Design and synthesis of ERα/ERβ selective coumarin and chromene derivatives as potential anti-breast cancer and anti-osteoporotic agents†
M. Kamil Hussaina,
M. Imran Ansaria,
N. Yadava,
Puneet K. Guptaa,
A. K. Guptaa,
R. Saxenab,
I. Fatimab,
M. Manoharb,
P. Kushwahab,
V. Khedgikarb,
J. Gautamb,
Ruchir Kantc,
P. R. Maulikc,
R. Trivedib,
A. Dwivedib,
K. Ravi Kumard,
A. K. Saxenaa and
K. Hajela*a
aMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram Extension, Sitapur Road, Lucknow-226031, India. E-mail: kanchan_hajela @cdri.res.in; Tel: +91-522-261241-18, PABX, 4679/4680
bEndocrinology Division, CSIR-Central Drug Research Institute, Lucknow-226031, India
cMolecular and Structural Biology Division, CSIR-Central Drug Research Institute, Lucknow-226031, India
dX-ray Crystallographic Division, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, India
First published on 27th November 2013
Abstract
Several new coumarin and chromene prototype derivatives have been synthesised and evaluated for their ERα and ERβ selective activity. Coumarin prototype compounds 18 & 19 were found to be ERα selective and the most active, exhibiting potential antiproliferative activity against both ER +ve & ER −ve breast cancer cell lines. The surprise finding of the series, however, are the novel prototype III chromenes 45 & 46, with aroyl substitution at the 6th position. Both the compounds have shown potent antiproliferative activity against both the breast cancer cell lines, promote alkaline phosphatase activity, enhance osteoblast mineralization in vitro, significantly decrease ERE–ERα dependent transactivation and induce ERβ activity. This specific upregulation of ERβ isoform activity of compound 45 may be responsible for the antiosteoporotic activity at picomolar concentration. In addition, both the compounds were also devoid of any estrogenic activity, which correlates to their antiestrogenic behaviour in the two breast cancer cell lines. Assessment of selectivity using specific SiRNAs for ERα and ERβ revealed that most of the compounds showed ERα and ERβ-mediated action, except compound 28, which showed selectivity to ERα only. Computational docking analysis of active compounds 18 and 45 was conducted to correlate the interaction with the two receptors and it was found that the docked conformations of the coumarin prototype, compound 18 at ERα and ERβ active sites were more or less superimposable on each other. However, the unique orientation of the aminoalkoxy side chain of novel chromene (prototype III) compound 45 in the ERβ binding cavity may be responsible for its potential biological response.
1. Introduction
Mammary carcinoma is the most common malignancy in women and the leading cause of cancer death among females, accounting for 23% of the total cancer cases and 14% of all cancer deaths. An estimated 1.7 million women will be diagnosed with breast cancer in 2020, which is a 26% increase from current levels.1,2 The steroid hormone estrogen mediates a number of biological processes, ranging from reproductive health to bone maintenance, through estrogen receptors (ER), a member of a large superfamily of nuclear receptors (NR).3 The same estrogen is also predominantly involved in the initiation and proliferation of ER +ve breast cancer and much effort is now being devoted to block estrogen formation and action. The ER exists in two isoforms, α (ERα) and β (ERβ), both of which are ligand-induced transcription factors that have different distributions in various estrogen target tissues and also have different functions, some of which have not yet been clarified. ERα and ERβ share modest overall sequence identity (47%), where the DNA and ligand-binding domains are highly conserved with only minor structural differences, but there is little or no detectable homology between their N-terminal transactivation (AF-1) domains.4–6 ERα, the predominant subtype expressed in breast cancer, induces proliferation in response to estrogen, while ERβ, the predominant subtype present in bone, colon and prostrate, inhibits proliferation of breast cancer cells by antagonizing the function of ERα and also checks the development of osteoporosis. Thus ERα and ERβ are potential targets for the treatment of breast and endometrial cancers. Mapping the distribution of ERα and ERβ mRNA in normal and neoplastic tissues has provided an intriguing picture of differential expression patterns in different tissue types. This existence of clear-cut differences in the two subtype receptor expressions suggests that tissues could be differentially targeted with receptor selective ligands.7–11 The most widely used strategy to disrupt estrogen mediated breast cancer proliferation is through targeted antagonism of estrogen receptors in the breast tissues by antiestrogens or SERMs, a new class of tissue selective therapeutic agents called “designer molecules” with specific interactions in the target cells leading to tissue selective action.12–15Coumarins, a class of naturally occurring benzopyrone derivatives, comprise a vast array of biologically active compounds found in a variety of plant sources. They display interesting pharmacological properties, which have encouraged medicinal chemists for decades to explore the natural coumarins or their synthetic analogues for their applicability as drugs.16a,b The coumarins are extremely variable in structure due to the presence of different types of substitutions in their basic structure, which can influence their biological, pharmacological and therapeutic applications.17,18 Hydroxycoumarins act as potent metal chelators and free radical scavengers and the active metabolite 7-hydroxycoumarin derivatives have shown sulfatase and aromatase inhibitory activities.19 Coumarin based selective estrogen receptor modulators (SERMs) and coumarin–estrogen conjugates have been described as potential anti-breast cancer agents.19 Recently, coumarin based new generation SERM, SP500263 is reported to be a highly potent antiestrogen currently under clinical investigation.20
With the emergence of drug resistance as a major new impediment in breast cancer treatment, combined with the problems of low tumor selectivity and toxicity, there is an urgent need for the discovery of less toxic and potent new anti-breast cancer drugs, which target the interactive mechanisms involved in growth and metastasis of breast cancer.21,22 The emerging role of ERβ in hormone related cancers has opened the possibility of developing new ERβ inducing agents. Therefore the search for novel pharmacophores eliciting opposing action on ERα or ERβ may provide a rationale for developing a drug that might have bone protective effects in addition to anti-cancer effects. In this paper, we report the structural design and synthesis of some novel coumarin/chromene based molecules of prototypes I, II and III and their biological efficacy for selective ER mediated anti-breast cancer and anti-osteoporotic activities.
2. Results and discussion
2.1. Chemistry
The synthesis of all the target molecules was achieved through efficient synthetic routes. For the synthesis of prototype molecules I, benzils 3 and 5, which formed the key precursors, were prepared as follows. Sonogashira coupling of 4-iodoanisole with commercially available 2-methoxy phenylacetylene conveniently formed the 2,4′-dimethoxy diphenyl acetylene,23 1 which on oxidation with PdI2 in DMSO gave the corresponding 2,4′-dimethoxybenzil 2 in >90% yields.24 Subsequent demethylation under basic conditions using ethanethiol–sodium hydride in dry DMF led to the hydroxy derivative 3. The product was isolated as a yellow solid in 95% yield, mp 162–163 °C.25a,b The synthesis of 2,4′-dihydroxy-4-methoxy benzil 5 was carried out by the acylation of 3-methoxyphenol with 4-hydroxy phenylacetic acid, followed by the oxidation of the resulting deoxybenzoin 4 with selenium dioxide in dioxane–water (Scheme 1).26,27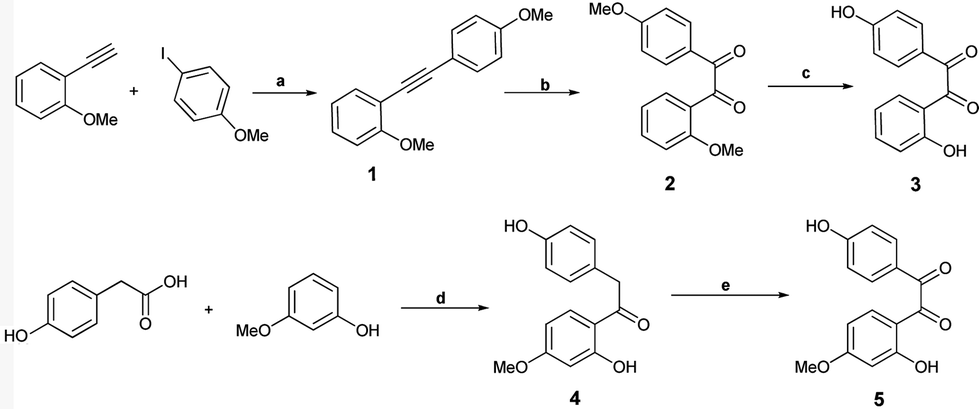 | ||
Scheme 1 Synthesis of 2,4′-dihydroxy and 2,4′-dihydroxy-4-methoxy benzils (compounds 3 and 5). Reagents and reaction conditions (a) Pd(PPh3)4Cl2, CuI, Et3N, N2, RT, 90%. (b) PdI2, DMSO,140 °C, 95%. (c) EtSH–NaH, DMF, 0 °C–120 °C, 95%. (d) BF3.OEt, N2, 85 °C, 90%. (e) SeO2, 1,4-dioxane/H2O; (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 110 °C, 90%. :1), 110 °C, 90%. | ||
For the synthesis of 4-aroylated coumarins, we have developed a simple methodology and report the synthesis of 3-aryl-4-aroyl coumarin, as the desired scaffold, for the first time through condensation of benzils with aryl acetyl chlorides (Scheme 2).
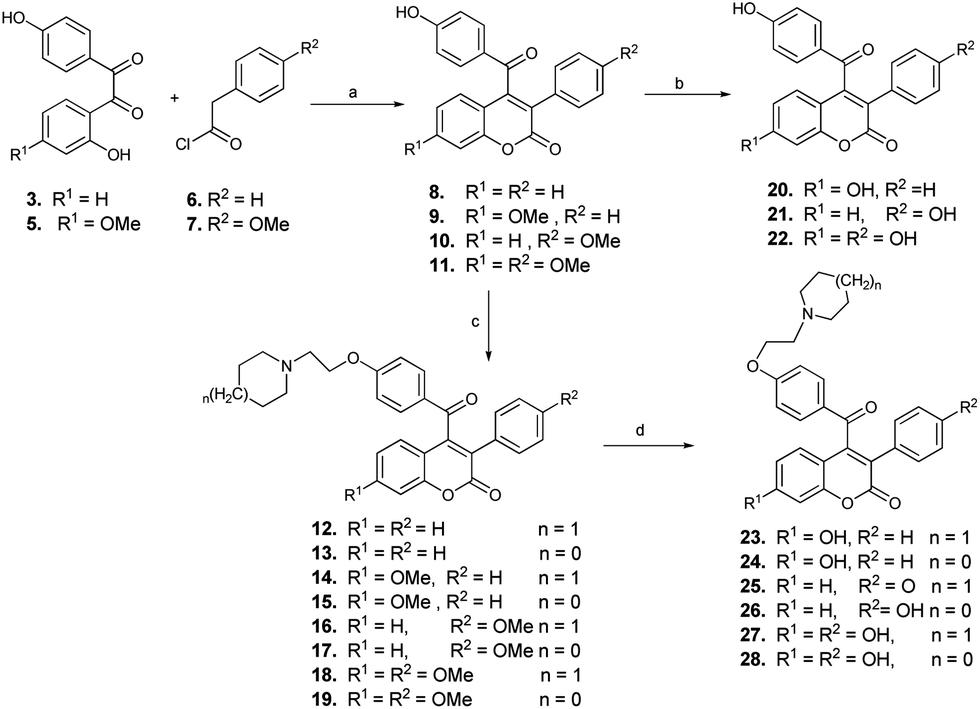 | ||
| Scheme 2 Synthesis of molecules of prototype I (compounds 8–28). Reagents and conditions: (a) acetone, K2CO3, reflux, 4–5 h, 90–95%. (b) EtSH, AlCl3, DCM, 0 °C, 3 h. (c) piperidine/pyrrolidine 1-(2-chloroethyl) monohydrochlorides, acetone, K2CO3, reflux, 3–4 h, 90–95%. (d) EtSH, AlCl3, DCM, 0 °C, 3 h, 90%. | ||
The reaction of benzils 3 and 5 with phenyl or 4-methoxyphenyl acetyl chlorides 6 or 7 under reflux in acetone and anhydrous potassium carbonate afforded coumarins (8–11) in pure form by simple crystallisation and without any chromatographic purification in excellent yields (90–95%). Subsequent base catalysed O-alkylation of the hydroxy group of compounds 8–11 with piperidine/pyrrolidine 1-(2-chloroethyl) monohydrochlorides formed the target compounds, 3-phenyl/4-methoxy-phenyl-7-methoxy-4-[-4-(2-piperidinyl/pyrrolidinyl) ethoxy benzoyl]coumarins (12–19) in good yields.
Since the presence of hydroxy groups at appropriate positions plays a prominent role in the binding of ligands to the biological target, the estrogen receptors, it was thought worthwhile to demethylate the methoxy derivatives to the corresponding hydroxy compounds. A combination of ethanethiol with anhydrous aluminium chloride in dry DCM at 0–10 °C was used for demethylation of (8–11 and 12–19), which conveniently gave the corresponding phenolic analogues (20–22 and 23–28) in high yields.
The chemical methodology, which was followed to synthesize compounds of prototype II, is shown in Scheme 3. The starting precursor, 7-methoxy isoflavanone 30, was easily prepared by base catalyzed condensation of 2-hydroxy-4-methoxy deoxybenzoin 29 with paraformaldehyde.28 The introduction of an aroyl group at the 4-position of the isoflavene by direct aroylation (obtained by reduction and subsequent dehydration of isoflavonone 30) was not successful. Subsequently, it was thought to introduce the aroyl functionality through formation of 4-bromo isoflavene 31. Refluxing of the compound 30 with PBr3 in dry benzene formed the desired 4-bromo isoflavene 31 in excellent yield and was isolated as a white solid on crystallisation from methanol. However, it was found to be unstable (decomposed at rt) and was therefore immediately subjected to anion formation with n-butyl lithium at 0 °C, followed by quenching with 4-methoxy benzonitrile or 4-chloroethoxy benzonitrile to afford the 7-methoxy-4-(4-methoxy/chloroethoxy) benzoylated chromenes 34 and 35 in good yields. Subsequent reaction of compound 35 with piperidine or pyrrolidine in the presence of TBAI in dry DMF formed the aminoalkoxy compounds 36 and 37. Cleavage of the methyl ether linkage of compound 35 using EtSH/anhy. AlCl3 formed the phenolic derivative 38, which on further reaction with piperidine and pyrrolidine in dry DMF–TBAI gave the target phenolic compounds 39 and 40.
 | ||
| Scheme 3 Synthesis of molecules of prototype II (compounds 34–40). Reagents and reaction conditions: (a) (HCHO)2, 25% aq. Me2NH, EtOH, reflux,75%, (b) PBr3, benzene, 80 °C, 85%, (c) 1.6 M n-butyllithium in hexane, dry ether, N2, 0–5 °C, 70–75%, (d) EtSH/AlCl3, DCM, 0 °C, 3 h, 85%, (e) piperidine/pyrrolidine, dry DMF, TBAI 80 °C, 90–95%. | ||
Having successfully achieved the synthesis of prototype compounds I and II, we next directed our efforts to synthesise prototype III molecules, as shown in Scheme 4. The envisaged precursor, 7-methoxy-3-phenyl-coumarin 41, was easily prepared in quantitative yields through the condensation of 2,4-dihydroxy benzaldehyde with phenyl acetic acid, followed by subsequent deacetylation of the 7-hydroxy group and finally its methylation. A gem-dimethyl group was introduced through Grignard reaction with an excess of freshly prepared methylmagnesium iodide at the 2nd position of 41, forming 7-methoxy-2,2-dimethy-3-phenyl-2H-chromene 42 in good yields.29 Again, for the introduction of the aroyl group at the 4-position of chromene 42, the synthetic plan through formation of 4-bromo derivative of isoflavene 42 was not successful.
