
The preparation of hierarchical tubular structures comprised of NiO nanosheets with enhanced supercapacitive performance
Abstract
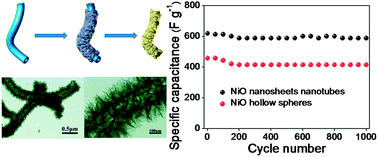
Hierarchically tubular structures comprised NiO nanosheets were successfully prepared through a mild solution route based on the template of polymeric nanotubes (PNT) followed by a thermal annealing treatment. The microstructure and chemical composition of NiO nanosheets nanotubes are investigated by SEM, TEM, HRTEM, SAED and XRD. The Brunauer–Emmett–Teller (BET) specific surface area of this sample is calculated to be 98 m2 g−1 and the majority of pores have a size in the range of 2–10 nm. The thermal behavior of Ni-precursor@PNT was studied by TGA and the weight fraction of NiO nanosheets nanotubes obtained by calcination is measured to be 57.0%. The specific capacitance of the unique NiO nanosheets nanotubes is 588 F g−1 at the end of 1000 cycles when the charge–discharge current density is 3 A g−1, leading to only 5.2% capacity loss. In addition, the NiO nanotubes coating by relatively sparse and thin nanosheets possess better electrochemical properties. The specific capacitance is 960 F g−1 at the end of 1000 cycles when the charge–discharge current density is 10 A g−1, leading to only 1.2% capacity loss. Broadly, the as-obtained NiO nanosheets nanotubes reveal relatively high capacitance and remarkable cycling stability in virtue of the hollow, porous, flaky and tubular nanostructures.
 Please wait while we load your content...
Please wait while we load your content...

