 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A divalent protecting group for benzoxaboroles†
Brett VanVeller‡b, Matthew R. Aronoff‡a and Ronald T. Raines*ab
aDepartment of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706-1322, USA. E-mail: rtraines@wisc.edu; Fax: +1 608 890 2583; Tel: +1 608 262 8588
bDepartment of Biochemistry, University of Wisconsin–Madison, 433 Babcock Drive, Madison, WI 53706-1544, USA
First published on 16th September 2013
Abstract
1-Dimethylamino-8-methylaminonaphthalene is put forth as a protecting group for benzoxaboroles. The ensuing complex is fluorescent, charge-neutral, highly stable under basic conditions, stable to anhydrous acid, and readily cleavable in aqueous acid to return the free benzoxaborole.
Oxaborole heterocycles are boronic acids that are receiving much attention for applications in drug discovery,1 synthetic methodology,2 molecular recognition,3 and biotechnology.4 Benzoxaborole (1; Fig. 1),5 which is characterized by a phenyl ring fused to a five-membered oxaborole, is the most widely employed oxaborole pharmacophore in medicinal chemistry.1 Compared to phenylboronic acid, the annulated benzylic alcohol in 1 confers high stability,6 low pKa,7 and superior polyol-binding (1 → 2) under physiological conditions (i.e., water near neutral pH),3a,b,f,8 favorable attributes for applications that entail binding to carbohydrates.3,4
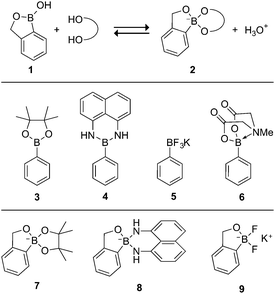 | ||
| Fig. 1 Structure of free benzoxaborole (1), its complexation with a polyol (2), and other general structures of protected boronic acids (3–6) and their unsuitable complexes (7–9) with 1. | ||
The vacant p-orbital on boron—essential for complexation with polyols—can confound multistep synthetic routes and the purification/isolation of derivatives. This issue is compounded further by the commercial availability of countless elaborately functionalized boronic acids but only a small number of simple benzoxaboroles. Our interest in benzoxaboroles for the cellular delivery of chemotherapeutic agents4a and other pharmacological applications drove us to develop a protecting group for 1 that would enhance its synthetic utility.
A variety of successful protecting group strategies have been developed to modulate the undesired reactivity of boronic acids. A common example is a pinacol ester (3), which sterically shields the p-orbital from reaction. Similarly, Suginome and coworkers demonstrated the reduced reactivity of boronic acids in a complex with 1,8-diaminonaphthalene (4).9 An often-employed strategy developed by Molander and coworkers capitalizes on the fluoro-affinity of boron to form a trifluoroboronate salt (5).10 While highly stable, these salts are incompatible with chromatography, limiting their utility in multistep synthetic routes. Burke and coworkers have popularized a trivalent N-methyliminodiacetic acid (MIDA)11 ligand for boronic acids that coordinates the vacant p-orbital with a trialkylamine through a dative bond to give a charge-neutral complex (6).12 This complex is broadly compatible with synthetic reagents and chromatographic purification.13 The trivalent MIDA group is not, however, suitable for protecting 1, which can coordinate to only two ligands. Divalent protecting groups like 3 and 4 are also not appropriate for 1 because they would lead to anionic complexes (7 and 8). Likewise, fluoride protection would yield an anionic difluoroborate salt (9).
We sought to apply the principle of charge neutrality, as demonstrated by the efficacious MIDA protecting group, to the divalent protection of benzoxaboroles. Inspired by the strong complexes that simple boronic acids form with 1,8-diaminonaphthalene (4),9 we devised a protecting group based on 1,8-bis(dimethylamino)naphthalene14 (Proton-sponge®) to generate charge-neutral complexes with 1 that maintain a Lewis basic site to promote acid-mediated cleavage (Scheme 1). We found that complexes 11a–e were fluorescent, stable to basic conditions, moderately stable to anhydrous acid, and readily deprotected under aqueous acidic conditions.