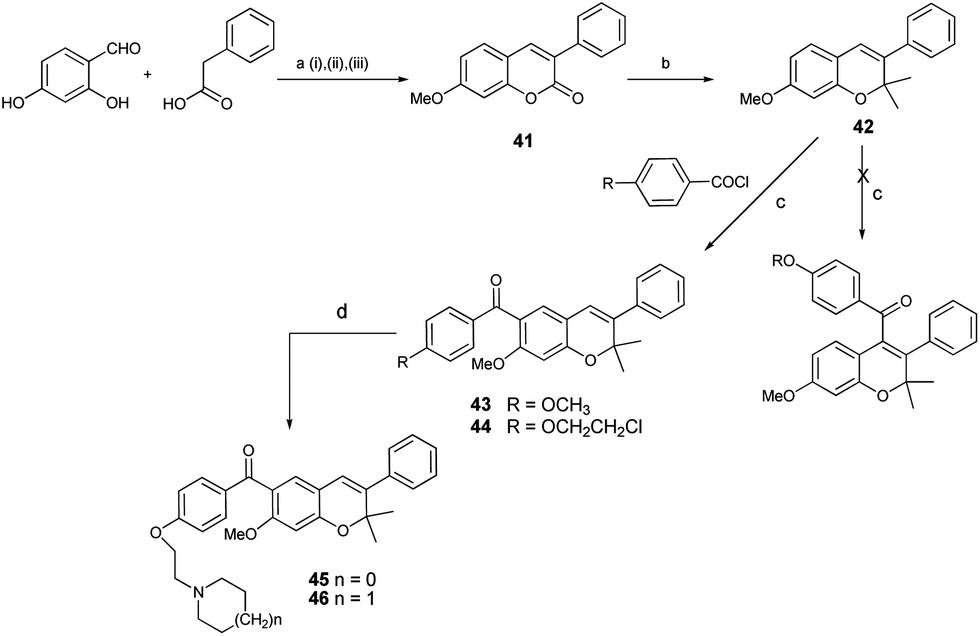 | ||
| Scheme 4 Synthesis of molecules of prototype III (compounds 43, 45 and 46). Reagents and conditions: (a) (i) Ac2O, TEA, reflux, 78%, (ii) MeOH, K2CO3 RT, 90%, (iii), K2CO3, acetone, MeI, reflux, 92%; (b) MeMgI, THF, reflux, conc. HCl, 70%; (c) SnCl4, DCM, 0 °C, 1 h 95%; (d) piperidine or pyrrolidine, dry DMF, TBAI 80 °C, 6 h, 92%. | ||
The next option of direct aroylation of 42 with 4-methoxy benzoyl chloride was carried out using SnCl4 in dry DCM and, surprisingly, the reaction was successful. Spectral analysis of the isolated product by 1H NMR showed three singlets, each integrating for one proton in the aromatic region. This indicated regioselective aroylation, probably occurring at the 6th position of the chromene 42 instead of at the 4th position. The structure was finally confirmed by HMBC spectral analysis. The proton at the C-5 position of the chromene ring showed three bond correlations with signals of the carbon of the carbonyl group as well as the C-7 carbon. On the other hand, the proton at the C-8 position showed a 3-bond correlation with the C-6 carbon signal and the proton at the C-4 position showed a 3-bond correlation with the C-2 carbon signal. These correlations confirmed the presence of an aroyl group at the 6th position, as shown in Fig. 1. It can be speculated that the presence of the methoxy group at the 7th position may be the driving force for the aroylation at the 6th position being more nucleophilic as compared to the 4th position. The 6-aroylated chromenes 43 and 44 were obtained in pure form by simple crystallization in excellent yield. Reaction of compound 44 with pyrrolidine or piperidine, respectively, in the presence of TBAI in dry DMF at 80 °C, formed the corresponding alkylaminoalkoxy derivatives 45 and 46, thus completing the synthesis of prototype III compounds.
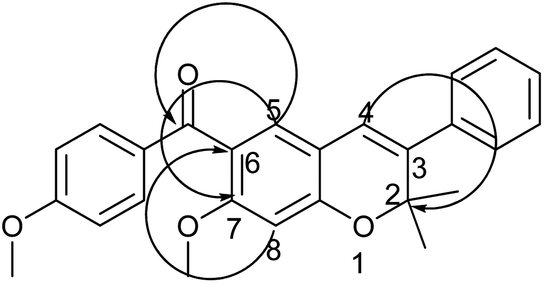 | ||
| Fig. 1 Significant HMBC (H → C) correlation of compound 43. | ||
2.2. Biological evaluation
Of the first prototype I, molecules 8–28, which had a basic coumarin nucleus substituted with variable 3-aryl and 4-aroyl groups, it was found that in general the compounds displayed anti-proliferative activity against the both ER +ve & ER −ve breast cancer cell lines and an almost negligible activity against the Ishikawa and PC-3 cell lines. Three compounds 9, 18 & 19, showed very good activity. Compound 18 was the most active, showing IC50 of 5.7 μM in MCF-7 cells, and 12.7 μM in MDA-MB 231 breast cancer cells, better than tamoxifen and raloxifene, used as a positive control, which showed IC50 of 13.7 in MCF-7 and 17.9 μM in MDA-MB 231 cells. Similarly, compound 19 also showed comparable inhibition of both breast cancer cell lines. In prototype II molecules, where the basic pharmacophore was changed from coumarin to chromene, the activity was considerably decreased in compounds 34, 36, 37, 39 and 40. Only two compounds, 34 and 36, showed growth inhibitions in both the cell lines. However, the introduction of a gem-dimethyl group at the 2nd position of the chromene pharmacophore, coupled with aroyl functionality at the 6th position instead of at the 4th position in the prototype III molecules, resulted in a better activity profile, and all three compounds 43, 45 and 46 showed substantial growth inhibition against both the breast cancer and Ishikawa cell lines. Compared to tamoxifen, compounds 45 and 46 showed potent inhibition in the range 6.8 μM to 8.1 μM against the MCF-7 cell line. The compounds did not show any significant antiproliferative activity against the prostate cancer cell line and only four compounds 17, 19, 23 & 25 were moderately active (Table 1 and Fig. 2a–c).
| IC50 values (μM) | |||||
|---|---|---|---|---|---|
| Compound no. | MCF-7 | MDA-MB 231 | Ishikawa | PC-3 | % RBA |
| 8 | 24.3 | >40 | >40 | >40 | 0.05 |
| 9 | 17.7 | 34.2 | ND | >40 | 0.02 |
| 10 | 27.9 | 37.8 | ND | >40 | 0.026 |
| 11 | >40 | >40 | ND | >40 | 0.05 |
| 12 | >40 | >40 | >40 | >40 | 3 |
| 13 | 33.7 | >40 | >40 | >40 | 0.3 |
| 14 | 29.3 | >40 | ND | >40 | 0.114 |
| 15 | 32.4 | 31.2 | ND | >40 | 0.285 |
| 16 | 19.7 | 20.5 | ND | >40 | 0.8 |
| 17 | 16.3 | 18.8 | ND | 22.8 | 1.3 |
| 18 | 5.7 | 12.7 | >40 | >40 | 0.16 |
| 19 | 12.8 | 18.4 | >40 | 22.5 | 0.1 |
| 20 | 22.1 | 34.3 | ND | >40 | 34 |
| 21 | >40 | >40 | ND | >40 | 0.08 |
| 22 | >40 | >40 | ND | >40 | 6 |
| 23 | >40 | >40 | >40 | 28.6 | 11.42 |
| 24 | >40 | >40 | >40 | >40 | 16 |
| 25 | 31.7 | >40 | ND | 18.8 | 2 |
| 26 | >40 | >40 | ND | >40 | 2.6 |
| 27 | 25.9 | >40 | ND | >40 | 12.3 |
| 28 | 27.1 | >40 | ND | >40 | 13.3 |
| 34 | 20.0 | >40 | ND | >40 | 0.05 |
| 36 | 34.5 | >40 | >40 | >40 | 0.3 |
| 37 | >40 | 17.5 | >40 | >40 | 0.6 |
| 39 | >40 | >40 | ND | >40 | 0.1 |
| 40 | >40 | >40 | ND | >40 | 2.25 |
| 43 | 19.8 | 20.2 | 15 | ND | 0.09 |
| 45 | 6.8 | 16.9 | 17.7 | >40 | 0.225 |
| 46 | 8.1 | 14.8 | 16.9 | >40 | 0.6 |
| Tamoxifen | 13.7 | 17.9 | ND | >40 | 2.25 |
| Raloxifene | 15.4 | 18.5 | ND | >40 | 15 |
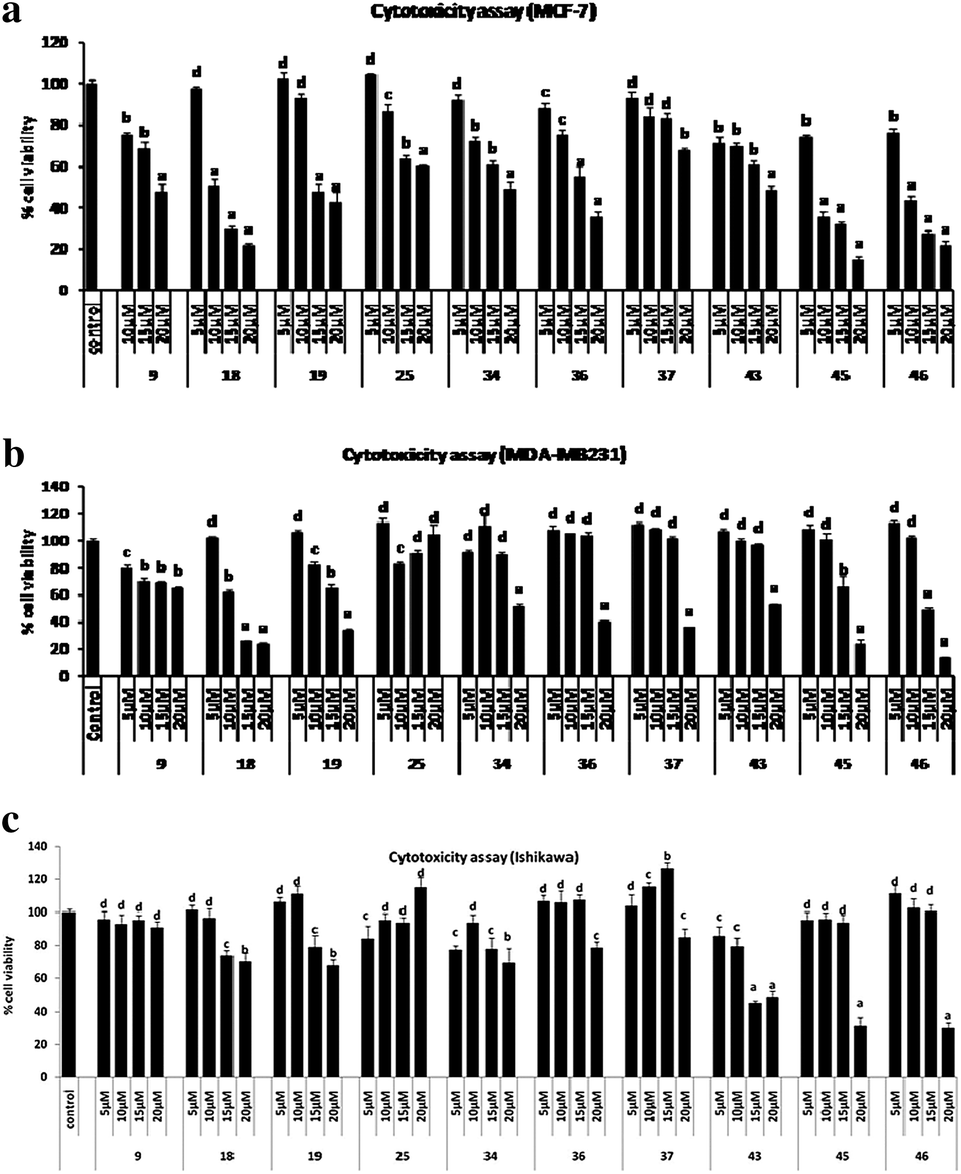 | ||
| Fig. 2 (a) MTT cell proliferation assay of some active compounds in MCF-7 cells. Results are expressed as mean ± SE, n = 5. P values are a – p < 0.001, b – p < 0.01, c – p < 0.05, d – p > 0.05 vs. control. (b) MTT cell proliferation assay of some active compounds in MDA-MB 231 cells. Results are expressed as mean ± SE, n = 5. P values are a – p < 0.001, b – p < 0.01, c – p < 0.05, d – p > 0.05 vs. control. (c) MTT cell proliferation assay of some active compounds in Ishikawa cells. Results are as expressed mean ± SE, n = 5. P values are a – p < 0.001, b – p < 0.01, c – p < 0.05, d – p > 0.05 vs. control. | ||
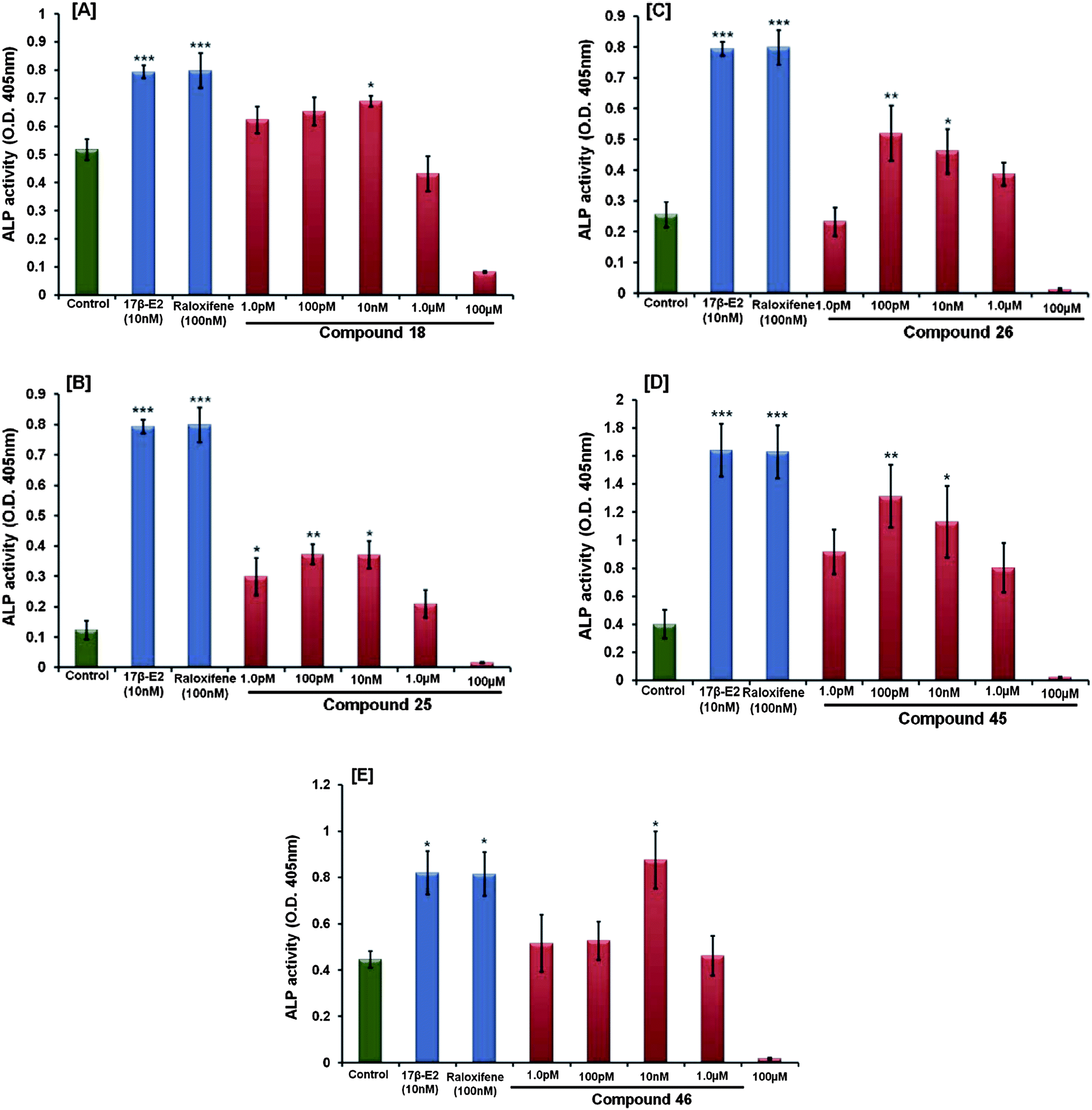 | ||
| Fig. 3 (A–E) The activity of compounds 18, 25, 26, 45 and 46 were assessed by measuring ALP activity in calvarial osteoblasts (for details see text). At the end of the experiment, ALP activity was measured colorimetrically, as described before. The data shows the mean ± SE of three independent experiments *p < 0.05 and **p < 0.01. | ||
17β-Estradiol and raloxifene were used as standard controls, which stimulated ALP activity at 10 nM, 100 nM conc., respectively (Fig. 4). It was observed from the data of this experiment that although the five active compounds have the potential to increase the proliferation and differentiation of osteoblast cells, compared to the positive control only compound 46 showed an increased proliferation comparable to raloxifene. The rest of the four compounds showed no significant change.
