Synthesis of 5,10,15,20-meso-unsubstituted and 5,10,15,20-meso-substituted-21,23-ditellura/diselena core-modified porphyrinogens: oxidation and detection of mercury(II)†
Sohail Ahmad,
Kumar Karitkey Yadav,
Sarangthem Joychandra Singh and
S. M. S. Chauhan*
Bio-organic Research Laboratory, Department of Chemistry, University of Delhi, Delhi-110 007, India. E-mail: smschauhan@chemistry.du.ac.in; Fax: +91-11-27666845; Tel: +91-11-27666845 Tel: +91-9871969266
First published on 2nd December 2013
Abstract
Tellurium and selenium incorporated 5,10,15,20-meso-unsubstituted-21,23-ditellura/diselena core-modified porphyrinogens (N2Te2 and N2Se2), 5,10,15,20-meso-unsubstituted-21-tellura/selena core-modified porphyrinogens (N3Te and N3Se) and fully substituted meso-carbons porphyrinogens (N2Te2, N2Se2 and higher analogs) are synthesized by 3 + 1 condensation of tellurophene/selenophene dipyrranes and their corresponding diols in the presence of BF3–etharate or BF3–methanol. The meso-unsubstituted and substituted porphyrinogens were oxidized with chloranil/0.1% aqueous FeCl3 in CHCl3 at room temperature to obtain the corresponding porphines and porphyrins which are further reduced to corresponding chlorin and bacteriochlorin, whereas the fully meso-substituted porphyrinogens were found to be good ligands for Hg2+. The structures of the products were characterized by IR, 1H, 13C, 125Te, 77Se NMR, CHN analysis, mass spectrometry and single-crystal XRD.
Introduction
Porphyrinogens (calixpyrroles) are a class of tetrapyrrolic macrocycles composed of four pyrrole rings linked through the meso-positions by carbon atoms. The β- and meso-positions in porphyrinogens are reactive positions at which suitable substituents can be introduced to tune the properties of the porphyrinogens for specific applications. Fully meso-substituted porphyrinogens are stable and have been used in anion binding,1 detection of various explosive1f and self-assembly,1 while meso-unsubstituted porphyrinogens have remained unexplored due to unstability and complexity in synthesis.2 Meso-unsubstituted porphyrinogens are highly desirable synthetic precursors for the construction of meso-unsubstituted porphines3,4 and other complex systems with special physical and chemical properties.3b,5 Porphines/porphyrins of porphyrinogens are used as a starting material for various newer porphyrins.5 These are further utilized for the synthesis of longer-wavelength absorbing5c–e and fluorescing molecules such as chlorins, bacteriochlorins and purpurins which are potentially used as fluorescence imaging or phototherapeutic agents.5f–iA wide variety of porphyrinogens with donor atoms (O, S, N and P) containing nonpyrrolic arene units,6–9 have been designed and synthesized to modify the binding ability of the porphyrinogens. The 5,10,15,20-meso-octamethyl-21,23-dithiaporphyrinogen (N2S2) and 5,10,15,20-meso-octamethyl-21-thiaporphyrinogen (N3S) ligands containing nitrogen–sulfur donor atoms are highly selective complexing agents towards heavy transition metal ions (HgII, CdII and AgI).10 Therefore, a similar development is anticipated for the porphyrinogen with heavier chalcogen donor atoms such as selenium and tellurium, due to their ‘softness’ and better sigma-donor capacity as compared to the lighter group 16 congeners.11 The synthesis of selenium and tellurium 21- and 21,23-core-modified porphyrins,12,3b in which one or more heteroatoms replace some of the four nitrogen atoms, has been already reported with altered core sizes, metal ion binding properties,12 reactivity12 and redox potentials.12
Herein we report the synthesis of novel meso-unsubstituted Te/Se core-modified porphyrinogens and meso-substituted Te/Se core-modified porphyrinogens via acid catalyzed 3 + 1 condensation of corresponding tellurophene/selenophene dipyrranes and diols. The meso-unsubstituted porphyrinogens are further oxidized to their corresponding porphyrins and porphines, whereas the meso-substituted porphyrinogens were utilized for the complexation of Hg2+.
Results and discussion
The desired 2,5-bis(2-pyrrolylmethyl)tellurophene (dipyrrane) 4a was synthesized in three steps starting from prop-2-yn-1-ol (1a). The reaction of prop-2-yn-1-ol 1a with CuI and NiCl2·6H2O, followed by the addition of TMEDA in THF under aerobic condition,13 proceeded smoothly to afford the diynediol 2a in 93% yield. The solution of diynediol 2a and AgOAc in MeOH was added to aqueous solution of Na2Te14 to afford tellurophene diol 3a in 85% yield. The tellurophene diol 3a reacted with excess pyrrole in the presence of BF3–etharate to afford dipyrrane 4a in 75% yield. The other dipyrranes (4b–h) were synthesized via similar route (Scheme 1). | ||
| Scheme 1 Synthesis of tellurophene and selenophene dipyrranes (4a–h). | ||
N2Te2 and N2Se2 5,10,15,20-meso-unsubstituted and 5,10,15,20-meso-substituted-21,23-ditellura/diselena core-modified porphyrinogens (5a–i) were synthesized by following the two methods A and B (Scheme 2).