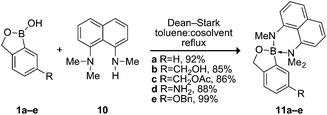 | ||
| Scheme 1 Protection of benzoxaborole derivatives. | ||
Following a previous report,15 1,8-bis(dimethylamino)naphthalene was mono-demethylated to give 1-dimethylamino-8-methylaminonaphthalene (10) in one step of >90% yield without chromatography. After azeotropic removal of water, 1a–e and 10 readily formed complexes 11a–e (Scheme 1). Excess 10 was necessary for high yields, but unreacted 10 was re-isolated quantitatively by chromatography (Fig. 2B, inset). Formation of the complex generated a tetrahedral boron center (1c → 11c11B NMR δ 33.2 sp2 → δ 9.1 sp3)11 that was stereogenic—the benzylic protons of 1 became nonequivalent (Fig. 2A) and served as signature of complex formation. The complex was characterized with X-ray crystallography (Fig. 2C).
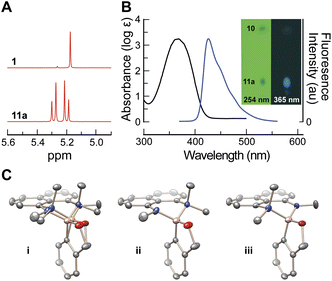 | ||
| Fig. 2 Characterization of complex 11a. (A) 1H NMR spectra of 1a and 11a showing the splitting of benzylic proton signals in 1 into two diasteriotopic doublets. (B) Absorbance (black) and fluorescence (blue) spectra of 11a in CHCl3. Inset: silica gel thin-layer chromatography plate (0.5% v/v MeOH in DCM, Rf = 0.7 separation between 10 and 11a) illuminated under short (254 nm) and long (365 nm) wavelength light. (C) X-ray crystal structure of racemic 11a with anisotropic thermal ellipsoids set at 50% probability and hydrogen atoms omitted. (i) 11a showing both components of positional disorder. (ii) and (iii) Disorder components (enantiomers) separated for clarity. | ||
Next, we investigated the generality of our protecting group design. First, we investigated a small library of compounds similar to 10 for protection of 1a (see ESI†). Only 10, however, led to complexes that were stable during chromatography. α-Amino acids (which resemble divalent versions of MIDA) are known to form stable complexes with dialkyl boranes (i.e., borinic acids).16 We found these complexes to be too fragile for effective protection of 1. From these data, we conclude that the stability of complexes 11a–e stems from both the rigid structure imposed by the diaminonaphthalene ring and the strongly donating nitrogen ligands. The ease of purification also relies on coordinating the vacant p-orbital through a dative bond supplied by the dimethyl amino ligand to create a charge-neutral complex. Finally, we note that 11a–e are highly fluorescent (11a, ΦF = 0.45, Fig. 2B and inset) allowing for easy tracking of product derivatives (11–18) during multi-step synthesis and purification using a standard long-wave (365 nm) bench-top lamp.17 Complex 11a was subjected to a screen of conditions to determine its stability (Table 1, representative time points, see ESI† for kinetic traces and further discussion). In general, 11a is deprotected readily under aqueous acidic conditions (entries 1–3), but exhibits modest (i.e., kinetic) stability under anhydrous acid (entries 4–6) (Table 1). This sensitivity to acid affirms an aspect of our design—protonation of the methylamino group leads to weaker binding and allows for hydrolytic cleavage. In contrast, 11a is highly stable under basic conditions (entries 7–10). This reactivity indicates that protecting group 10 is well suited for solid-phase peptide synthesis,3c in which amino acids are coupled under basic conditions and subsequently cleaved from a solid-support using acid. Finally, the complex tolerated strong reducing reagents, such as LiAlH4 (entry 11). We do note that oxidative conditions were not compatible with 11b and lead to complex mixtures. This sensitivity is not surprising due to the electron-rich nature of the diaminonaphthalene moiety, which is prone to oxidation.18