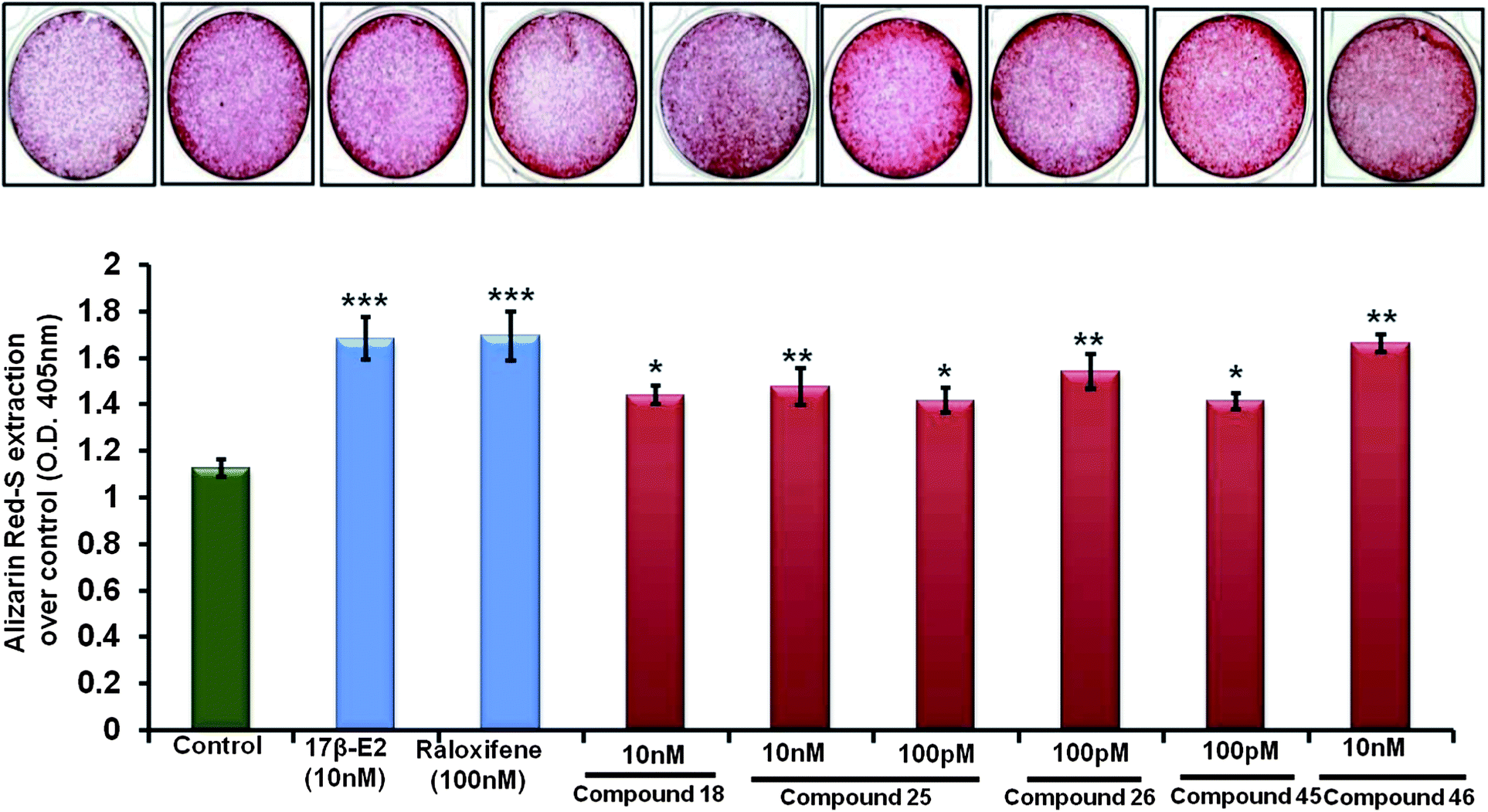 | ||
| Fig. 4 Compounds 18, 25, 26, 45 and 46 increased osteoblast mineralization. Calvarial osteoblasts were grown in the osteoblast differentiation medium as described before (see text for details). At the end of the experiments, cells were stained with alizarin red-S. Photomicrographs show increased formation of mineralized nodules by compound treatment compared to the vehicle treated cells. The bar diagram shows quantification of mineralization by the extraction of alizarin red-S dye. The data represents the mean ± SE of three independent experiments *p < 0.05 and ***p < 0.001. | ||
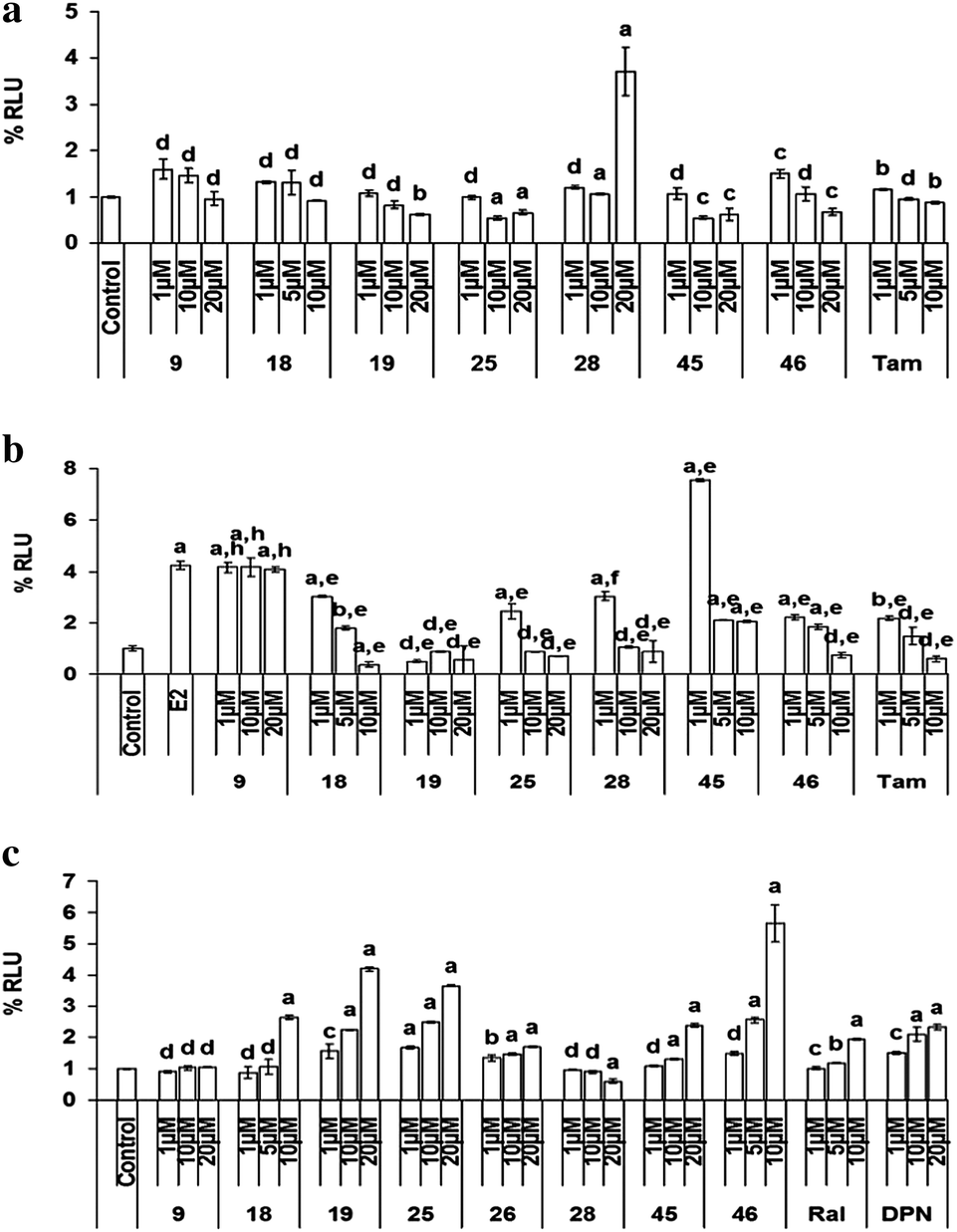 | ||
| Fig. 5 (a) ERE–ERα-mediated transcriptional activation. (b) ERE–ERα mediated transcriptional activation in the presence of E2. Transcription activation of compounds via ERα–ERE in COS-1 cells in (6a) the absence of E2 and (5b) in the presence of 10 nM of E2. Results are expressed as mean ± SE, n = 3. P values are a – p < 0.001, b – p < 0.01, c – p < 0.05, d – p > 0.05 vs. control, e – p < 0.001, f – p < 0.01, g – p < 0.05, h – p > 0.05 vs. E2. (c) The effect of compounds on transcription activation via ERβ–ERE in COS-1 cells. Results are expressed as mean ± SE, n = 3. P values are a – p < 0.001, b – p < 0.01, c – p < 0.05, d – p > 0.05 vs. control. | ||
 | ||
| Fig. 6 MCF-7 cells were transiently transfected with ERα (A) or ERβ (B) or non-specific scrambled SiRNA and treated with the compounds for 48 h. Cell viability was measured using MTT cell viability assay. The percentage of viable cells was calculated as the ratio of treated cells to control cells. Results are expressed as mean ± SE, n = 5. p values are a – p < 0.001, b < 0.01, c – p < 0.05 and d – p > 0.05 vs. control. | ||
As shown in Fig. 6(B), in the presence of ERβ SiRNA, significant induction of cellular proliferation was observed either alone or in presence of compounds 18, 19, 25, 28, 45 and 46 suggesting the role of ERβ in the action of these compounds. However, it was interesting to note that ERβ knockdown did not significantly alter the decrease in cell viability in the case of compound 28, demonstrating that this compound acts via ERα only and ERβ is not involved in the anti-proliferative action of this compound. These results are in accordance with the results observed in ERα knockdown experiments. In the presence of DPN, the ERβ agonist, cells showed less viability as compared to the control group, which was transfected with scrambled SiRNA.
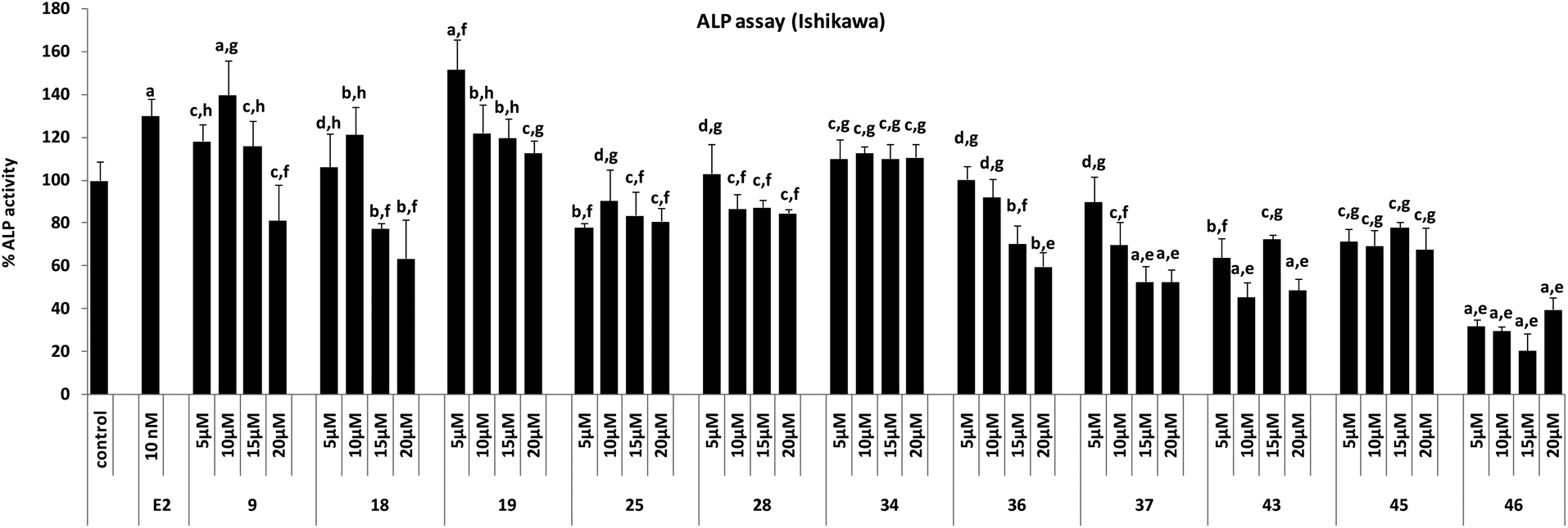 | ||
| Fig. 7 ALP assay in Ishikawa cells. Results are expressed as mean ± SE, n = 5. P values are a – p < 0.001, b – p < 0.01, c – p < 0.05, d – p > 0.05 vs. control, e – p < 0.001, f – p < 0.01, g – p < 0.05, h – p > 0.05 vs. E2. | ||
2.3. Docking protocol
The docking experiments were carried our using the GOLD docking program. For docking studies, the atomic co-ordinates of ERα (PDB id: 1ERR)30 as well as ERβ (PDBid: 1QKN)31 were downloaded from the Brookhaven Protein Data Bank (www.rcsb.org) and prepared using the protein preparation wizard implemented in the Schrödinger software package,32 where bond orders were assigned, water and other residues except bound ligands were deleted, and finally the protein was backbone constrained minimized using OPLS-2005 force field implemented in Schrödinger software package. The bound conformation of the co-crystallized ligands was used in order to define the LBD domain of ERα and ERβ. The optimized 3D-structures of new ligands were docked within a 10 Å radius by running 20 genetic algorithm (GA) steps for each. The docked poses of geometry optimized ligands were ranked using the GoldScore (GS), to find the most optimal binding pose of each ligand. In the GOLD program, the default parameters: population size (100); selection-pressure (1.1); number of operations (10000); number of islands (1); niche size (2); and operator weights for migrate (0), mutate (100) and crossover (100) were applied. Finally, the top five binding poses of each ligand were analyzed to select the best binding pose of each ligand in order to untangle the essential parameters in terms of direct- (H-bonds) and indirect (hydrophobic) interactions governing binding disparities among the series of these compounds.
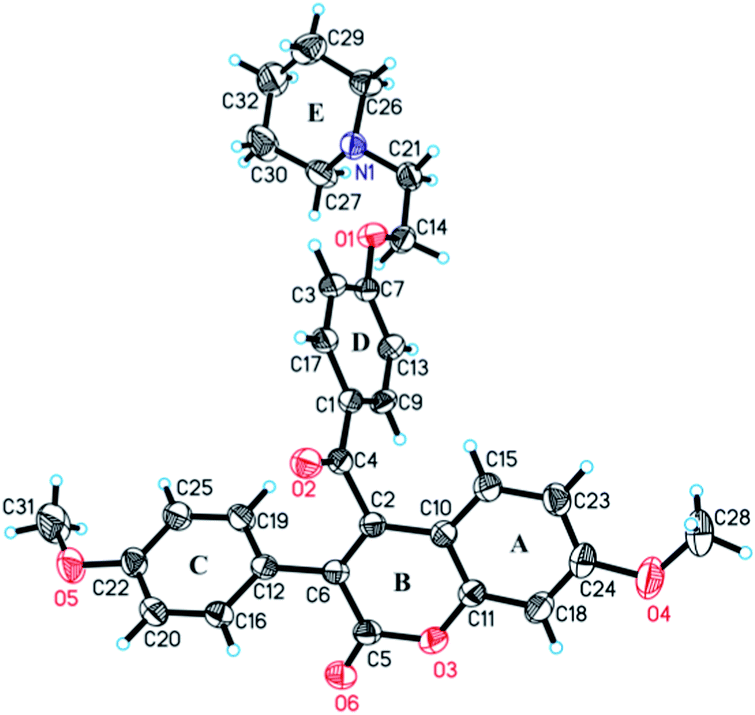 | ||
| Fig. 8 ORTEP diagram (at 30% probability) of 18, along with atomic numbering scheme for the non-hydrogen atoms. | ||
The binding site analyses of a number of co-crystallized ER–LBDs have revealed that all of them share a common hydrophobic cavity at the ER–LBD. The ER agonist binding induces a conformational rearrangement in the LBD,33 resulting in the facilitation of nuclear coactivators binding,34 while the ER antagonists sterically prevent the correct assembly of the AF-2 region and the NR-box binding cleft.35 A third category, termed selective ER modulators (SERMS), act both as agonists and antagonists, depending on their tissue locations and the ER isoform.36 The first SERM tamoxifen (TAM) approved for breast cancer is effective against all stages of breast cancer, whereas raloxifene (RAL) is indicated for the prevention and treatment of osteoporosis in postmenopausal women.37 These SERMs have beneficial effects on bone and lipid metabolism while antagonizing the effects of estrogens on the uterus and breasts.
In the present study molecular docking was performed through the GOLD38 docking program to gain insight into the probable nature of the binding and to understand the activity variation among the highly active representatives (compound 18 and 45) of synthesized prototypes, as well as the least active compound (compound 20) reported herein, along with the control drug RAL (to date RAL is the only SERM that has been co-crystallized with both ERs, the ERα (PDB id: 1ERR) and ERβ (PDB id: 1QKN)) and TAM at the LBD domain of ERα as well as ERβ.
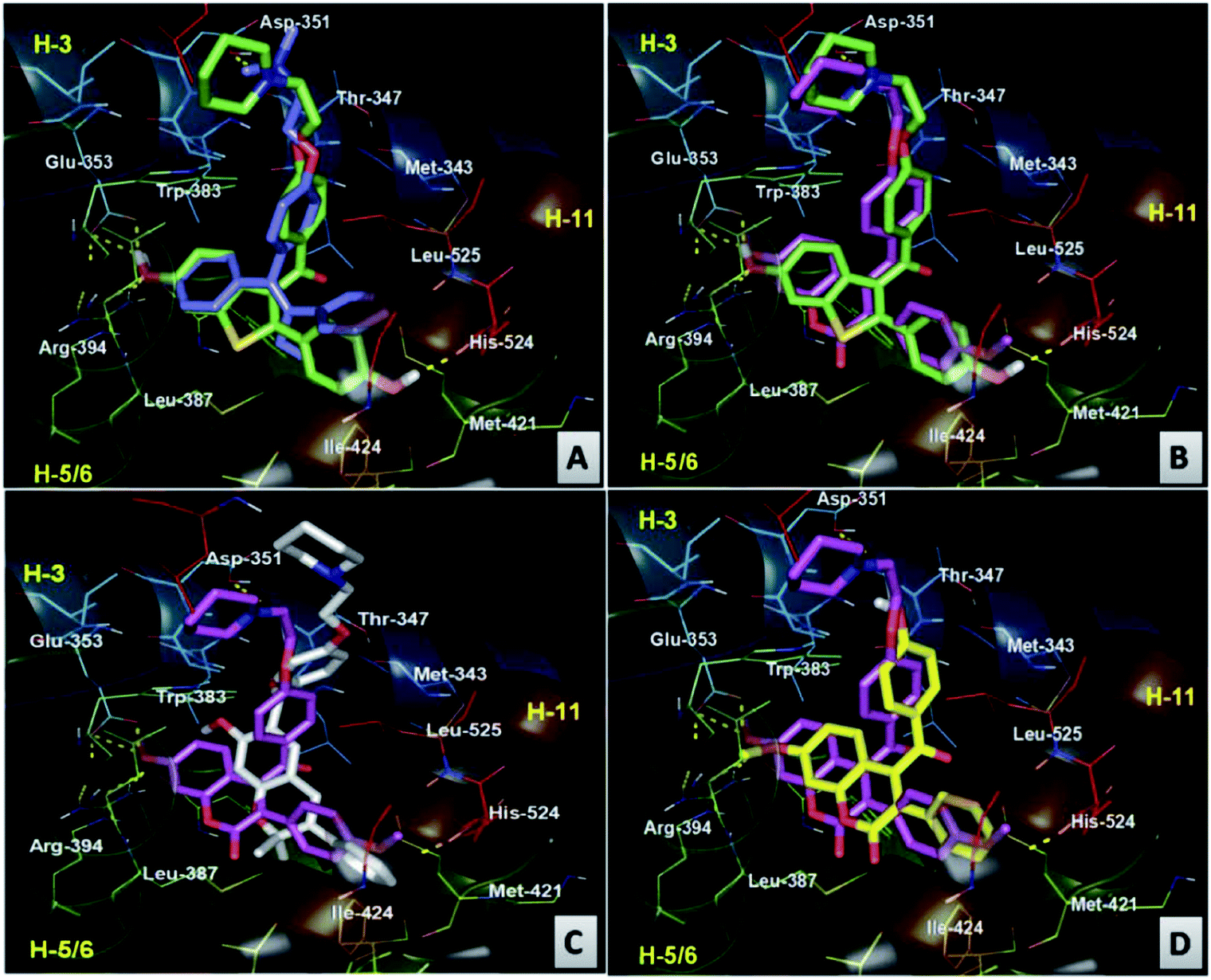 | ||
| Fig. 9 Comparative 3D binding view of (A) RAL (green carbon) and TAM (blue carbon) (B), RAL (green carbon) and compound 18 (pink carbon) (C), compound 18 (pink carbon) and compound 45 (white carbon) (D) compound 18 (pink carbon) and compound 20 (yellow carbon) at the LBD domain of ERα. | ||
The binding analysis of compound 45 showed that the chromene moiety of the molecule occupied the hydrophobic cavity made by the residues Phe404, Leu391, Met388, and Glu353, while the phenyl group linked with the chromene moiety is accommodated in the cavity made by the residues Met421, His524, Gly521, Ile424 and Leu428. The binding cavity of the aroyl chromene core of compound 45 occupied the hydrophobic space similar to RAL but less efficiently (cavity diagonally across the cavity between H11, H3 and H6). However, this less efficient hydrophobic occupancy has been compensated by the H-bond interaction of ketonic oxygen attached to the chromene moiety. The side chain of compound 45, though, showed a different orientation than the side chain orientation of compound 18 and RAL but it efficiently displaces the H12 and protrudes from the pocket between H3 and H11, resulting in ER antagonism better than RAL and comparable to compound 18 (Fig. 10C). The weak antagonism/activity of compound 20 can be easily explained by the absence of the aminoalkoxy side chain, thereby resulting in the non-displacement of H12 (Fig. 9D).
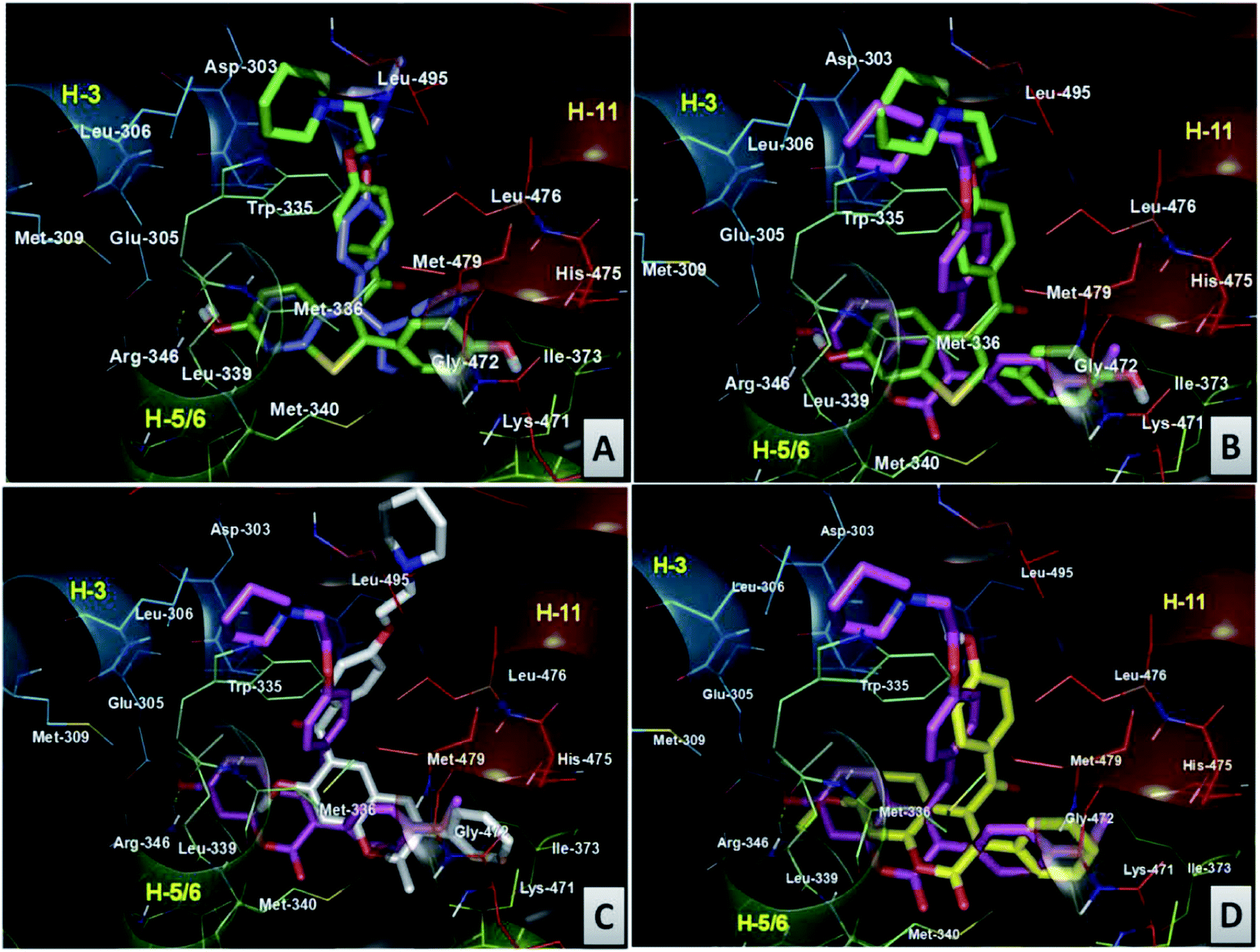 | ||
| Fig. 10 Comparative 3D binding view of (A) RAL (green carbon) and TAM (blue carbon) (B), RAL (green carbon) and compound 18 (pink carbon) (C), compound 18 (pink carbon) and compound 45 (white carbon) (D) compound 18 (pink carbon) and compound 20 (yellow carbon) at the LBD domain of ERβ. | ||
A comparison of the binding of RAL with TAM in ERβ binding cavity revealed both compounds comparably accommodated in the same manner, as observed in the ERα complex (Fig. 10A). Furthermore, it was also observed that compound 18 in ERβ followed a similar binding pattern to RAL (Fig. 10B). The basic pharmacophore, made of a coumarin core (compound 18), occupied the hydrophobic cavity made by the residues Glu305, Arg346, Leu301, Leu339, and Leu343, which were different from the amino acid residues as observed in ERα. The 4-methoxyphenyl group attached at the 4th position of the coumarin nucleus occupied the hydrophobic cavity made from the residues Met421, Gly472, His575, Leu298, Phe356, Met340, Ile376 and Ile373, the characteristic amino acid residue for ERβ affinity. The side chain of compound 18 is anchored by hydrophobic contacts made by residues Leu476, Leu495, Leu306, Met494, Trp335, and Leu491 around the piperidine ring. However, the length of the side chain of compound 18 is almost superimposable on the side chain of RAL, efficiently displacing the H12, and protrudes from the pocket between H3 and H11, giving credence to the partial agonism and antagonism (SERM like behavior) shown by compound 18. Next, a comparative binding study of compound 18 with that of compound 45 at the LBD domain of ERβ (Fig. 10C) revealed that the binding orientation of the side chain of compound 45 is slightly moved towards H11, compared to its binding orientation as observed in the ERα complex; still it efficiently displaces H12 and protrudes from the pocket between H3 and H11. This explains its ER antagonism being better than that of RAL and comparable to that of compound 18. The aroylchromene cores of the molecule occupy a similar hydrophobic space, diagonally across the cavity between H11, H3 and H6, as observed in the ERα complex. The binding orientation of the non active compound 20 in the binding domain of ERβ was the same as that observed in the ERα complex (Fig. 10D). The key observation is that the binding orientation of the side chain of compound 45 in ERβ is different from that of 18 and RAL; it induces maximum ERβ specific activity and thereby enhances bone specific osteoblast differentiation at 100 pM.
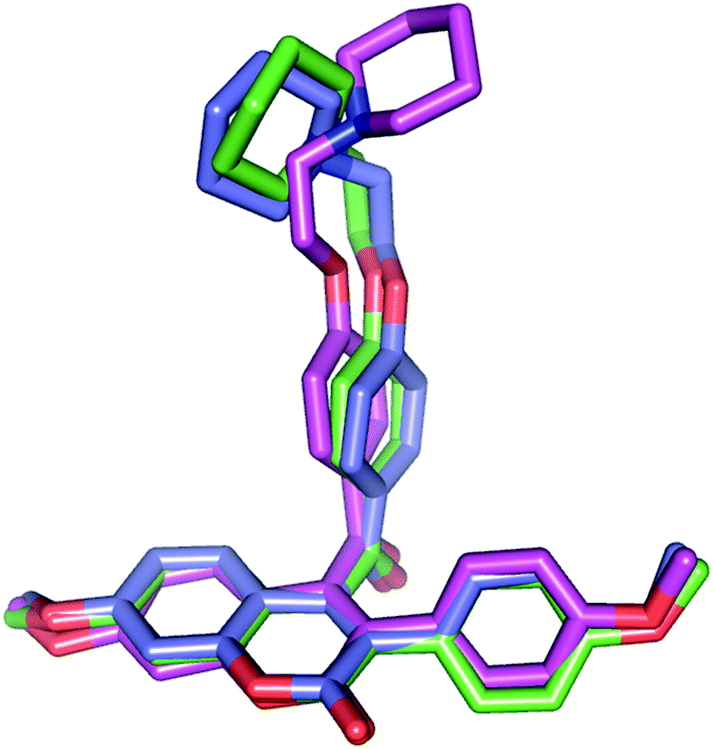 | ||
| Fig. 11 Comparison of conformation for the compound 18 in the crystallized form (pink colored carbon) with docked conformation at the ERα (green colored carbon) and ERβ (blue colored carbon) binding site. | ||
3. Conclusions
In conclusion, it can be clearly seen that the carbon chain of active compounds 18 and 45 is oriented perpendicular to the bicyclic coumarin/chromene pharmacophore, giving the favorable conformation shown by most of the estrogen antagonists. Docking of 18 in the ligand binding cavity of ERα, showed that the molecule favorably fits in the ligand binding space and makes hydrophobic interactions with the salient amino acids (Glu353, Arg394, His524) known to anchor both the estrogenic and antiestrogenic ligands in the binding pocket of LBD, and the side chain is projected towards Asp351. In prototype II molecules, where the carbonyl functional group is reduced to methylene (CH2), due to weak hydrophobic interactions biological activity is substantially decreased. This reinforces the hypothesis that a polar atom or a substituent is an essential requisite for making hydrophobic contacts within the receptor pocket and the potential biological efficacy. The surprise discovery of the present study is the novel prototype III molecules 45 & 46 with aroyl substitution at the 6th position. Both the compounds have shown potent antiproliferative activity against both the breast cancer cell lines, promote alkaline phosphatase activity, enhance osteoblast mineralization in vitro, significantly decrease ERE–ERα dependent transactivation and induce ERβ activity. The upregulation of ERβ specific isoform activity of 45 may be responsible for the antiosteoporotic activity of this molecule at picomolar concentrations. In addition, both the compounds were also devoid of any estrogenic activity, which correlates to their antiestrogenic behaviour in the two breast cancer cell lines.4. Experimental
4.1. Materials and methods
All the glass apparatus was oven dried prior to use. All chemicals and reagents were purchased from Sigma-Aldrich or ACROS ORGANICS and were used without further purification. Solvents THF, DCM, DMF, DMSO, diethyl ether, acetone were purified using standard methods. 100–200 mesh silica gel was used for column chromatography, and TLC was performed on Merck-precoated silica gel 60-F254 and aluminum oxide 60-F254 plates. Melting points were recorded with COMPLAB melting point apparatus and are uncorrected. The chromatographic solvents are mentioned as v/v ratios. All the synthesized compounds were fully characterized by 1H, 13C NMR, IR, and further confirmed through ESI-MS, ESI-HRMS analysis. IR spectra were recorded on a Perkin-Elmer FT-IR RXI spectrophotometer and the values were reported in cm−1. 1H NMR and 13C NMR spectra were recorded on Bruker DRX-300 (300 MHz for 1H) and at 75 MHz for 13C or DPX-200 (at 50 MHz for 13C) spectrometers using CDCl3, DMSO-d6, CD3OD or acetone-d6 as solvents using tetramethylsilane as the internal standard. Chemical shifts are reported in parts per million. ESI-MS spectra were obtained on a LCQ Advantage Ion trap mass spectrometer (Finnigan thermo Fischer scientific) and High-resolution mass spectra (ESI-HRMS) were recorded on an Agilent 6520 ESI-QTOP mass spectrometer. All products reported showed 1H NMR and 13C NMR spectra in agreement with the assigned structures. The purity of compounds was determined by HRMS, and all tested compounds yielded data consistent with a purity of at least 95% as compared with the theoretical values.1-(2-Hydroxy-4-methoxyphenyl)-2-(4-hydroxyphenyl)ethane-1,2-dione (compound 5). Yellow solid, mp 172 °C; IR (KBr) ν 3342, 1652, 1623 1525, cm−1; 1H NMR (300 MHz, DMSO d6) δ 11.00 (s, 1H), 10.61 (s, 1H), 7.68 (d, J = 8.41 Hz, 3H), 23 (m, 2H), 6.90 (d, J = 8.5 Hz, 2H); 6.61–6.57 (m, 1H) 6.41 (d, J = 1.92 Hz, 1H); 13C NMR (50 MHz), 195.0, 166.9, 163.5, 132.4, 132.2, 127.5, 124.8, 116.2, 114.2, 108.4, 101.2, 56.1. ESI-MS: (m/z) 273 [M + H].+
Coumarins 8–11 were synthesised following the representative procedure as described for compound 8 and compounds 12–19 were synthesized by using same procedure described for compound 12.
Synthesis of 4-(4-hydroxybenzoyl)-3-phenyl-2H-chromen-2-one (compound 8). To a stirred solution of benzil 3 (0.726 g, 3 mmol) in anhydrous acetone (20 ml) was added anhydrous K2CO3 (0.690 g, 5 mmol) followed by slow addition of phenylacetyl chloride (0.541 g, 3.5 mmol). The reaction mixture was then stirred vigorously at 80 °C for 4 h. After completion of the reaction, excess solvent was removed under vacuum and 5% aq. cold HCl (30 ml) was added. The solid precipitate was filtered, washed with water, dried and crystallized from diethyl ether to afford a white crystalline compound; yield: 0.923 g, 95%; mp 168 °C; IR (KBr) ν 3208, 1684, 1656 1595, cm−1; 1H NMR (300 MHz, CDCl3 + acetone d6) δ 7.61 (d, J = 8.7 Hz, 2H), 7.51–7.45 (m, 1H), 7.33 (d, J = 8.1 Hz, 1H) 7.27–7.23 (m, 2H), 7.20–7.11 (m, 5H), 6.70 (d, J = 8.7 Hz, 2H); 13C NMR (50 MHz, DMSO-d6) δ 191.2, 163.6, 159.8, 152.9, 148.2, 132.9, 132.3, 132.0, 129.7, 128.4, 127.7, 126.4, 126.0, 124.7, 117.4, 116.7, 115.7; ESI-MS: (m/z) 343 [M + H]+; HRMS-ESI: [M + H]+ for C22H15O4 calcd 343.0970; found 343.0965%.
4-(4-Hydroxybenzoyl)-7-methoxy-3-phenyl-2H-chromen-2-one (compound 9). In accordance to general procedure benzil 5 and phenylacetyl chloride were used as reactants; white solid; yield: 1.06 g, 95%; mp 174 °C; IR (KBr) ν 3238, 1689, 1594, 1506,cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.65 (hump, 1H), 7.73 (d, J = 8.7 Hz, 2H) 7.26–7.22 (m, 5H), 7.14 (d, J = 2.3 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H), 6.89 (dd, J = 8.8, 2.3 Hz, 1H), 6.73 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H); 13C NMR (DMSO-d6, 75 MHz), δ 191.9, 164.0, 162.9, 155.3, 149.0, 133.6, 132.8, 130.4, 128.6, 128.2, 127.6, 126.9, 121.7, 116.2, 113.2, 111.3, 101.6, 56.5; ESI-MS: (m/z) 372; found 373 [M + H ]+; HRMS-ESI: [M + H]+ for C23H16O5 calcd 373.1076; found 373.1066%.
4-(4-Hydroxybenzoyl)-3-(4-methoxyphenyl)-2H-chromen-2-one (compound 10). In accordance to general procedure benzil 3 and 4-methoxy phenylacetyl chloride were used as reactants; white solid; yield: 1.004 g, 90%; mp 162 °C; IR (KBr) ν 3278, 1691, 1651, 1602, 1577, cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.73 (d, J = 8.7 Hz, 2H), 7.66–7.60 (m, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.30–7.21 (m, 3H), 7.15–7.12 (m, 1H), 6.82 (d, J = 8.8 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 3.69 (S, 3H); 13C NMR (DMSO-d6, 75 MHz) δ 191.7, 165.0, 160.5, 159.7, 153.2, 148.2, 132.8, 132.3, 131.7, 126.4, 126.3, 125.5, 125.2, 124.8, 118.1, 117.2, 116.4, 113.7, 55.5; ESI-MS: (m/z) 372; found, 373 [M + H]+; HRMS-ESI: [M + H]+ for C23H17O5 calcd 373.1076; found 373.1057%.
4-(4-Hydroxybenzoyl)-7-methoxy-3-(4-methoxyphenyl)-2H-chromen-2-one (compound 11). In accordance with general procedure benzil 5 and 4-methoxy phenylacetyl chloride were used as reactants; yellow solid; yield: 1.08 g, 90%; mp 180 °C, IR (KBr) ν 3221, 1686, 1646, 1592, 1509, cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.73 (d, J = 8.7 Hz, 2H), 7.20 (d, 8.7 Hz, 2H), 7.12 (d, J = 2.3 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H), 6.88 (dd, J = 8.8, 2.4 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 6.73 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H), 3.68 (s, 3H); 13C NMR (DMSO-d6, 75 MHz), δ 192.1, 164.1, 162.7, 160.8, 159.5, 155.1, 148.5, 132.82, 131.7, 127.5, 126.9, 125.6, 121.4, 116.2, 113.7, 111.4, 101.5, 56.5, 55.4; ESI-MS (m/z): 402; found 403 [M + H]+; HRMS-ESI: [M + H]+ for C24H19O6 calcd 403.1182; found 403.1054%.
Synthesis of 3-phenyl-4-(4-(2-(piperidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 12). To the stirred solution of compound 8 (0.513 g, 1.5 mmol) in dry acetone (20 ml) anhydrous K2CO3 (0.690 g, 4.5 mmol) and 1-(2-chloroethyl) piperidine hydrochloride (0.322 g, 1.75 mol) were added. The reaction mixture was refluxed with stirring until complete consumption of the starting material. The excess solvent was removed under vacuum and cold water (30 ml) was added to the residue. It was extracted with DCM (20 ml × 3) and the combined organic layers were dried over Na2SO4. The crude product obtained on removal of the solvent was purified by chromatography on a silica gel column (chloroform
:methanol 50:1) to give a white crystalline solid; yield: 0.625 g, 92%; mp 180 °C; IR (KBr) ν 1725, 1658, 1598, cm−1; 1H NMR (300 MHz, CDCl3) δ 7.74 (d, J = 8.8 Hz, 2H), 7.59–7.54 (m, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.37–7.34 (m, 2H), 7.30–7.19 (m, 5H), 6.82 (d, J = 8.8 Hz, 2H) 4.11 (t, J = 5.9 Hz, 2H), 2.74 (t, J = 5.9 Hz, 2H), 2.48 (t, J = 4.8 Hz, 4H) 1.62–1.58 (m, 4H), 1.45 (m, 2H); 13C NMR (CDCl3, 75 MHz), δ 192.1, 163.9, 160.5, 153.4, 148.9, 132.4, 131.41, 131.8, 129.9, 128.9, 126.5, 124.7, 117.8, 117.0, 114.7, 66.4, 57.6, 55.0, 29.7, 25.8, 24.0; ESI-MS (m/z): 453; found 454 [M + H]+; HRMS–ESI; [M + H]+ for C29H28NO4 calcd 454.2018; found 454.2006%.
