I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101, 3706–3712 CrossRef CAS.
- D. Ensign, M. Young and T. Douglas, Inorg. Chem., 2004, 43, 3441–3446 CrossRef CAS PubMed.
- Nanosensors, Biosensors, and Info-Tech Sensors and Systems 2010, edited by Vijay K. Varadan, Proc. of SPIE Vol. 7646, 76460J · © 2010 SPIE · CCC code: 0277-786X/10/$18 · DOI: 10.1117/12.858830 Search PubMed.
- Nanosensors, Biosensors, and Info-Tech Sensors and Systems 2010, edited by Vijay K. Varadan, Proc. of SPIE Vol. 7646, 764607 · © 2010 SPIE · CCC code: 0277-786X/10/$18 · DOI: 10.1117/12.847660 Search PubMed.
- M. L. Marin, K. L. McGilvray and J. C. Scaiano, J. Am. Chem. Soc., 2008, 130, 16572–16584 CrossRef CAS PubMed.
- K. L. McGilvray, C. Fasciani, C. J. Bueno-Alejo, R. Schwartz-Narbonne and J. C. Scaiano, Langmuir, 2012, 28, 16148–16155 CrossRef CAS PubMed.
- K. Mallick, Z. L. Wang and T. Pal, J. Photochem. Photobiol., A, 2001, 140, 75–80 CrossRef CAS.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed.
- G. Frens, Prog. Colloid Polym. Sci., 1972, 250, 736–741 CAS.
- S. Guerin, B. E. Hayden, D. Pletcher, M. E. Rendall and J.-P. Suchsland, J. Comb. Chem., 2006, 8, 679–686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra46520a |
| This journal is © The Royal Society of Chemistry 2014 |