| Entry | Conditions | % Cleaved | Time (h) |
|---|---|---|---|
| a Average of two experiments at 0.1 M 11a with 4-bromo-2,6-dimethylaniline as an internal standard.b Analyzed with LC-MS.c Analyzed with 1H NMR spectroscopy. | |||
| 1 | 0.5 M HCl in (THF–H2O, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)b :1)b | 94 | 4 |
| 2 | 0.5 M TFA in (THF–H2O, 1:1)b | 99 | 4 |
| 3 | 0.5 M AcOH in (THF–H2O, 1:1)b | 90 | 4 |
| 4 | 0.5 M TFA in DCMb | 82 | 4 |
| 5 | 0.5 M BF3OEt2 in DCMb | 43 | 4 |
| 6 | 4 M HCl in dioxaneb | 30 | 4 |
| 7 | 0.1 M pH = 7 PBS in THF (1:1)c | 0 | 36 |
| 8 | 0.5 M NaOH in (THF–H2O, 1:1)c | 0 | 72 |
| 9 | 20% v/v piperidinec in CH2Cl2 | 0 | 72 |
| 10 | 20% v/v DIEAc in CH2Cl2 | 0 | 72 |
| 11 | 0.5 M LiAlH4 in THFb,c | 0 | 2 |
To explore further the stability of the protecting group, we evaluated 11b, 11d, and 11e under a series of synthetic transformations (Scheme 2 and 3, see ESI† for 11b). As expected, the complex was able to tolerate reducing conditions such as H2/Pd (11e → 12). Conversion of 12 to triflate 13 and subsequent Suzuki–Miyaura coupling using an XPhos palladacyclic precatalyst,19 provided 14 in high yield. Notably, 10 prevented reaction at the benzoxaborole center.20 Recent data suggest that Suzuki–Miyaura reactions necessitate a vacant boron p-orbital,21 validating our protecting group design.22
The base stability of the complex allowed for Buchwald–Hartwig C–N cross-coupling of the protected boron center to give 15 from 11d.23 As noted above, protecting group 10 is compatible with organic amine bases associated with peptide bond formation reagents to provide 16 in high yield (94%). In comparison, a similar peptide coupling reaction attempted with unprotected 1d provided only a 23% yield of the amide 18. Notably, in both the Pd-catalyzed and peptide-coupling reactions, the methylamino groups in 11d are unreactive. Finally, evincing the utility of the protecting group under anhydrous acid, selective deprotection of the amino group in 16 with HCl in dioxane gave 17 in good yield (78%), while the benzoxaborole moiety could be deprotected selectively with aqueous acetic acid to return 18 (Scheme 2).
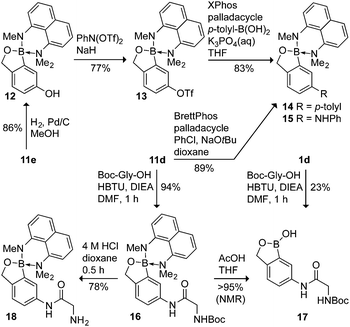 | ||
| Scheme 2 Synthetic evaluation of protected benzoxaborole derivatives; yields are isolated but not optimized. | ||
In conclusion, benzoxaborole 1 has become a privileged entity in medicinal chemistry and for carbohydrate recognition. Its continued development will rely on the efficiency of its derivatization. We have demonstrated the stability and utility of 1 and other benzoxaboroles when protected with 10. Protecting group 10 occupies the vacant p-orbital on boron while creating charge neutrality in the final product. Its complexes are formed readily and are compatible with synthetic reagents and separation/purification techniques employed commonly in multistep syntheses to enable or improve the efficiency of manipulating benzoxoborole.