3-Phenyl-4-(4-(2-(pyrrolidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 13). Compound 8 and 1-(2-chloroethyl) pyrrolidine hydrochloride were used as reactants; white solid; yield: 0.593 g, 90%; mp 186 °C; IR (KBr) ν, 1722, 1659, 1597, 1424 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.73 (d, J = 8.7, 2H), 7.59–7.55 (m, 1H), 7.45 (d, J = 8.1 Hz, 1H), 7.34–7.28 (m, 2H), 7.24–7.19 (m, 5H), 6.84 (d, J = 8.7 Hz, 2H), 4.11 (t, J = 5.7 Hz, 3H), 2.88 (t, J = 5.7 Hz, 2H), 2.60 (m, 4H), 1.80 (m, 4H); 13C NMR (CDCl3, 75 MHz), δ 192.1, 163.9, 153.4, 148.9, 132.4, 131.9, 131.8, 129.9, 128.8, 128.3, 128.1, 126.53, 125.2, 124.7, 117.8, 117.0, 114.6, 67.5, 54.7, 23.5; ESI-MS (m/z): 439; found 440 [M + H]+; HRMS-ESI: [M + H]+ for C28H26NO4 calcd 440.1862; found 440.1844%.
7-Methoxy-3-phenyl-4-(4-(2-(piperidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 14). Compound 9 and 1-(2-chloroethyl) piperidine hydrochloride were used as reactants. Creamy solid; yield: 0.689 g, 95%; mp 192 °C; IR (KBr) ν, 1714, 1653, 1603, 1509, cm−1; 1H NMR (300 MHz, CDCl3) δ 7.73 (d, J = 8.8 Hz, 2H), 7.35–7.32 (m, 2H), 7.26–7.16 (m, 4H), 6.94 (d, J = 2.4 Hz, 1H), 6.82–6.76 (m, 3H), 4.10 (t, J = 6.0 Hz, 2H), 3.89 (s, 3H), 2.74 (t, J = 5.9 Hz, 2H), 2.48 (t, J = 4.8 Hz, 4H) 1.63–1.56 (m, 4H), 1.48–1.45 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ 192.3, 163.9, 162.9, 160.9, 155.2, 149.2, 132.7, 131.8, 130.0, 128.5, 128.1, 127.5, 121.8, 114.6, 112.9, 111.3, 100.9, 66.4, 57.61, 55.5, 55.1, 25.8, 24.1; ESI-MS (m/z): 483; found 484 [M + H ]+; HRMS-ESI: [M + H]+ for C30H30NO5 calcd 484.2124; found 484.2116%.
7-Methoxy-3-phenyl-4-(4-(2-(pyrrolidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 15). Compound 9 and 1-(2-chloroethyl) pyrrolidine hydrochloride were used as reactants; creamy solid; yield: 0.634 g, 90%; mp 188 °C; IR (KBr) ν, 1713, 1653, 1603, 1508, cm−1; 1H NMR (300 MHz, CDCl3) δ 7.75 (dd, J = 8.9, 2.6 Hz, 2H), 7.36–7.30 (m, 2H), 7.25–7.18 (m, 4H), 6.95 (d, J = 2.5 Hz, 1H), 6.86–6.78 (m, 3H), 4.13 (t, J = 5.6 Hz, 2H), 3.91 (s, 3H), 2.91 (t, J = 3.0 Hz, 2H), 2.62 (m, 4H) 1.82 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 192.4, 163.9, 162.9, 161.0, 155.3, 149.2, 132.7, 131.7, 130.0, 128.6, 128.4, 127.5, 121.8, 114.6, 112.95, 111.3, 101.0, 67.5, 55.8, 54.7, 29.7, 23.50; ESI-MS (m/z):469; found 470 [M + H]+; HRMS-ESI: [M + H]+ for C29H28NO5 calcd 470.1967; found = 470.1975%.
3-(4-Methoxyphenyl)-4-(4-(2-(piperidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 16). Compound 10 and 1-(2-chloroethyl) piperidine hydrochloride were used as reactants; creamy solid; yield: 0.689 g, 95%; mp 190 °C; IR (KBr) ν, 1720, 1655, 1598, 1570, 1511, cm−1; 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.6 Hz, 2H), 7.56–7.51 (m, 1H), 7.43 (d, J = 8.1 Hz, 1H), 7.32–7.26 (m, 3H), 7.23–7.20 (m, 1H), 6.82 (d, J = 8.7 Hz, 2H), 6.77 (d, J = 8.6 Hz, 2H), 4.10 (t, J = 5.7 Hz, 2H), 3.74 (s, 3H) 2.74 (t, J = 5.7 Hz, 2H), 2.48 (m, 4H), 1.59–1.57 (m, 4H); 1.46–1.44 (m, 2H); 13C NMR (CDCl3, 75 MHz), δ 192.4, 163.9, 160.8, 159.9, 153.3, 148.1, 131.8, 131.6, 131.4, 128.2, 126.3, 12 4.9, 124.7, 118.0, 116.9, 114.7, 113.6, 66.5, 57.6, 55.1, 25.9, 24.1; ESI MS (m/z): 483; found 484 [M + H]+; HRMS-ESI: [M + H]+ for C30H30NO5 calcd 484.2124; found 484.2229%.
3-(4-Methoxyphenyl)-4-(4-(2-(pyrrolidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 17). Compound 10 and1-(2-chloroethyl) pyrrolidine hydrochloride were used as reactants. Creamy solid; yield: 0.633 g, 90%; mp 186 °C; IR (KBr) ν, 1721, 1652, 1597, cm−1; 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.7 Hz, 2H), 7.56–7.52 (m, 1H), 7.43 (d, J = 8.1 Hz, 1H), 7.32–7.26 (m, 3H), 7.22–7.20 (m, 1H), 6.84 (d, J = 8.7 Hz, 2H), 6.77 (d, J = 8.6 Hz, 2H), 4.11 (t, J = 5.6 Hz, 2H), 3.74 (s, 3H) 2.89 (t, J = 5.7 Hz, 2H), 2.61 (m, 4H), 1.80 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 192.41, 163.94, 160.89, 159.89, 153.25, 148.17, 131.86, 131.69, 131.37, 128.27, 126.37, 124.90, 124.72, 124.66, 117.98, 116.97, 114.70, 113.69, 67.49, 55.15, 54.74, 30.89, 23.49; ESI-MS (m/z): 469; found 470 [M + H]+; HRMS-ESI: [M + H]+ for C29H28NO5 calcd 470.1967; found 470.1982%.
7-Methoxy-3-(4-methoxyphenyl)-4-(4-(2-(piperidin-1-yl) ethoxy)benzoyl)-2H-chromen-2-one (compound 18). Compound 11 and 1-(2-chloroethyl) piperidine hydrochloride were used as reactants; white solid; yield: 0.731 g, 95%; mp 210 °C; IR (KBr) ν, 1726, 1652, 1602, 1508, cm−1; 1H NMR (300 MHz, CDCl3) δ 7.73 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 7.16 (d, J = 8.8 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.83–6.74 (m, 5H), 4.10 (t, J = 5.6 Hz, 2H), 3.88 (s, 3H), 3.72 (s, 3H), 2.74 (t, J = 5.7, 2H), 2.48 (m, 4H) 1.59–1.57 (m, 4H); 1.46–1.44 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ 192.6, 163.8, 162.7, 161.2, 159.6, 155.0, 148.4, 131.8, 131.3, 128.2, 127.3, 124.9, 121.5, 114.6, 113.6, 112.8, 111.4, 100.9, 66.40, 5.60, 55.8, 55.1, 55.0, 30.8, 25.8, 24.0; ESI-MS (m/z): 513; found 514 [M + H]+; HRMS-ESI: [M + H]+ for C31H32 NO6 calcd 514.2230; found 514.2231%.
7-Methoxy-3-(4-methoxyphenyl)-4-(4-(2(-pyrrolidin-1-yl) ethoxy)benzoyl)-2H-chromen-2-one (compound 19). Compound 11 and 1-(2-chloroethyl) pyrrolidine hydrochloride were used as reactants; white solid; 0.674 g, 90%; mp 203 °C; IR (KBr), 1728, 1653, 1600, 1508 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.74 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 8.7 Hz, 2H), 7.16 (d, J = 8.8 Hz, 1H), 6.92 (d, J = 2.3 Hz, 1H), 6.85–6.74 (m, 5H), 4.15 (t, J = 5.6 Hz, 2H), 3.88 (s, 3H) 3.73 (s, 3H), 2.94 (t, J = 5.6 Hz, 2H), 2.68 (m, 4H) 1.83 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 192.6, 163.6, 162.7, 161.2, 159.6, 155.0, 148.4, 131.86, 131.4, 128.4, 127.3, 124.9, 121.5, 114.6, 113.6, 112.8, 111.4, 100.9, 67.0, 55.82, 55.1, 54.6, 54.6, 30.8, 23.4, ESI-MS (m/z) 499; found 500 [M + H]+; HRMS-ESI: for C30H30 NO6 [M + H]+ calcd 500.2073; found 500.2093%.
Demethylation of compound 9 to 7-hydroxy-4-(4-hydroxybenzoyl)-3-phenyl-2H-chromen-2-one (compound 20). To a mixture of dry dichloromethane (2 ml) and anhydrous aluminum chloride (0.160 g, 1.5 mmol) was added dry ethanethiol (0.5 ml) at 0 °C. The resulting solution was stirred for 10 minutes and 4-(4-hydroxybenzoyl)-7-methoxy-3-phenyl-2H-chromen-2-one 9 (0.186 g, 0.5 mmol) was added at 0 °C. The reaction mixture was stirred for 3 h at room temperature. After completion, it was quenched with ice cold water (5 ml) and acidified with 10% aq. HCl (15 ml). The solid precipitate was filtered and purified by column chromatography, eluting with CHCl3–MeOH (25
:1) to give the title compound; pale yellow solid; yield: 0.170 g, 95%; mp 215 °C; IR (KBr) ν 3481, 3359, 1685, 1596, 1507 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.65 (brs, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.24–7.20 (m, 5H), 6.98 (d, J = 8.7 Hz, 1H), 6.85 (d, J = 2.2 Hz, 1H), 6.76–6.71 (m, 3H); 13C NMR (DMSO-d6, 75 MHz), δ 192.0, 163.9, 161.8, 160.8, 155.3, 149.4, 133.7, 132.7, 130.4, 128.53, 128.2, 127.9, 126.9, 120.6, 116.1, 114.0, 110.1, 102.9; ESI-MS (m/z): 358 found 359 [M + H]+; HRMS-ES [M + H]+: for C22H15O5 calcd 359.0919; found 359.0878%.
4-(4-Hydroxybenzoyl)-3-(4-hydroxyphenyl)-2H-chromen-2-one (compound 21). Pale yellow solid; yield: 0.165 g, 92%; mp 205 °C; IR (KBr) ν 3310, 1700, 1684, 1575 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.63 (brs 1H), 9.57 (brs, 1H) 7.74 (d, J = 8.6 Hz, 2H), 7.64–7.60 (m, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.29–7.25 (m, 1H), 7.15–7.09 (m, 3H), 6.73 (d, J = 8.6 Hz, 2H), 6.62 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 75 MHz) δ 192.1, 163.9, 160.6, 158.0, 153.2, 147.6, 132.7, 132.18, 131.7, 126.9, 126.3, 125.2, 125.2, 123.8, 118.1, 117.1, 116.1, 115.0; ESI-MS (m/z): 358 found 359 [M + H]+; HRMS-ESI: [M + H]+ for C22H15O5 calcd 359.0919, found = 359.0807%.
7-Hydroxy-4-(4-hydroxybenzoyl)-3-(4-hydroxyphenyl)-2H-chromen-2-one (compound 22). Yellow solid; yield: 0.168 g, 90%; mp 240 °C; IR (KBr) ν 3407, 3111, 1663, 1603, 1510, cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.65 (s, 1H), 10.61 (s, IH), 9.50 (s, 1H), 7.69 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.4 Hz 2H), 6.96 (d, J = 8.7 Hz, 1H), 6.82 (d, J = 1.8 Hz, 1H), 6.74–6.72 (m, 3H), 6.59 (d, J = 8.4 Hz, 2H); 13C NMR (DMSO-d6, 50 MHz) δ 191.8, 163.3, 161.00, 160.5, 157.2, 154.5, 147.9, 132.2, 131.2, 127.19, 126.4, 123.6, 120.2, 115.6, 114.5, 113.4, 109.8; ESI-MS (m/z): 374 found 375 [M + H]+; HRMS-ESI: [M + H]+ for C22H15O6 calcd 375.0869; found 375.0864%.
7-Hydroxy-3-phenyl-4-(4-(2-(piperidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 23). White solid; yield: 0.211 g, 90%; mp 212 °C; IR (KBr) ν 3557, 3165, 1694, 1657, 1598, 1510 cm−1; 1H NMR (300 MHz, CDCl3 + DMSO-d6) δ 10.48 (s, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.26–7.21 (m, 5H), 7.007–6.87 (m, 4H), 6.73 (d, J = 8.6 Hz, 1H), 4.10 (t, 2H), 2.61 (t, 2H), 2.39 (m, 4H), 1.46–1.36 (m, 6H); 13C NMR (DMSO-d6, 50 MHz) δ 192.0, 162.4, 161.9, 161.5, 154.8, 148.5, 133.1, 131.9, 129.9, 128.3, 127.7, 127.2, 120.3, 114.9, 113.6, 109.4, 102.6, 62.6, 54.46, 52.5, 22.6, 22.2, 21.1; ESI-MS: (m/z) 469; found 470 [M + H]+; HRMS-ESI: [M + H]+ for C29H28NO5 470.1967; found 470.1955%.
7-Hydroxy-3-phenyl-4-(4-(2-(pyrrolidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 24). White solid; yield: 0.194 g, 85%; mp 204 °C; IR (KBr) ν 695, 1659, 1597 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.63 (brs, 1H), 7.62 (d, J = 8.7 Hz, 2H), 7.00–6.97 (m, 5H), 6.76–6.65 (m, 4H), 6.53–6.49 (m, 1H), 4.15 (t, 2H), 2.83 (t, 2H), 2.63 (m, 4H) 1.69 (m, 4H); 13C NMR (DMSO-d6, 75 MHz) δ 192.5, 163.0, 161.9, 160.7, 155.3, 148.9, 133.6, 132.5, 130.4, 128.7, 128.6, 128.2, 127.7, 120.8, 115.4, 114.1, 109.9, 103.1, 64.1, 54.1, 52.8, 23.1; ESI-MS: m/z 455; found 456 [M + H]+; HRMS-ESI: [M + H]+ for C28H26NO5 [M + H]+ calcd 456.1811 found 456.1806%.
3-(4-Hydroxyphenyl)-4-(4-(2-(piperidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 25). Yellow solid; yield: 0.212 g, 90%; mp 224 °C; IR (KBr) ν 3430, 1717, 1663, 1600, 1510 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.04 (s, 1H),7.64 (d, J = 7.8 Hz, 2H), 7.46–7.38 (m, 1H), 7.38–7.31 (m, 1H), 7.14–7.06 (m, 3H), 6.76 (d, J = 8.0 Hz, 2H), 6.62 (d, J = 8.0 Hz 2H), 4.23 (t, 2H), 2.78 (t, 2H) 2.46 (m, 4H), 1.48–1.44 (m, 6H); 13C NMR (DMSO-d6, 50 MHz), δ 192.1, 162.5, 160.0, 157.7, 152.7, 146.6, 131.99, 131.2, 128.2, 125.6, 124.9, 123.1, 117.5, 116.7, 114.9, 62.6, 54.4, 52.5, 22.2, 21.1; ESI-MS: (m/z) 469; found 470 [M + H]+; HRMS-ESI: [M + H]+ for C29H28NO5 calcd 470.1967; found 470.2065%.
3-(4-Hydroxyphenyl)-4-(4-(2-(pyrrolidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 26). Yellow solid; yield: 0.201 g, 88%; mp 230 °C; IR (KBr) ν 3498, 1726, 1661, 1598, cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.10 (hump, 1H) 7.65 (d, J = 8.7 Hz, 2H), 7.43–7.38 (m, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.07–7.02 (m, 1H), 6.91–6.87 (m, 3H), 6.76 (d, J = 8.8 Hz, 2H), 6.43 (d, J = 8.5 Hz, 2H), 4.20 (t, 2H), 2.77 (t, 2H) 2.64 (m, 4H), 1.69 (m, 4H); 13C NMR (DMSO-d6, 50 MHz) δ 192.1, 162.5, 160.89, 157.6, 152.6, 146.6, 132.0, 131.1, 128.1, 124.9, 124.7, 123.1, 117.50, 116.7, 114.9, 63.6, 53.4, 52.2, 22.5; ESI-MS: (m/z) 455 found 456 [M + H]+, HRMS-ESI: [M + H]+ for C28H26NO5 calcd 456.1811; found 4456.1452%.
7-Hydroxy-3-(4-hydroxyphenyl)-4-(4-(2-(piperidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 27). Yellow solid; yield 0.220 g, 90% mp 256 °C; IR (KBr) ν 3400, 1699, 1657, 1599, cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.49 (brs, 1H), 9.54 (s, 1H), 7.76 (d, J = 7.8 Hz, 2H), 6.99 (d, J = 7.6 Hz, 2H), 6.91–6.80 (m, 4H), 6.66 (d, J = 7.5 Hz, 1H), 6.53 (d, J = 7.6 Hz, 2H), 4.11 (t, 2H), 2.63 (t, 2H), 2.40 (m, 4H) 1.46–1.37 (m, 6H); 13C NMR (DMSO-d6, 50 MHz) δ 192.8, 162.8, 161.6, 160.9, 157.7, 155.1, 147.9, 132.4, 131.7, 128.7, 127.4, 124.0, 120.8, 115.39, 114.0, 110.1, 103.0, 63.1, 53.0, 49.0, 31.1, 22.7, 21.6; ESI-MS: (m/z) 485 found 486 [M + H]+; HRMS-ESI: [M + H]+ for C29H28NO6 calcd 486.1917; found 486.1942%.
7-Hydroxy-3-(4-hydroxyphenyl)-4-(4-(2-(pyrrolidin-1-yl)ethoxy)benzoyl)-2H-chromen-2-one (compound 28). Yellow solid; yield: 0.213 g, 90%; mp 248 °C; IR (KBr) ν 3455, 3418, 1704,1663, 1600 cm−1; 1H NMR (300 MHz, CDCl3 + DMSO-d6) δ 10.18 (brs, 1H), 9.09 (hump, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 8.0 Hz, 2H), 6.98 (d, J = 8.6 Hz, 1H), 6.87–6.85 (m, 3H), 4.11 (t, 2H), 2.76 (t, 2H), 2.38 (m, 4H) 1.66 (m, 4H); 13C NMR (DMSO-d6, 50 MHz) δ 192.3, 162.4, 161.1, 160.4, 157.2, 154.5, 147.5, 131.9, 131.2, 128.3, 126.9, 123.5, 120.3, 114.9, 114.6, 113.5, 109.6, 102.5, 63.5, 53.5, 52.5, 22.2; ESI-MS: (m/z) 471 found 472 [M + H]+; HRMS-ESI: [M + H]+ for C28H26NO6 calcd 472.1760; found 472.1799%.
Synthesis of 4-bromo-7-methoxy-3-phenyl-2H-chromene (compound 31). To the solution of isoflavone 30 (5.080 g, 20 mmol) in dry benzene (25 ml), phosphorus tribromide (16.26 g, 60 mmol) was added carefully, and the resulting solution was stirred at 85 °C for 2 h. It was cooled and cautiously poured onto crushed ice (200 g). The aqueous solution was extracted with benzene (50 ml × 3) and the combined organic layers were washed with a saturated aqueous solution of sodium bicarbonate (4 × 40 ml) and brine (2 × 100 ml), dried over Na2SO4 and evaporated at reduced pressure. The colourless oil crystallized from methanol at 0 °C on standing for 12 h. The white solid of 4-bromo chromene 31 (5.38 g, 85%) was filtered, dried and used immediately in the next reaction.
Synthesis of (7-methoxy-3-phenyl-2H-chromen-4-yl)-(4-methoxyphenyl)methanone (compound 34). To a stirred solution of 31 (1.59 g, 5 mmol) in dry diethyl ether (15 ml) at 0 °C under nitrogen 1.6 M n-butyllithium in hexane (5 ml, 8 mmol) was added slowly via syringe in a single portion. The orange solution was stirred at 0 °C for 1.5 h, after which a solution of anisonitrile 32 (0.865 g, 6.5 mmol) in anhydrous ether (10 ml) was added slowly via syringe in a single portion. The solution was stirred for 1.5 h at 10 °C. After completion of the reaction the reaction mixture was poured into water and extracted with ethyl acetate (30 ml × 4), dried over Na2SO4 and concentrated at reduced pressure to give a brown semisolid, which was purified through column chromatography with hexane ethylacetate (10
:1); yellow solid; yield: 1.39 g, 75% mp 130 °C; IR (KBr) 1620, 1534 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.80 (d, J = 8.7 Hz, 2H), 7.22–7.19 (m, 5H),6.87–6.83 (m, 3H) 6.5 (d, J = 2.4 Hz, 1H), 6.39 (dd, J = 8.5 Hz, J = 2.43 Hz, 1H), 5.11 (s, 2H), 3.81 (s, 3H), 3.79 (s, 3H); 13C NMR (75 MHz, CDCl3) 174.2, 161.9, 160.9, 154.6, 136.8, 132.5, 130.1, 129.6, 128.4, 127.7, 127.2, 126.9, 126.8, 115.5, 113.8, 107.6, 101.9, 69.20, 55.4, 55.3.0; ESI-MS: (m/z) 372; found 372[M]+; HRMS-ESI: [M]+ for C24H20O4 372.1362 found 372.1641%.
(4-(2-Chloroethoxy)phenyl)(7-methoxy-3-phenyl-2H-chromen-4-yl)methanone (compound 35). Compound 35 was synthesized from compound 31 and nitrile 33. Yellow solid; yield: 1.470 g, 70%; mp 135 °C; IR, (KBr), ν 1640, 1544 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 8.46 Hz, 2H), 7.22 (m, 5H), 6.86–6.84 (m, 3H) 6.53 (d, J = 2.46 Hz, 1H), 6.39 (dd, J = 8.55 Hz, J = 2.46 Hz, 1H), 5.10 (s, 2H), 4.23 (t, J = 5.82 Hz, 2H), 3.83–3.81 (t, J = 5.79, 2H), 3.79 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 174.1, 160.9, 160.4, 154.6, 136.7, 132.3, 130.7, 129.7, 128.4, 127.8, 127.2, 126.7, 115.4, 114.4, 107.6, 101.9, 69.1, 67.9, 55.4, 41.7; ESI-MS: (m/z) 420; found 420 [M]+.
(4-(2-Chloroethoxy)phenyl)(7-hydroxy-3-phenyl-2H-chromen-4-yl)methanone (compound 38). Compound 38 was synthesized from compound 35 (1.260 g, 3 mmol) according to the procedure of demethylation used for the synthesis of compound 20. Solid residue after vacuum drying was purified by chromatography on a silica gel column (chloroform–methanol, 50
:1) to give a pale yellow solid; yield: 0.820 g, 70%; mp 142 °C; IR KBr ν 3318, 1721, 1605, 1503 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.30 (brs, 1H), 7.70 (d, J = 7.6 Hz, 2H), 7.29–7.19 (m, 5H), 6.92 (d, J = 8.8 Hz, 2H), 6.57 (d, J = 7.1 Hz, 1H), 6.34 (d, J = 2.2 Hz, 1H), 6.27 (m, 1H) 5.05 (s, 2H), 4.24 (t, J = 4.6 Hz, 2H), 3.92 (t, J = 5.16 Hz, 2H); 13C NMR (50 MHz, DMSO-d6) δ 171.4, 159.9, 158.8, 154.2, 136.9, 131.5, 130.24, 129.0, 128.1, 127.4, 126.8, 114.3, 113.8, 108.8, 102.8, 68.3, 67.9, 42.8; ESI-MS: (m/z) 390; found 390 [M]+.
(7-Methoxy-3-phenyl-2H-chromen-4-yl)(4-(2-(piperidin-1-yl) ethoxy)phenyl)methanone (compound 36). To a stirred solution of compound 35 (0.420 g, 1 mmol) in dry DMF (10 ml) was added TBAI (6 mg) and piperidine (0.3 ml) and the resulting solution was stirred at 80 °C for 6 h. After completion of the reaction, the reaction mixture was poured into ice water, the solid precipitate was filtered and after vacuum drying it was crystallized from hexane to afford the title compound as a white solid; yield: 0.445 g, 95%; mp 156 °C. IR (KBr) ν 1730, 1615, 1503, cm−1. 1H NMR (300 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 2H), 7.22–7.20 (m, 5H), 6.87–6.82 (m, 3H) 6.53 (d, J = 2.4 Hz, 1H), 6.38 (dd, J = 8.5 Hz, J = 2.4 Hz, 1H), 5.10 (s, 2H), 4.10 (t, J = 6.1 Hz), 3.79 (s, 3H), 2.76 (t, J = 6.0 Hz, 2H), 2.50 (t, J = 4.59, Hz 4H), 1.46–1.45 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 174.2, 161.1, 160.9, 154.6, 136.8, 132.5, 130.1, 129.5, 127.7, 127.19, 126.9, 115.5, 114.4, 107.6, 101.8, 69.2, 65.9, 57.7, 55.38, 55.0, 25.8, 24.1; ESI-MS: (m/z) 469; found 469 [M]+; HRMS-ESI: [M]+ for C30H31NO4 calcd 469.2253; found 469.2477%.
(7-Methoxy-3-phenyl-2H-chromen-4-yl)(4-(2-(pyrrolidin-1-yl) ethoxy) phenyl)methanone (compound 37). White solid; yield: 0.409 g, 90%; mp 152 °C; IR (KBr), ν 3142, 2918, 2872, 2271, 1722, 1605, 1503, 1461, 1362, 1254, 1159, 1023, 850, 761 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.78 (d, J = 7.2 Hz, 2H), 7.21–7.19 (m, 5H), 6.86–6.84 (m, 3H) 6.53 (d, J = 2.4 Hz, 1H), 6.38 (dd, J = 8.7 Hz, J = 2.6 Hz, 1H), 5.10 (s, 2H), 4.11 (t, J = 6.0 Hz, 2H), 3.79 (s, 3H), 2.90 (t, J = 5.9 Hz, 2H), 2.62 (m, 4H), 1.86–1.81 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 174.26, 161.19, 160.95, 154.61, 136.84, 130.11, 129.56, 128.42, 127.76, 127.19, 126.81, 115.52, 114.38, 107.65, 101.88, 69.20, 67.10, 55.39, 54.94, 54.71, 23.49; ESI-MS: (m/z) 455; found 455 [M]+; HRMS-ESI: [M]+ for C29H29NO4 calcd 455.2097; found 455.2322%.
(7-Hydroxy-3-phenyl-2H-chromen-4-yl)(4-(2-(piperidin-1-yl) ethoxy)phenyl)methanone (compound 39). White solid; yield: 0.432 g, 95%; mp 168 °C. IR (KBr), ν 3442, 2934, 1718, 1605, cm−1. 1H NMR (300 MHz, CDCl3 + CD3OD) δ 7.75 (d, J = 8.5 Hz, 2H), 7.19 (m, 5H), 6.7–6.69 (m, 3H), 6.41 (d, J = 2.1 Hz, 1H), 6.22 (dd, J = 8.3 Hz, J = 2.1 Hz, 1H), 5.01 (s, 2H), 4.11 (t, J = 5.6 Hz, 2H), 2.80 (t, J = 5.4 Hz, 2H), 2.57 (m, 4H), 1.65–1.63 (m, 4H), 1.48–1.39 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 174.2, 164.4, 161.9, 157.8, 140.1, 135.5, 133.0, 131.67.4, 130.4, 130.2, 130.0, 117.7, 112.4, 106.5, 72.3, 68.5, 60.8, 58.0, 28.5, 27.1; ESI-MS: (m/z) 455; found 455 [M]+, HRMS-ESI: [M]+ for C29H29NO4 calcd 455.2097; found 455.2036%.
(7-Hydroxy-3-phenyl-2H-chromen-4-yl)(4-(2-(pyrrolidin-1-yl) ethoxy)phenyl)methanone (compound 40). White solid; yield: 0.397 g, 90%; mp 160 °C; IR (KBr), ν 3552, 1722, 1596 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.6 Hz, 2H), 7.19 (m, 5H), 6.77 (d, J = 8.7 Hz, 2H) 6.68 (d, J = 8.4 Hz, 1H), 6.39 (d, 2.6 Hz, 1H), 6.19 (dd, J = 8.3 Hz, J = 2.0 Hz, 1H), 5.05 (s, 2H), 4.11 (t, J = 5.4 Hz, 2H), 2.96 (t, J = 5.5 Hz, 2H), 2.78–2.71 (m, 4H), 1.85 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 175.4, 161.2, 158.88, 154.7, 137.0, 131.9, 129.5, 128.0, 127.4, 127.2, 127.1, 126.4, 114.3, 114.2, 108.7, 102.9, 68.8, 66.2, 54.4, 54.2, 22.9; ESI-MS (m/z): 441 found 441 [M]+; HRMS-ESI: [M]+ for C28H27NO4 calcd 441.1940; found 441.2179%.
(7-Methoxy-2,2-dimethyl-3-phenyl-2H-chromen-6-yl)(4-methoxyphenyl)methanone (compound 43). To the ice cooled stirred solution of 7-methoxy-2,2-dimethyl-isoflavene 42 (0.532 g, 2 mmol), and anisoyl chloride (0.341 g, 2 mmol) in dry dichloromethane (10 ml) was added SnCl4 (0.782 g, 3 mmol) slowly. The reaction mixture was stirred with cooling until the starting material was consumed and then cold 2N HCl (10 ml) was added to the reaction mixture. The organic phase was separated and the aqueous phase was further extracted once with dichloromethane (15 ml). The collected organic layers were washed with water (20 ml × 2) and finally with brine (20 ml × 3). The combined organic layer was dried over Na2SO4. After filtration and evaporation, the solid was crystallized from hexane to afford the pure product. White solid; yield: 0.760 g, 95%; mp 126 °C; IR (KBr), ν 1670, 1607, 1496 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.82 (d, J = 8.8 Hz, 2H), 7.35–7.30 (m, 5H), 7.12 (s, 1H) 6.91 (d, J = 8.8 Hz, 2H), 6.52 (s, 1H), 6.28 (s, 1H), 3.87 (s, 3H), 3.73 (s, 3H), 1.58 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 194.5, 163.5, 159.2, 156.2, 140.0, 139.5, 132.4, 131.5, 128.5, 128.4, 128.3, 127.7, 122.3, 121.5, 115.6, 113.5, 100.2, 80.0, 55.9, 55.6, 29.9, 27.4; ESI-MS: (m/z) 400, found; 401 [M + H]+ HRMS-ESI calcd for C26H25O4 [M + H]+ = 401.1753 found 401.1747%.
(4-(2-Chloroethoxy)phenyl)(7-methoxy-2,2-dimethyl-3-phenyl-2H-chromen-6yl)methanone (compound 44). White solid; yield: 0.852 g, 95%; mp 128 °C; IR (KBr), ν 1680, 1630, 1512 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 8.8 Hz, 2H), 7.35–7.31 (m, 5H), 7.14 (s, 1H) 6.94 (d, J = 8.8 Hz, 2H), 6.53 (s, 1H), 6.29 (s, 1H), 4.31 (t, J = 5.8 Hz, 2H), 3.86 (t, J = 5.8 Hz, 2H), 3.87 (s, 3H), 3.74 (s, 3H), 1.59 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 194.5, 163.5, 159.2, 156.2, 140.0, 139.5, 132.4, 131.5, 128.5, 128.4, 128.3, 127.7, 122.3, 121.5, 115.6, 113.5, 100.2, 80.0, 55.9, 55.6, 29.9, 27.4; ESI-MS: (m/z) 448; found 449; [M + H]+.
(7-Methoxy-2,2-dimethyl-3-phenyl-2H-chromen-6-yl)(4-(2-(piperidin-1-yl)ethoxy)phenyl) methanone (compound 45). Compound 45 was prepared from compound 44 (0.448 g, 1 mmol), according to the procedure which was used for the synthesis of compound 36. White solid; yield: 0.473 g, 95%; mp 132 °C; IR (KBr), ν 1643, 1604, 1495 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 8.7 Hz, 2H), 7.35–7.33 (m, 5H), 7.12 (s, 1H), 6.92 (d, J = 8.7 Hz, 2H), 6.52 (s, 1H), 6.29 (s, 1H), 4.18 (t, J = 5.9 Hz, 2H), 3.78 (s, 3H), 2.80 (t, J = 5.9 Hz, 2H), 2.53 (m, 4H), 1.64–1.58 (m, 12H); 13C NMR (50 MHz, CDCl3) δ 194.2, 162.5, 158.9, 156.0, 139.8, 139.2, 132.2, 131.3, 128.2, 127.4, 122.0, 121.3, 115.4, 113.8, 100.0, 9.80, 66.1, 55.7, 55.50, 55.0, 27.2, 25.8, 24.1; ESI-MS: (m/z) 497; found 498 [M + H]+; HRMS-ESI: [M + H]+ for C32H36NO4 calcd 498.2644; found 498.2648%.
(7-Methoxy-2,2-dimethyl-3-phenyl-2H-chromen-6-yl)(4-(2-(pyrrolidin-1-yl) ethoxy)phenyl)methanone (compound 46). Compound 46 was prepared from compound 44 according to the procedure used for the synthesis of compound 36. White solid; 0.445 g, 92%; mp 125 °C; IR (KBr), ν 1642, 1601, 1494 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 2H), 7.35–7.33 (m, 5H), 7.13 (s, 1H) 6.94 (d, J = 8.6 Hz, 2H), 6.52 (s, 1H), 6.29 (s, 1H), 4.19 (t, J = 5.8 Hz, 2H), 3.74 (s, 3H), 2.94 (t, J = 5.7 Hz, 2H), 2.65 (m, 4H), 1.83 (m, 4H), 1.59 (s, 6H) 13C NMR (50 MHz, CDCl3) δ 194.2, 162.5, 158.9, 155.9, 139.8, 139.2, 132.1, 131.4, 128.2, 127.4, 122.0, 121.3, 115.4, 113.8, 100.0, 79.7, 67.2, 55.7, 55.7, 54.8, 54.7, 27.2, 23.4; ESI-MS: (m/z) 483; found 484 [M + H]+; HRMS-ESI: [M + H]+ for C31H34NO4 calcd 484.2488; found 484.2485%.
4.2 Materials and methods
4.3 Anti-osteoporotic activity
Cell culture media and supplements were purchased from Invitrogen (Carlsbad, CA). All fine chemicals were purchased from Sigma-Aldrich (St. Louis, MO). HPLC grade acetonitrile was obtained from Merck India Ltd (Mumbai, India).Acknowledgements
The authors (M. K. H., M. I. A., N. Y., P. K. G., R. S., M. M., P. K.) are thankful for financial assistance in the form of SRF by CSIR, New Delhi, India, (I. F.,V. K., J. G.) UGC India, A. K. G ICMR, New Delhi and PRM thanks to CSIR, New Delhi, for the Grant-in-aid of Emeritus Scientist, Scheme [no. 21(0766)/09/EMR-II]. Furthermore, all are grateful to the Sophisticated Analytical Instrument Facility (SAIF) CDRI, Lucknow, India, for providing spectral analysis/data of pure compounds and the Ministry of Health and Family Welfare for financial assistance. CDRI communication no. 8568.Notes and references
- A. Jemal, F. Bray, M. Melissa, J. Ferlay, E. Ward and D. Forman, Global Cancer statistics, Ca-Cancer J. Clin., 2011, 61, 69 CrossRef PubMed.
- American Cancer Society, Cancer Facts & Figures, 2012, http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941.pdf Search PubMed.
- B. S. Katzenellenbogen, Biol. Reprod., 1996, 54, 287 CrossRef CAS.
- P. S. Danielian, R. White, J. A. Lees and M. G. Parker, EMBO J., 1992, 11, 1025 CAS.
- P. Huang, V. Chandra and F. Rastinejad, Annu. Rev. Physiol., 2010, 72, 247 CrossRef CAS PubMed.
- J. S. Lewis and V. C. Jordan, Mutat. Res., 2005, 591, 247 CrossRef CAS PubMed.
- G. G. Kuiper, B. Carlsson, K. Grandien, E. Enmark, J. Haggblad, S. Nilsson and J. Å. Gustafsson, Endocrinology, 1997, 138, 863 CAS.
- S. Nilsson and J. Å. Gustafsson, Clin. Pharmacol. Ther., 2011, 89, 44 CrossRef CAS PubMed.
- C. Thomas and J. Å. Gustafsson, Nat. Rev. Cancer, 2011, 11, 597 CrossRef CAS PubMed.
- V. Speirs, G. P. Skliris, S. E. Burdall and P. J. Carder, J. Clin. Pathol., 2002, 55, 371 CrossRef CAS.
- E. S. Manas, Z. B. Xu, R. J. Unwalla and W. S. Somers, Structure, 2004, 12, 2197–2207 CrossRef CAS PubMed.
- J. A. Katzenellenbogen, J. Med. Chem., 2011, 54, 5271 CrossRef CAS PubMed.
- S. Nilsson, K. F. Koehler and J. Å. Gustafsson, Nat. Rev. Drug Discovery, 2011, 10, 778 CrossRef CAS PubMed.
- S. Ali, L. Buluwela and R. C. Coombes, Annu. Rev. Med., 2011, 62, 217 CrossRef CAS PubMed.
- (a) V. C. Jordan, J. Med. Chem., 2003, 46, 883 CrossRef CAS PubMed; (b) V. C. Jordan, J. Med. Chem., 2003, 46, 1081 CrossRef CAS PubMed.
- (a) I. Kostova, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 29 CrossRef CAS; (b) D. Egan, E. O'Kennedy, E. Moran, D. Cox, E. Prosser and R. D. Thornes, Drug Metab. Rev., 1990, 22, 503 CrossRef CAS PubMed.
- M. A. Musa, J. S. Cooperwood and M. O. F. Khan, Curr. Med. Chem., 2008, 15, 2664 CrossRef CAS.
- J. R. S. Hoult and M. Paya, Gen. Pharmacol., 1996, 27, 713 CrossRef CAS.
- L. W. L. Woo1, A. Purohit, B. Malini, M. J. Reed and B. V. L. Potter, Chem. Biol., 2000, 7, 773 CrossRef.
- H. Brady, M. Doubleday, L. M. Gayo-Fung, M. Hickman, S. Khammungkhune, A. Kois, S. Lipps, S. Pierce, N. Richard, G. Shevlin, M. K. Sutherland, D. W. Anderson, S. Bhagwat and B. Stein, Mol. Pharmacol., 2002, 61, 562 CrossRef CAS.
- N. S. I. Wind and I. Holen, Int. J. Breast Cancer, 2011, 2011, 967419 CAS.
- J. Liang and Y. Shang, Annu. Rev. Physiol., 2013, 75, 225 CrossRef CAS PubMed.
- D. Yue, T. T. Tuanli and R. C. Larock, J. Org. Chem., 2005, 70, 10292 CrossRef CAS PubMed.
- C. Mousset, O. Provot, A. Hamze, J. Bignon, J. D. Brion and M. Alami, Tetrahedron, 2008, 64, 4287 CrossRef CAS PubMed.
- (a) J. A. Dodge, M. G. Stocksdale, K. J. Fahey and C. D. Jones, J. Org. Chem., 1995, 60, 739 CrossRef CAS; (b) C. J. Lion, D. A. Vasselin, C. H. Schwalbe, C. S. Matthews, M. F. G. Stevensa and A. D. Westwell, Org. Biomol. Chem., 2005, 3, 3996 RSC.
- (a) K. Wähälä and A. Hase, J. Chem. Soc., Perkin Trans. 1, 1991, 3005–3008 RSC; (b) S. Balasubramanian and M. G. Nair, Synth. Commun., 2000, 30, 469 CrossRef CAS.
- K. S. Singh, V. Saibaba, V. Ravikumar, V. Santosh, Rudrawar, P. Daga, C. S. Rao, V. Akhila, P. Hegde and K. Rao, Bioorg. Med. Chem. Lett., 2004, 12, 1881 CrossRef PubMed.
- R. Gandhidasan, S. Neelkantan and P. V. Raman, Synthesis, 1982, 1110 CrossRef CAS.
- S. D. Prasad, (A2) WO 2009078029, 2009.
- A. M. Brzozowski, A. C. W. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, L. E. O. Ohman, G. L. Greene, J.-Å. Gustafsson and M. Carlquist, Nature, 1997, 389, 753 CrossRef CAS PubMed.
- A. C. W. Pike, A. M. Brzozowski, R. E. Hubbard, T. Bonn, A. G. Thorsell, J. E. O. Ljunggren, J. Å. Gustaffson and M. Carlquist, EMBO J., 1999, 17, 4608 CrossRef PubMed.
- Schrödinger, version 9.1, Schrödinger, LLC, New York, 2005 Search PubMed.
- B. S. Katzenellenbogen, Biol. Reprod., 1996, 54, 287 CrossRef CAS.
- P. S. Danielian, R. White, J. A. Lees and M. G. Parker, EMBO J., 1992, 11, 1025 CAS.
- M. Beato and A. Sanchez-Pacheco, Endocr. Rev., 1996, 17, 587 CAS.
- E. A. Ariazi, J. L. Ariazi, F. Cordera and V. C. Jordan, Curr. Top. Med. Chem., 2006, 6, 181 CrossRef CAS.
- J. L. Plouffe and C. M. Facoq, J. Soc. Gynecol. Invest., 2000, 7, S38–S46 CrossRef.
- GOLD, Version 3.1, Cambridge Crystallographic Data Centre, Cambridge, UK Search PubMed.
- I. Fatima, V. Chandra, R. Saxena, M. Manohar, Y. Sanghani, K. Hajela, M. P. S. Negi, P. L. Sankhwar, S. K. Jain and A. Dwivedi, Mol. Cell. Endocrinol., 2012, 348, 198 CrossRef CAS PubMed.
- C. S. Blesson, S. Awasthi, G. Kharkwal, A. Daverey and A. Dwivedi, Steroids, 2006, 71, 993 CrossRef CAS PubMed.
- R. Saxena, I. Fatima, V. Chandra, C. S. Blesson, G. Kharkwal, M. K. Hussain, K. Hajela, B. G. Roy and A. Dwivedi, Steroids, 2013, 78, 1071 CrossRef CAS PubMed.
- K. V. Sashidhara, M. Kumar, V. Khedgikar, P. Kushwaha, R. K. Modukuri, A. Kumar, J. Gautam, D. Singh, B. Sridhar and R. Trivedi, J. Med. Chem., 2013, 56, 109 CrossRef CAS PubMed.
- G. L. Wong and D. V. Cohn, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3167 CrossRef CAS.
- J. A. Siddiqui, G. Swarnkara, K. Sharana, B. Chakravartia, G. Sharmaa, P. Rawatb, M. Kumarb, F. M. Khan, D. Pierrozc, R. Maurya and N. Chattopadhyaya, Mol. Cell. Endocrinol., 2010, 323, 256 CrossRef CAS PubMed.
- (a) C. A. Gregory, W. G. Gunn, A. Peister and D. J. Prockop, Anal. Biochem., 2004, 329, 77 CrossRef CAS PubMed; (b) J. Gautam, P. Kushwaha, G. Swarnkar, V. Khedgikar, G. K. Nagar, D. Singh, V. Singh, M. Jain, M. Barthwal and R. Trivedi, Phytomedicine, 2012, 19, 1134 CrossRef CAS PubMed; (c) V. Khedgikar, J. Gautam, P. Kushwaha, A. Kumar, G. K. Nagar, P. Dixit, R. Chillara, Voruganti, S. Singh, Wahajuddin, R. Maurya, N. Chattopadhyay and R. Trivedi, Menopause, 2012, 19, 1336 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray Crystallographic data of compound 18, spectroscopic data, copies of 1H and 13C NMR spectra of compounds and 2D binding pose view of ligands at the binding site of ER-α and ER-β. CCDC 962314. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra45749d |
| This journal is © The Royal Society of Chemistry 2014 |