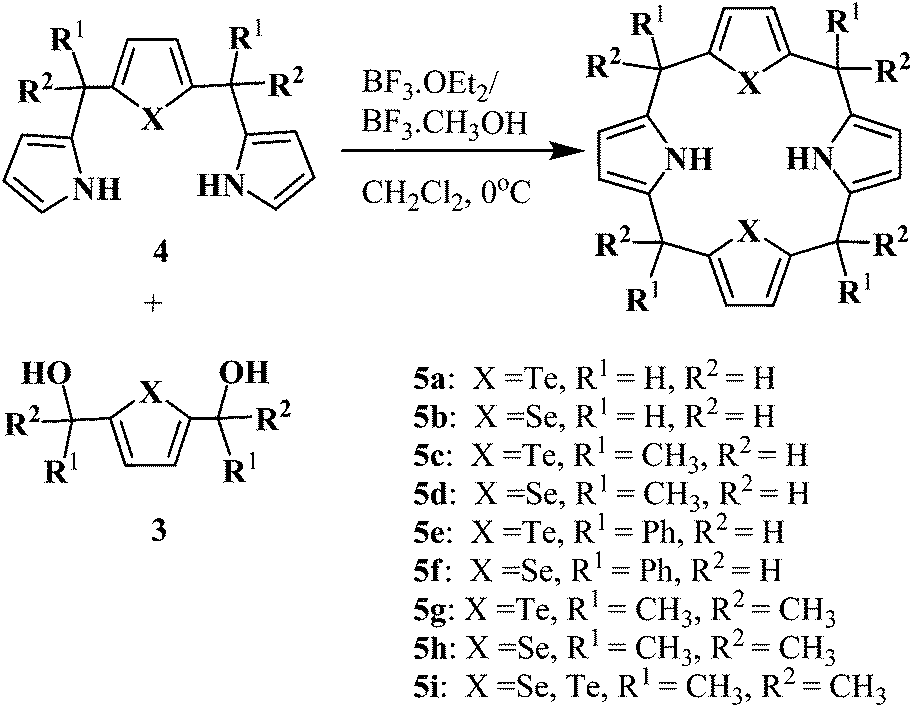 | ||
| Scheme 2 Synthesis of core-modified porphyrinogens. | ||
The meso-unsubstituted and meso-tetramethyl core-modified porphyrinogens (5a–d) were prepared by 3 + 1 condensation of one equivalent of dipyrranes (4a–d) with one equivalent of corresponding diols (3a–d) in the presence of BF3–methanol using dichloromethane as a solvent (3.2 mM) at 0 °C for 1 h, followed by stirring the reaction mixture to room temperature for 45 min (method A). The crude porphyrinogens were purified by silica gel column chromatography to afford white solids 5a in 15%, 5b in 11%, 5c in 27% and 5d in 22% yields respectively. The appearance of meso-methylene protons (meso-CH2 for 5a, 5b and meso-CH for 5c, 5d) at about δ 3.92–4.04 ppm as a sharp singlet in the 1H NMR spectra and a corresponding m/z peak in the mass spectra confirmed the formation of ditellura/diselena core-modified porphyrinogens (5a–d). An attempt to collect X-ray diffraction data was unsuccessful because of the poor quality of crystals. Four unsubstituted meso-carbons porphyrinogens (5a and 5b) are highly unstable and are very difficult to handle and become dark at room temperature in comparison to 5c and 5d porphyrinogens. Interestingly the above reaction conditions favor the formation of cyclic tetramers but their higher cyclic oligomers and N-confused products were not obtained. To obtain the higher analogue, the reaction of dipyrranes (4a–d) with corresponding diols (3a–d) in higher reactant concentration (5.3 mM and 16 mM) in dichloromethane or acetonitrile at 0 °C were carried out and it afforded a polymeric oxidized products along with small amount of porphyrinogens. In most cases, the N-confused cyclic tetramer, hexaphyrinogen and octaphyrinogen could be detected only on TLC as they are readily oxidized to the dyes without isolation. Porphyrinogens with phenyl group (5e–f) were detected only on TLC and were not isolated, as they are readily oxidized to corresponding porphyrins during the workup procedure and mixtures of porphyrinogens and porphyrins were obtained. Even isolated porphyrinogens from column purification are readily oxidized to corresponding porphyrins.
The porphyrinogens 5a–f were oxidized with chloranil in CHCl3 at room temperature for 10 min to obtain the corresponding porphines and porphyrins or by shaking the porphyrinogens solution with 0.1% aqueous FeCl3 solution for 10 min (Scheme 3). Prolonged shaking or stirring the reaction mixture caused decomposition of the product, whereas the porphyrinogens with 0.1% aqueous FeCl3 solution in shorter exposure times were not oxidized to the product. However, attempts to oxidize the porphyrinogens 5a–f with DDQ led to complete decomposition. The high resolution mass spectrometry showed corresponding m/z peak to porphyrin/porphine along with porphyrinogen peak (see ESI†). The corresponding porphyrins of porphyrinogens 5a–c are unstable and readily undergo to decomposition at room temperature. Porphyrinogen 5b on oxidation with 0.1% aqueous FeCl3 solution gave unstable isolable novel 21,23-diselenaporphine in 5% yield, while oxidation with chloranil gave 2% of porphine. In the 1H NMR spectrum of the 21,23-diselenaporphine, the meso proton appeared at δ 10.49 ppm, while pyrrolic and selenophene protons were appeared at δ 9.36 ppm and 10.29 ppm, respectively. The electronic absorption spectra of 21,23-diselenaporphine was recorded in CHCl3 and a soret along with Q bands were observed at 415, 474, 504 and 535 nm. Porphyrinogen 5d on oxidation with chloranil gave stable novel 5,10,15,20-meso-tetramethyl-21,23-diselenaporphyrin in 76% yield. In the 1H NMR spectrum of the 5,10,15,20-meso-tetramethyl-21,23-diselenaporphyrin, the meso methyl group (CH3) appeared at δ 4.5 ppm, while pyrrolic and selenophene protons were appeared as singlet at δ 9.2 ppm and 10.1 ppm, respectively.15 The electronic absorption spectra of 5,10,15,20-meso-tetramethyl-21,23-diselenaporphyrin was recorded in CHCl3 and four Q bands along with one intense soret band were observed at 693.5, 630.0, 554.1, 521.8 and 437.8 nm. The electronic absorption spectra of corresponding porphyrins of porphyrinogens 5a–d are given in ESI.† Porphyrinogens with phenyl group (5e and 5f) were oxidized with DDQ to corresponding porphyrins in 86–90% yield, which were characterized and matched with the spectrum of 5,10,15,20-meso-tetraphenyl-21,23-ditellura/diselenaporphyrin.12
 | ||
| Scheme 3 | ||
The N3Te and N3Se porphyrinogens with four meso-unsubstituted carbons can be used in constructing highly desirable novel 21-tellura/selenaporphyrinogens systems. The N3Te porphyrinogens 9a (Scheme 4) were synthesized from the unsubstituted diols (3a) and 2,5-bis(2-pyrrolylmethyl)pyrrole (tripyrrane) in the concentration of a 3.2 mM scale (method A), affording 9a (7% yield) and 9b (8% yield). 2,5-Bis(2-pyrrolylmethyl)pyrrole (tripyrrane) was obtained by formylation of pyrrole followed by condensation with pyrrole of the resulting 2,5-bis(hydroxymethyl)pyrrole in the presence of acid.2a In the 1H NMR spectrum of 9a in CDCl3 at room temperature, meso-methylene protons connected with a tellurophene ring and connected with a pyrrole ring appeared at δ 3.99 and δ 3.86 ppm as a sharp singlet, respectively. The N3Se porphyrinogen 10a was prepared in the same manner, affording 10a (11% yield) and 10b (traceable amount). An attempt to obtain 21-tellura/selenaporphine by oxidation of 9a and 10a were carried out by shaking the porphyrinogens (9a and 10a) solution with 0.1% aqueous FeCl3 or addition of chloranil directly to the reaction mixture at room temperature for 10 min, but it led to the formation of a complex mixture and even in HRMS no peak corresponding to 21-tellura/selenaporphine were detected.
 | ||
| Scheme 4 | ||
The absence of steric hindrance in meso-unsubstituted selenaporphine and meso-tetramethylselenaporphyrin offers the further utilization of these compounds. The reduction of selenaporphine/porphyrin were carried out by using p-toluenesulfonhydrazide in the presence of pyridine and potassium carbonate to synthesize novel phototherapeutic model compounds diselenachlorin and diselenabacteriochlorin (Scheme 5). Reaction of the 5,10,15,20-meso-tetramethyl-21,23-diselenaporphyrin with 10 equivalent p-toluenesulfonhydrazide and 10 equivalent K2CO3 in pyridine for 24 h, gave meso-tetramethyldiselenachlorin and meso-tetramethyldiselenabacteriochlorin in 5% and 2% yield respectively. The corresponding chlorin and bacteriochlorin of meso-tetramethylselenaporphyrin are unstable and readily undergo decomposition at room temperature. The formation of meso-tetramethyldiselenachlorin was confirmed by the appearance of reduced pyrrolic proton at about δ 4.29 ppm and δ 4.60 ppm as triplet in the 1H NMR spectra. The selenophene and pyrrole protons of diselenachlorin are upfield shifted with respect to selenaporphyrin. The selenophene protons were appeared as doublets at δ 9.27 and 9.64 ppm (J = 4.8 Hz) while pyrrolic protons appeared as singlet at δ 9.07 ppm. The electronic absorption spectra of meso-tetramethyldiselenachlorin (soret and Q bands were observed at 437.9, 522.6, 550.8, 631.7 and 689.0 nm) was more similar to the starting material. However, the longest wavelengths absorption (689 nm) is more intensified and 4 nm hypsochromically shifted compared to the starting meso-tetramethylselenaporphyrin. The 1H NMR spectrum of meso-tetramethyldiselenabacteriochlorin is much simplified due to symmetrical structure of bacteriochlorin. The selenophene signal is downfield shifted and appeared at δ 8.92 ppm as singlet. A comparative shift for the reduced pyrrolic protons was also observed. The UV-vis spectrum of the meso-tetramethyldiselenabacteriochlorin, shows three-bands at 408, 535, 739 nm, in which the longest wavelengths absorption is significantly more red-shifted (739 nm) as compared to the corresponding chlorin (689 nm) and selenaporphyrin (693 nm).
 | ||
| Scheme 5 | ||
An attempt to obtain chlorin by reduction of 21,23-diselenaporphine (meso-unsubstituted porphyrin) using p-toluenesulfonhydrazide, led to the decomposition of starting material or product and no corresponding chlorin was detected in UV-Vis spectrum. Selena/telluraporphyrins with phenyl group at meso position are less prone to reduction and require excess of the reducing agent and longer reaction time. The reaction was monitored by TLC and UV-Vis spectroscopy.
The fully meso-substituted core-modified porphyrinogens (5g–i) were synthesized by condensation of dipyrranes (4g–h) with corresponding diols (3g–h) in the presence of BF3–etharate in dichloromethane (3.2 mM concentration) at room temperature for 30 min (method B). These porphyrinogens (5g–i) are highly stable and can be stored for over 1 year at room temperature. To obtain the higher analogue (Fig. 1), similar reactions were carried out in acetonitrile at 0 °C with the same concentration (∼3.2 mM) but not much difference in cyclic tetramer yields were observed and no higher ordered species were detected (Table 1). The increase in reactant concentration (∼16 mM) favored the formation of higher ordered species and gave 5g in 23% yield, N-confused cyclic tetramer (6g) in 5% yield, hexaphyrinogen (7g) in 13% yield and octaphyrinogen (8g) in 11% yield. Increasing the concentration upto 81 mM gave hexaphyrinogen (7g) and octaphyrinogen (8g) as the major product. The formation of hexaphyrinogen (7g) involves the condensation of diol (3g) with two molecules of 4g, in which one of the 4g undergo pyrrolic group cleavage under acidic condition and followed by subsequent cyclization gave hexamer.16 Further the nature of other acid catalysts were investigated in two different concentrations 3.2 mM and 16 mM. In 3.2 mM concentration, the above reaction in halogenated acid such as TFA and in nonhalogenated acid such as methane sulfonic acid, exclusively gave 5g. While the same reaction in 16 mM concentration, 5g, 6g, 7g and 8g were obtained in 19%, 3%, 16%, 10% yields respectively in TFA, whereas with methane sulfonic acid 5g and 8g were obtained as the major products (Table 1). These results imply that acid-catalysed cleavage of pyrryl group in the reaction increases on increasing the concentration of reactant and the template effect of trihalogenated acid (TFA and BF3–etharate) assists the formation of hexaphyrinogen. The template effect of trihalogenated acid in the cyclization reactions, have been previously reported with dipyrromethane and acetone.17 Similar concentration studies were carried out with selenophene diol (3h) and dipyrrane (4h) to get 5h, 6h, 7h and 8h. The yield of these reactions did not vary much in comparison to tellurophene condensation reactions as reported above.
 | ||
| Fig. 1 Structure of N-confused and expanded porphyrinogens. | ||
| Entry | Conc. of reactant (mM) | Catalyst | 5gb | 6gb | 7gb | 8gb |
|---|---|---|---|---|---|---|
| a Reaction condition: 3 g (0.5 mmol), 4 g (0.5 mmol) and acid catalyst (0.092 mmol) at 0 °C for 30 min.b Isolated yields in percentage (%). | ||||||
| 1 | 3.2 | BF3·OEt2 | 41 | 0 | 0 | 0 |
| 2 | 5.3 | BF3·OEt2 | 42 | 0 | 0 | 3 |
| 3 | 16 | BF3·OEt2 | 23 | 5 | 13 | 11 |
| 4 | 40 | BF3·OEt2 | 15 | 7 | 19 | 17 |
| 5 | 81 | BF3·OEt2 | 10 | 10 | 26 | 21 |
| 6 | 3.2 | TFA | 42 | 0 | Trace | 0 |
| 7 | 16 | TFA | 19 | 3 | 16 | 10 |
| 8 | 3.2 | MeSO3H | 43 | 6 | 0 | 0 |
| 9 | 16 | MeSO3H | 41 | 8 | 3 | 16 |
The structures of all the products were characterized by IR, 1H NMR, 13C NMR, CHN analysis and mass spectrometry (ESI†). In the 1H NMR spectra of 5a, 5c and 5g in CDCl3, tellurophene protons are more deshielded and appeared at δ 7.24–7.14 ppm compared to the selenophene protons (δ 6.84–6.82 ppm) of 5b, 5d and 5h due to better acceptor ability of σ*(Te–carbon) bond of tellurium than selenium.11a In the 125Te NMR (ESI†), chemical shift values of porphyrinogens 5a, 5c and 5g were appeared as singlet at δ 758.91, 755.08 and 754.88 ppm respectively. These values are more similar to the 125Te chemical shift for tellurophene (δ 775.1 ppm).18 While in case of 77Se NMR of porphyrinogen 5b, 5d and 5h, it showed upfield as compared to corresponding tellurophene porphyrinogens and exhibited 77Se NMR signal at δ 601.02, 596.16 and 593.75 ppm respectively. Suitable crystal of 5g for single-crystal X-ray diffraction was obtained as shown in Fig. 2.19 The analysis of the above result revealed that 5g adopt an 1,3-alternate conformation in the solid state. Porphyrinogen 5g has a Te⋯Te distance of 4.61 °A and the two Te atoms occupy the same side of core and no flipped tellurophene was found in the crystal.12c
 | ||
| Fig. 2 X-ray crystal structure of 5g (ORTEP plot). | ||
The electronic absorption spectroscopic titration of 5,10,15,20-meso-octamethyl-21,23-ditelluraporphyrinogen (5g) with “soft” Lewis acid, mercuric perchlorate Hg(ClO4)2 were performed in dry acetonitrile at room temperature. Porphyrinogen 5g gives a characteristic absorption maxima at 284 nm, on the addition of Hg2+ in to acetonitrile solution of 5g (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex), the absorption peak was bathochromically shifted to 296 nm with the increase in relative intensity (Fig. 3). The binding constant Ka of 5g–Hg2+ complex was found to be 1.45 × 105 M−1. The method of continuous variation (Job's plot) was also used to prove the 1:1 stoichiometry (ESI†). A comparative study for the binding of Hg2+ cation with porphyrinogen 5g and 5,10,15,20-meso-octamethyl-21,23-dithiaporphyrinogen (N2S2) was performed and it was observed that porphyrinogen 5g showed more bathochromic shift (12 nm) than 5,10,15,20-meso-octamethyl-21,23-dithiaporphyrinogen (3 nm). The electronic absorption spectroscopic titration of Hg2+ with 7g and 8g were also performed and their respective preliminary peaks were shifted by 10–12 nm. UV-Vis spectra of 7g and 8g with Hg2+ are given in ESI.†
:1 complex), the absorption peak was bathochromically shifted to 296 nm with the increase in relative intensity (Fig. 3). The binding constant Ka of 5g–Hg2+ complex was found to be 1.45 × 105 M−1. The method of continuous variation (Job's plot) was also used to prove the 1:1 stoichiometry (ESI†). A comparative study for the binding of Hg2+ cation with porphyrinogen 5g and 5,10,15,20-meso-octamethyl-21,23-dithiaporphyrinogen (N2S2) was performed and it was observed that porphyrinogen 5g showed more bathochromic shift (12 nm) than 5,10,15,20-meso-octamethyl-21,23-dithiaporphyrinogen (3 nm). The electronic absorption spectroscopic titration of Hg2+ with 7g and 8g were also performed and their respective preliminary peaks were shifted by 10–12 nm. UV-Vis spectra of 7g and 8g with Hg2+ are given in ESI.†
 | ||
| Fig. 3 The absorption spectra of 5g (4.2 × 10−6) in acetonitrile upon the addition of 20 μL stock solution (4.2 × 10−6) of mercuric perchlorate. | ||
The 1H NMR data of Hg2+ with tellurium and selenium porphyrinogen at 298 K are reported in Table 2. A significant downfield chemical shift change was observed for tellurophene and selenophene protons of 5g and 5h, which suggests that the interaction takes place through the tellurium and selenium donor atom of the tellurophene and selenophene rings. Furthermore, the involvement of tellurium of tellurophene and selenium of selenophene in binding with Hg2+ ion, was confirmed by 125Te and 77Se NMR. The 5g exhibited a signal at 754.88 ppm in 125Te NMR which was significantly downfield shifted by 1.21 ppm and appeared at 756.09 ppm (ESI†). Similarly selenium of porphyrinogen 5h in the 77Se NMR showed downfield shift by 0.71 ppm in the presence of Hg2+. The calix[4]pyrrole was also investigated for its interaction with the Hg2+ cation at 298 K and no significant chemical shift change was observed for the NH of calix[4]pyrrole, suggesting the absence of complexation between pyrrolic ligand and Hg2+ cation. It indicates the incapability of the NH groups of the pyrrolic units to bind with the Hg2+ cation.
| Porphyrinogen | 1H | 125Te | 77Se | |||
|---|---|---|---|---|---|---|
| HMethyl | HPyrrole | HTelluro/Seleno | HNH | |||
| 5g | 1.63 | 5.81 | 7.24 | 7.26 | 754.88 | — |
| 5g + Hg2+ | −0.03 | +0.11 | +0.31 | +0.11 | +1.21 | — |
| 5h | 1.61 | 5.85 | 6.83 | 7.19 | — | 593.75 |
| 5h + Hg2+ | −0.02 | +0.09 | +0.24 | +0.09 | — | +0.71 |
| Calix[4]pyrrole | 1.51 | 5.91 | — | 7.06 | — | — |
| Calix[4]pyrrole + Hg2+ | −0.04 | +0.02 | — | +0.06 | — | — |
Conclusion
In summary, the selenium and tellurium N2Te2, N2Se2, N3Te and N3Se meso-unsubstituted core-modified porphyrinogens and N2Te2, N2Se2, N2TeSe meso-substituted core-modified porphyrinogens have been synthesized for the first time. The meso-unsubstituted porphyrinogens are oxidized by chloranil to corresponding porphyrins and porphines which are easily reduced to chlorin and bacteriochlorin. The meso-substituted porphyrinogens are used for the complexation of Hg2+ ion. The reaction approach using readily available conjugated diynes as starting materials make this strategy highly attractive in diversity oriented synthesis.Experimental section
General
Melting points were determined on a capillary melting point apparatus and are uncorrected. The 1H NMR and 13C NMR spectra were recorded on Jeol (400 MHz, 75 MHz) spectrometers at room temperature using TMS as an internal standard. The chemical shifts (δ ppm) are referenced to the respective solvents and splitting patterns are designed as s (singlet), d (doublet), t (triplet), m (multiplet), dt (double triplet), br (broad) and brs (broad singlet). The 77Se NMR and 125Te NMR spectra were recorded on a DPX-300 Bruker spectrometer at 57.24 and 94.69 MHz and the chemical shifts were reported relative to Me277Se and Me2125Te. The mass spectra of selected compounds were recorded on Agilent tech. high resolution Q-TOF mass spectrometry. The column chromatography was carried out using silica gel (100–200 mesh). Solvents used were of analytical grade and were dried before use. Pyrrole was distilled before use. All other chemicals were purchased in reagent quality and were used as received. All processes and reactions were carried out under argon and protected from light. 2,5-Bis(hydroxymethyl)pyrrole and 2,5-bis(2-pyrrolylmethyl)pyrrole were prepared according to the literature method.2a:1, 150 mL). The organic layer was washed with water (2 × 20 mL), dried over Na2SO4 and evaporated under reduced pressure to give (3a–h) as a off white solid (86–91%).:30 hexanes–EtOAc) to give (89–94%) of (4a–h) as a light brown solid.Synthesis of porphyrinogens (5a–i)
:1) to afford two products, (5g) and (6g). N-confused cyclic tetramer (6g): white solid 7% yield, mp 229 °C (decompose); Rf: 0.81 (hexane–CH2Cl2 1:1); 1H NMR (CDCl3, 400 MHz): δ = 7.69 (br, 1H, NH), 7.49 (br, 1H, NH), 7.20–7.16 (m, 4H, tellurophene), 6.37 (s, 1H, pyrrol), 5.78 (m, 2H, pyrrol), 5.61 (s, 1H, pyrrol), 1.63–1.61 (m, 24H, CH3); 13C NMR (CDCl3, 75 MHz): δ = 163.6, 161.9, 141.9, 139.6, 131.8, 130.0, 103.0, 101.0, 42.2, 32.5, 30.8, 29.6; 125Te NMR (CDCl3): δ = 753.11 (s); IR (KBr) νmax 3436, 3331 2959, 2928, 2854, 1638, 1490, 1449, 1420, 1254, 1221, 1121, 1038, 996, 780, 760, 714, 701, 583, 521; HR Q-TOF MS, m/z 659.114 (M + 1) (calcd for C28H34N2Te2 658.0846); analysis: calcd for C28H34N2Te2 C, 51.44; H, 5.24; N, 4.28; found: C, 51.41; H, 5.19; N, 4.23. Hexamer (7g): white solid 15% yield, mp 231 °C (decompose); Rf: 0.65 (hexane–CH2Cl2 1:1); 1H NMR (CDCl3, 400 MHz): δ = 7.69 (br, NH), 6.95 (s, 6H, tellurophene), 5.94–5.93 (d, J = 2.24 Hz, 6H, pyrrol), 1.59 (s, 36H, CH3); 13C NMR (CDCl3, 75 MHz): δ = 162.8, 141.5, 129.4, 99.8, 42.8, 31.5; 125Te NMR (CDCl3): δ = 754.65 (s); IR (KBr) νmax 3436, 2954, 2914, 2878, 1649, 1485, 1428, 1421, 1230, 1228, 1104, 1025; HR Q-TOF MS, m/z 987.11 (M+) (calcd for C42H51N3Te3 987.12); analysis: calcd for C42H51N3Te3 C, 51.44; H, 5.24; N, 4.28; found: C, 51.33; H, 5.19; N, 4.52%. Octamer (8g): white solid 8% yield, mp 223 °C (decompose); Rf: 0.54 (hexane–CH2Cl2 1:1); 1H NMR (CDCl3, 400 MHz): δ = 7.64 (br, NH), 6.98 (s, 8H, tellurophene), 5.88 (d, J = 2.9, 8H, pyrrol), 1.57 (s, 48H, CH3); 13C NMR (CDCl3, 75 MHz): δ = 163.0, 142.4, 129.6, 101.0, 42.5, 30.1; 125Te NMR (CDCl3): δ = 754.11 (s); IR (KBr) νmax 3435, 2944, 2985, 2756, 1652, 1478, 1436, 1391, 1241, 1254, 1110, 995, 780; HR Q-TOF MS, m/z 1316.32 (M+) (calcd for C56H68N4Te4 1316.169); analysis: calcd for C56H68N4Te4 C, 51.44; H, 5.24; N, 4.28; found: C, 51.61; H, 5.29; N, 4.46%.:1) and heated at 100 °C for 5 min. After cooling, the organic phase was separated and washed twice with aqueous HCl (2 M), twice with water then saturated aqueous sodium hydrogen carbonate solution. Organic layer was concentrated under vacuum. The crude product was chromatograph on silica gel eluted with a mixture of ethylacetate–hexane (0.1:9). Meso-tetramethyldiselenachlorin: Rf: 0.25 (ETOAc–hexane 0.1:9); brown-orange solid (5%), mp 48 °C (decompose); 1H NMR (CDCl3, 400 MHz): δ = 9.64–9.63 (d, J = 4.8, 2H, selenophene), 9.27–9.26 (d, J = 4.8, 2H, selenophene), 9.07 (s, 2H, pyrrol), 4.61–4.58 (t, 2H, reduced pyrrol), 4.40 (s, 6H, CH3), 4.31–4.27 (t, 2H, reduced pyrrol), 4.01 (s, 6H, CH3); 13C NMR (CDCl3, 75 MHz): δ = 181.8, 180.7, 161.2, 158.5, 152.4, 143.5, 135.9, 134.7, 127.6, 73.1, 31.0, 29.5; HR Q-TOF MS, m/z 499.015 (M + 1) (calcd for C24H22N2Se2 498.011). Meso-tetramethyldiselenabacteriochlorin: Rf: 0.20 (ETOAc–hexane 0.1:9); brown solid (2%), mp 46 °C (decompose); 1H NMR (CDCl3, 400 MHz): δ = 8.92 (s, 4H, selenophene), 4.20 (s, 8H, reduced pyrrol), 3.90 (s, 12H, CH3); 13C NMR (CDCl3, 75 MHz): δ = 179.6, 155.7, 143.5128.7, 69.2, 28.1; HR Q-TOF MS, m/z 501.045 (M + 1) (calcd for C24H24N2Se2 500.027).![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; unit cell dimensions a = 10.361(2) Å, b = 11.5721(3) Å, c = 11.8258(15) Å, alpha = 101.396(15)°, beta = 90.972(13)°, gamma = 104.51(2)°; Z = 2; calculated density = 1.613 mg m−3; goodness-of-fit on F2 = 0.656; total of 13052 reflections were measured, 6096 unique (Rint = 0.0748); final R indices R = 0.0713, Rw = 0.1865; F(000) = 636; the structure was refined on full-matrix least-squares on F2. Complete crystallographic data are given in ESI.†
; unit cell dimensions a = 10.361(2) Å, b = 11.5721(3) Å, c = 11.8258(15) Å, alpha = 101.396(15)°, beta = 90.972(13)°, gamma = 104.51(2)°; Z = 2; calculated density = 1.613 mg m−3; goodness-of-fit on F2 = 0.656; total of 13052 reflections were measured, 6096 unique (Rint = 0.0748); final R indices R = 0.0713, Rw = 0.1865; F(000) = 636; the structure was refined on full-matrix least-squares on F2. Complete crystallographic data are given in ESI.†Acknowledgements
We thank the Department of Science and Technology and University of Delhi, Delhi for financial assistance. S. Ahmad and K. K. Yadav are thankful to UGC, New Delhi for senior research fellowship and junior research fellowship. S. J. Singh is thankful to UGC, New Delhi for Dr D. S. Kothari Post-doctoral fellowship.Notes and references
- (a) S. K. Kim, D. E. Gross, D. G. Cho, V. M. Lynch and J. L. Sessler, J. Org. Chem., 2011, 76, 1005 CrossRef CAS PubMed; (b) J. Yoo, M. S. Kim, S. J. Hong, J. L. Sessler and C. H. Lee, J. Org. Chem., 2009, 74, 1065 CrossRef CAS PubMed; (c) J. L. Sessler, W. S. Cho, D. E. Gross, J. A. Shriver, V. M. Lynch and M. Marquez, J. Org. Chem., 2005, 70, 5982 CrossRef CAS PubMed; (d) B. Garg, T. Bisht and S. M. S. Chauhan, New J. Chem., 2010, 34, 1251 RSC; (e) B. Garg, T. Bisht and S. M. S. Chauhan, J. Inclusion Phenom. Macrocyclic Chem., 2011, 70, 249 CrossRef CAS; (f) D. S. Kim, V. M. Lynch, K. A. Nielsen, C. Johnsen, J. O. Jeppesen and J. L. Sessler, Anal. Bioanal. Chem., 2009, 395, 393 CrossRef CAS PubMed; (g) T. Guchhait, V. K. Jha and G. Mani, Org. Biomol. Chem., 2013, 11, 2818 RSC.
- (a) S. Taniguchi, H. Hasegawa, S. Yanagiya, Y. Tabeta, Y. Nakano and M. Takahashi, Tetrahedron, 2001, 57, 2103 CrossRef CAS; (b) Y. Matano, T. Shibano, H. Nakano and H. Imahori, Chem.–Eur. J., 2012, 18, 6208 CrossRef CAS PubMed.
- (a) N. Agarwal, C. H. Hung and M. Ravikanth, Eur. J. Org. Chem., 2003, 3730 CrossRef CAS; (b) I. Gupta and M. Ravikanth, Coord. Chem. Rev., 2006, 250, 468 CrossRef CAS PubMed.
- (a) H. Uno, T. Inoue, Y. Fumoto, M. Shiro and N. Ono, J. Am. Chem. Soc., 2000, 122, 6773 CrossRef CAS; (b) H. Uno, K. Inoue, T. Inoue and N. Ono, Org. Biomol. Chem., 2003, 1, 3857 RSC.
- (a) S. Neya, J. Quan, M. Hata, T. Hoshino and N. Funasaki, Tetrahedron Lett., 2006, 47, 8731 CrossRef CAS PubMed; (b) T. Okujima, T. Kikkawa, H. Nakano, H. Kubota, N. Fukugami, N. Ono, H. Yamada and H. Uno, Chem.–Eur. J., 2012, 18, 12854 CrossRef CAS PubMed; (c) E. R. Ranyuk, M. A. Filatov, A. D. Averin, A. V. Cheprakov and I. P. Beletskaya, Synthesis, 2012, 393 CAS; (d) M. A. Filatov and A. V. Cheprakov, Tetrahedron, 2011, 67, 3559 CrossRef CAS PubMed; (e) M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchenkov and A. V. Cheprakov, J. Org. Chem., 2012, 77, 11119 CrossRef CAS PubMed; (f) H. W. Whitlock, R. Hanauer, M. Y. Oester and B. K. Bower, J. Am. Chem. Soc., 1969, 91, 7485 CrossRef CAS; (g) M. Varamo, B. Loock, P. Maillard and D. S. Grierson, Org. Lett., 2007, 9, 4689 CrossRef CAS PubMed; (h) Q. Ouyang, K. Q. Yan, Y. Z. Zhu, C. H. Zhang, J. Z. Liu, C. Chen and J. Y. Zheng, Org. Lett., 2012, 14, 2746 CrossRef CAS PubMed; (i) L. P. Samankumara, M. Zeller, J. A. Krause and C. Bruckner, Org. Biomol. Chem., 2010, 8, 1951 RSC.
- (a) N. Arumugam, Y. S. Jang and C. H. Lee, Org. Lett., 2000, 2, 3115 CrossRef CAS PubMed; (b) J. L. Sessler, D. An, W. S. Cho, V. Lynch, D. W. Yoon, S. J. Hong and C. H. Lee, J. Org. Chem., 2005, 70, 1511 CrossRef CAS PubMed; (c) S. K. Kim, V. M. Lynch, N. J. Young, B. P. Hay, C. H. Lee, J. S. Kim, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 20837 CrossRef CAS PubMed.
- (a) E. C. Lee, Y. K. Park, J. H. Kim, H. Hwang, Y. R. Kim and C. H. Lee, Tetrahedron Lett., 2000, 43, 9493 CrossRef; (b) J. L. Sessler, D. An, W. S. Cho and V. Lynch, J. Am. Chem. Soc., 2003, 125, 13646 CrossRef CAS PubMed.
- (a) V. Kral, P. A. Gale, P. Anzenbacher Jr, K. Jursikova, V. Lynch and J. L. Sessler, Chem. Commun., 1998, 9 RSC; (b) J. L. Sessler, W. S. Cho, V. Lynch and V. Kral, Chem.–Eur. J., 2002, 8, 1134 CrossRef CAS.
- (a) Y. Matano, T. Miyajima, N. Ochi, T. Nakabuchi, M. Shiro, Y. Nakao, S. Sakaki and H. Imahori, J. Am. Chem. Soc., 2008, 130, 990 CrossRef CAS PubMed; (b) Y. Matano, T. Nakabuchi, T. Miyajima and H. Imahori, Organometallics, 2006, 25, 3105 CrossRef CAS; (c) T. Nakabuchi, Y. Matano and H. Imahori, Org. Lett., 2010, 12, 1112 CrossRef CAS PubMed; (d) Y. Matano, K. Matsumoto, H. Hayashi, Y. Nakao, T. Kumpulainen, V. Chukharev, N. V. Tkachenko, H. Lemmetyinen, S. Shimizu, N. Kobayashi, D. Sakamaki, A. Ito, K. Tanaka and H. Imahori, J. Am. Chem. Soc., 2012, 134, 1825 CrossRef CAS PubMed.
- (a) A. F. D. D. Namor and I. Abbas, J. Phys. Chem. B, 2007, 111, 5803 CrossRef PubMed; (b) A. F. D. D. Namor, I. Abbas and H. H. Hammud, J. Phys. Chem. B, 2006, 110, 2142 CrossRef PubMed.
- (a) A. Panda, S. Panda, K. Srivastava and H. B. Singh, Inorg. Chim. Acta, 2011, 372, 17 CrossRef CAS PubMed; (b) T. Chakrabortya, K. Srivastava, S. Pandaa, H. B. Singha and R. J. Butcher, Inorg. Chim. Acta, 2010, 363, 2905 CrossRef PubMed; (c) P. Singh and A. K. Singh, Inorg. Chim. Acta, 2012, 387, 441 CrossRef CAS PubMed; (d) P. Singh and A. K. Singh, Organometallics, 2010, 29, 6433 CrossRef CAS.
- (a) M. Abe, M. R. Detty, O. O. Gerlits and D. K. Sukumaran, Organometallics, 2004, 23, 4513 CrossRef CAS; (b) E. Pacholska, L. L. Grazynski and Z. Ciunik, Angew. Chem., Int. Ed., 2001, 40, 4466 CrossRef CAS; (c) B. Sathyamoorthy, A. Axelrod, V. Farwell, S. M. Bennett, B. D. Calitree, J. B. Benedict, D. K. Sukumaran and M. R. Detty, Organometallics, 2010, 29, 3431 CrossRef CAS; (d) M. Abe, D. G. Hilmey, C. E. Stilts, D. K. Sukumaran and M. R. Detty, Organometallics, 2002, 21, 2986 CrossRef CAS.
- W. Yin, C. He, M. Chen, H. Zhang and A. Lei, Org. Lett., 2009, 11, 709 CrossRef CAS PubMed.
- T. M. McCormick, A. A. Jahnke, A. J. Lough and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 3542 CrossRef CAS PubMed.
- B. M. Smith, S. D. Kean, M. F. Wyatt and A. E. Graham, Synlett, 2008, 1953 CAS.
- (a) Y. S. Jang, H. J. Kim, P. H. Lee and C. H. Lee, Tetrahedron Lett., 2000, 41, 2919 CrossRef CAS; (b) A. Nagarajan, J. W. Ka and C. H. Lee, Tetrahedron, 2001, 57, 7323 CrossRef CAS; (c) V. Kral, J. L. Sessler, R. S. Zimmerman, D. Seidel, V. Lynch and B. Andrioletti, Angew. Chem., Int. Ed., 2000, 39, 1055 CrossRef CAS.
- B. Turner, A. Shterenberg, M. Kapon, K. Suwinska and Y. Eichen, Chem. Commun., 2002, 404 RSC.
- T. B. Schroeder, C. Job, M. F. Brown, R. S. Glass, N. You and E. Block, Magn. Reson. Chem., 1997, 35, 752 CrossRef CAS.
- ESI.†.
- (a) M. J. Dabdoub, V. B. Dabdoub, P. G. Guerrero and C. C. Silveira, Tetrahedron, 1997, 53, 4199 CrossRef CAS; (b) A. Ulman, J. Manassen, F. Frolow and D. Rabinovich, Tetrahedron Lett., 1978, 19, 167 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds. 125Te and 77Se NMR spectra of all porphyrinogens. UV-Vis spectra of porphyrinogens in the presence of Hg2+. 125Te, 77Se NMR of 5g and 5h in the presence of Hg2+. CCDC 932049. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra46491a |
| This journal is © The Royal Society of Chemistry 2014 |