Acknowledgements
We are grateful to Dr Ilia Guzei for X-ray diffraction analysis of complex 11a. B.V. was supported by postdoctoral fellowship 289613 (CIHR). This work was supported by grant R01 GM044783 (NIH), and made use of the National Magnetic Resonance Facility at Madison, which is supported by grants P41 RR002301 and P41 GM066326 (NIH).Notes and references
- (a) F. L. Rock, W. Mao, A. Yaremchuk, M. Tukalo, T. Crépin, H. Zhou, Y. K. Zhang, V. Hernandez, T. Akama, S. J. Baker, J. J. Plattner, L. Shapiro, S. A. Martinis, S. J. Benkovic, S. Cusack and M. R. K. Alley, Science, 2007, 316, 1759–1761 CrossRef CAS PubMed; (b) S. J. Baker, C. Z. Ding, T. Akama, Y. K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed; (c) T. Akama, S. J. Baker, Y. K. Zhang, V. Hernandez, H. Zhou, V. Sanders, Y. Freund, R. Kimura, K. R. Maples and J. J. Plattner, Bioorg. Med. Chem. Lett., 2009, 19, 2129–2132 CrossRef CAS PubMed; (d) D. Ding, Y. Zhao, Q. Meng, D. Xie, B. Nare, D. Chen, C. J. Bacchi, N. Yarlett, Y.-K. Zhang, V. Hernandez, Y. Xia, Y. Freund, M. Abdulla, K.-H. Ang, J. Ratnam, J. H. McKerrow, R. T. Jacobs, H. Zhou and J. J. Plattner, ACS Med. Chem. Lett., 2010, 1, 165–169 CrossRef CAS; (e) X. Li, Y. K. Zhang, Y. Liu, C. Z. Ding, Q. Li, Y. Zhou, J. J. Plattner, S. J. Baker, X. Qian, D. Fan, L. Liao, Z. J. Ni, G. V. White, J. E. Mordaunt, L. X. Lazarides, M. J. Slater, R. L. Jarvest, P. Thommes, M. Ellis, C. M. Edge, J. A. Hubbard, D. Somers, P. Rowland, P. Nassau, B. McDowell, T. J. Skarzynski, W. M. Kazmierski, R. M. Grimes, L. L. Wright, G. K. Smith, W. Zou, J. Wright and L. E. Pennicott, Bioorg. Med. Chem. Lett., 2010, 20, 3550–3556 CrossRef CAS PubMed; (f) D. Obrecht, F. Bernardini, G. Dale and K. Dembowsky, in Annual Reports in Medicinal Chemistry, ed. E. M. John, Academic Press, 2011, vol. 46, pp. 245–262 Search PubMed; (g) D. Ding, Q. Meng, G. Gao, Y. Zhao, Q. Wang, B. Nare, R. Jacobs, F. Rock, M. R. K. Alley, J. J. Plattner, G. Chen, D. Li and H. Zhou, J. Med. Chem., 2011, 54, 1276–1287 CrossRef CAS PubMed; (h) Y. Xia, K. Cao, Y. Zhou, M. R. K. Alley, F. Rock, M. Mohan, M. Meewan, S. J. Baker, S. Lux, C. Z. Ding, G. Jia, M. Kully and J. J. Plattner, Bioorg. Med. Chem. Lett., 2011, 21, 2533–2536 CrossRef CAS PubMed; (i) S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC; (j) Z. Qiao, Q. Wang, F. Zhang, Z. Wang, T. Bowling, B. Nare, R. T. Jacobs, J. Zhang, D. Ding, Y. Liu and H. Zhou, J. Med. Chem., 2012, 55, 3553–3557 CrossRef CAS PubMed.
- D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432–8435 CrossRef CAS PubMed.
- (a) M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226–4227 CrossRef CAS PubMed; (b) M. Bérubé, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471–6479 CrossRef PubMed; (c) A. Pal, M. Bérubé and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 1492–1495 CrossRef CAS PubMed; (d) S. Schumacher, M. Katterle, C. Hettrich, B.-R. Paulke, A. Pal, D. G. Hall, F. W. Scheller and N. Gajovic-Eichelmann, Chem. Sens., 2011, 1, 1–7 Search PubMed; (e) S. Schumacher, M. Katterle, C. Hettrich, B. R. Paulke, D. G. Hall, F. W. Scheller and N. Gajovic-Eichelmann, J. Mol. Recognit., 2011, 24, 953–959 CrossRef CAS PubMed; (f) A. Adamczyk-Woźniak, K. M. Borys, I. D. Madura, A. Pawelko, E. Tomecka and K. Żukowski, New J. Chem., 2013, 37, 188–194 RSC.
- (a) G. A. Ellis, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 3631–3634 CrossRef CAS PubMed; (b) H. Kim, Y. J. Kang, S. Kang and K. T. Kim, J. Am. Chem. Soc., 2012, 134, 4030–4033 CrossRef CAS PubMed; (c) H. Li, H. Wang, Y. Liu and Z. Liu, Chem. Commun., 2012, 48, 4115–4117 RSC; (d) Y. Kotsuchibashi, R. V. C. Agustin, J. Y. Lu, D. G. Hall and R. Narain, ACS Macro Lett., 2013, 2, 260–264 CrossRef CAS; (e) S. Biswas, K. Kinbara, T. Niwa, H. Taguchi, N. Ishii, S. Watanabe, K. Miyata, K. Kataoka and T. Aida, Nat. Chem., 2013, 5, 613–620 CrossRef CAS PubMed.
- A. Adamczyk-Woźniak, M. K. Cyrański, A. Żubrowska and A. Sporzyński, J. Organomet. Chem., 2009, 694, 3533–3541 CrossRef PubMed.
- H. R. Snyder, A. J. Reedy and W. J. Lennarz, J. Am. Chem. Soc., 1958, 80, 835–838 CrossRef CAS.
- J. W. Tomsho, A. Pal, D. G. Hall and S. J. Benkovic, ACS Med. Chem. Lett., 2011, 3, 48–52 CrossRef.
- J. W. Tomsho and S. J. Benkovic, J. Org. Chem., 2012, 77, 11200–11209 CrossRef CAS PubMed.
- (a) H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS PubMed; (b) H. Noguchi, T. Shioda, C. M. Chou and M. Suginome, Org. Lett., 2008, 10, 377–380 CrossRef CAS PubMed.
- (a) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed; (b) G. A. Molander and L. Jean-Gérard, in Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 507–550 Search PubMed.
- T. Mancilla, R. Contreras and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 1–6 CrossRef CAS.
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed; (b) E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17–27 CAS; (c) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed.
- R. W. Alder, P. S. Bowman, W. R. S. Steele and D. R. Winterman, Chem. Commun., 1968, 723–724 RSC.
- V. A. Ozeryanskii, A. F. Pozharskii, M. G. Koroleva, D. A. Shevchuk, O. N. Kazheva, A. N. Chekhlov, G. V. Shilov and O. A. Dyachenko, Tetrahedron, 2005, 61, 4221–4232 CrossRef CAS PubMed.
- W. H. Dent III, W. R. Erickson, S. C. Fields, M. H. Parker and E. G. Tromiczak, Org. Lett., 2002, 4, 1249–1251 CrossRef PubMed.
- Protonated proton sponge is known to be highly fluorescent, See: A. Szemik-Hojniak, W. Rettig and I. Deperasińka, Chem. Phys. Lett., 2001, 343, 404–412 CrossRef CAS , and references therein.
- (a) X. G. Li, M. R. Huang and S. X. Li, Acta Mater., 2004, 52, 5363 CrossRef CAS PubMed; (b) R. Davis and N. Tamaoki, Org. Lett., 2005, 7, 1461–1464 CrossRef CAS PubMed.
- T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073–14075 CrossRef CAS PubMed.
- (a) Y. Yamamoto, J. Ishii, H. Nishiyama and K. Itoh, J. Am. Chem. Soc., 2005, 127, 9625–9631 CrossRef CAS PubMed; (b) D. S. Gunasekera, D. J. Gerold, N. S. Aalderks, J. S. Chandra, C. A. Maanu, P. Kiprof, V. V. Zhdankin and M. V. R. Reddy, Tetrahedron, 2007, 63, 9401–9405 CrossRef CAS PubMed.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed , and references therein.
- Attenuating the reactivity of boronic acids by rehybridization of the boron center from sp2 to sp3 using a dative nitrogen ligand was demonstrated initially by Burke and coworkers (ref. 12a).
- (a) M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6686–6687 CrossRef CAS PubMed; (b) B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and kinetic data for Table 1. CCDC 949879. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra44385j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |