 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural analysis of glycoprotein sialylation – part II: LC-MS based detection
Giuseppe Palmisano*ab, Martin R. Larsena, Nicolle H. Packerc and Morten Thaysen-Andersenc
aDepartment of Biochemistry and Molecular Biology, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark. E-mail: giuseppe@bmb.sdu.dk; Fax: +45 6550 2467; Tel: +45 6550 2342
bInstitute of Biomedical Sciences, Department of Parasitology – USP, São Paulo, Brazil
cDepartment of Chemistry and Biomolecular Sciences, Macquarie University, Sydney, Australia
First published on 16th September 2013
Abstract
Structural characterisation of sialylated glycoproteins in time and space is a requirement for further understanding of their involvement in biology. In this second of two related reviews, our focus is on the liquid chromatography (LC) and mass spectrometry (MS) based techniques, which have become the golden analytical tools in recent decades for the analysis of N- and O-linked glycoprotein sialylation. Analytical strategies for the analysis of N- and O-linked sialoglycoproteins and pre-LC-MS aspects including enrichment, derivatisation and metabolic labelling techniques were covered in the first review. Acknowledging that glycoprotein sialylation can be studied on multiple analyte levels, LC-MS detection of sialoglycans, sialoglycopeptides and intact sialoglycoproteins are separatedly discussed. All levels have benefitted from continuous improvements of LC-MS technologies, which have gradually pushed the boundaries for separation and detection capabilities to finally allow characterisation of sialoglycoproteins more directly from biological samples. Although still not achieved from a single analysis, LC-MS facilitates the characterisation of many aspects of the sialoglycoprotein structure including the identification and quantitation of the protein carrier, the underlying glycan structure and the sialyl linkage as well as the sialic acid speciation and its further modifications. It is evident from the body of literature that the analytical glycoscientist now has a much improved, though not yet fully mature, toolbox for the analysis of glycoprotein sialylation. This capacity enables structural based investigations of the functional relevance of sialoglycoproteins. Understanding the chemistry and biology of the conjugated sialic acids is essential in order to interpret the complex protein glycosylation code.
 Giuseppe Palmisano | Dr Giuseppe Palmisano is currently appointed professor at the Department of Parasitology, University of Sao Paulo, Brazil. He obtained his Ph.D. degree in mitochondrial physiopathology in 2008 from University of Bari under the supervision of Prof. Sergio Papa and then completed a postdoctoral fellowship at the University of Southern Denmark with Prof. Martin Larsen, developing methods for PTMs analysis in different biological samples. He obtained the Young Investigator Award at HUPO 2011 in Geneva. His current area of research is proteomics applied to cancer metastasis and parasite infections with a particular attention on regulatory mechanisms of PTMs-mediated cellular signals. |
 Martin R. Larsen | Prof. Martin R. Larsen is currently professor in clinical proteomics at the University of Southern Denmark and head of the Centre for Clinical Proteomics in Odense, Denmark. He obtained his Ph.D. degree in 2000 from University of Southern Denmark and then completed a postdoctoral fellowship at the APAF at Macquarie University in Sydney, Australia. In 2009 he obtained the Lundbeck Junior Group Leader Fellowship. His area of research is proteomics applied to biomedical and biological systems. He has developed several methods for the mass spectrometric characterization of phosphorylated proteins and glycosylated proteins, which are used by researchers worldwide. |
 Nicolle H. Packer | Prof. Nicki Packer was part of the team that established the Australian Proteome Analysis Facility (APAF) and co-founded Proteome Systems Limited, an Australian biotechnology company in which her group developed a platform of glycoanalytical technology and informatics tools. She is now Professor of Glycoproteomics, Director of the MQ Biomolecular Frontiers Research Centre and Consultant to APAF at Macquarie University, Sydney, Australia. She has gained an international and national profile by linking glycomics (the glycosylation profile of the expressed genome) with the proteomics and genomics approaches to biological functional research. |
 Morten Thaysen-Andersen | Dr Morten Thaysen-Andersen currently holds a three-year ARC Super Science Research Fellowship position at Macquarie University, Sydney, Australia. He obtained his Ph.D degree in glycoproteomics in 2009 from University of Southern Denmark, and then completed a two-year post doctoral fellowship in glycobiology at Macquarie University with Prof. Nicolle H. Packer funded by the Danish Research Agency. His current research is conducted in the area of structural glycobiology with a strong focus on understanding how human N- and O-linked protein glycosylation affects glycoprotein function in a variety of biological systems, including hormone binding/delivery and pathogenic-host interactions in the context of cystic fibrosis. |
1. Introduction
Mass spectrometry (MS) based molecular mass determination in structural and dynamical biology is sensitive, accurate, fast and robust, and has the ability to be coupled to a range of separation techniques.1 These properties have made MS the analytical detection tool of choice to structurally analyse the multitude of biomolecules encountered in nature including sialylated N- and O-linked glycoproteins,2 which are the focus of these two related reviews. The general structures and diverse biological functions of the most common mammalian sialic acids (i.e. N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc)) were introduced in Part I of the two reviews.3 The importance of characterising the structures of sialic acids and their conjugates in order to increase our understanding of their chemistry and biology and to take further steps towards being able to interpret the complex protein glycosylation code were stressed.Modern day mass spectrometers are capable of handling the large mass range spanning the spectrum from the relative low molecular mass of released sialoglycans (typically 0.5–3 kDa) over proteolytically generated sialoglycopeptides (typically 2–10 kDa) to intact sialoglycoproteins (typically >10 kDa) with high resolution, sensitivity and mass accuracy, in particular in the lower m/z end of the spectrum.4 In addition, free sialic acids (e.g. Neu5Ac or Neu5Gc) and their modified counterparts,5 sialic acid sugar nucleotides (e.g. CMP–Neu5Ac or CMP–Neu5Gc)6 and free (non-protein linked) sialoglycans7 can also be detected by MS, but will not be covered here as we will focus strictly on the analysis of the sialic acids carried by N- and O-linked glycoproteins. Although the linear repeats of sialic acid residues, termed oligo- and poly-sialic acids, can be carried by N-glycoproteins, this review will only briefly touch on these structures since very limited literature is available regarding their analysis.8,9
Depending on the purity of the sialylated glycoconjugate(s) of interest it is often desirable to separate the analytes prior to MS detection to avoid ion suppression and to maximise the number of molecular precursors that can be fragmented by tandem MS in order to facilitate further structural characterisation and confirmation. This is particularly useful for sialylated glycoconjugates where multiple isobaric molecules may be hidden under an identical molecular mass. Although other techniques are showing promising potential for the separation of sialylated compounds including capillary electrophoresis (CE) and ion mobility spectrometry (IMS) as discussed briefly in this review, separation is most commonly performed using a variety of liquid chromatography (LC) techniques. On-line LC separation, where the sialylated analytes are eluted directly from the LC column into the hyphenated mass spectrometer, is preferred when detection is performed using electrospray ionisation (ESI) MS. In contrast, different types of off-line LC separation techniques are usually favoured, or even avoided, when using matrix assisted laser desorption ionisation (MALDI) MS detection. Although MALDI MS has certain advantages including higher speed of data acquisition and ease of data interpretation, ESI MS is the most used detection technique in glycosicence due to the potential for LC hyphenation.10,11 The LC method should be carefully chosen by considering the physicochemical or structural properties of the sialylated analytes of interest. The negative charge of sialoglycoconjugates is naturally a useful molecular feature that can be used to separate asialo and sialo-compounds as well as differently sialylated species. Loss of sialic acids from the sialylated analyte during LC is usually not a problem even when the chromatography is carried out in very acidic solvents, however, it is recommended that excessive column heating is avoided and that acidic solvents are evaporated if the sialylated analytes are stored for an extended period post separation.12–14 The conditions applied during sample preparation are also very important to consider in order to keep the sialoglycoconjugates intact and avoid undesired derivatisation.3
Following on from the first of these two related reviews, which described the analytical strategies and pre-LC-MS tools for the analysis of glycoprotein sialylation,3 this second part summarises the present LC-MS based techniques in the context of glycoprotein sialylation analysis. Here released sialoglycans and sialoglycopeptides are covered separately due to the dedicated workflows and considerations required for their respective analyses (see overview of topics covered by parts I and II in Fig. 2 (ref. 3)). Relatively little analytical work has been presented for sialoglycoproteins; techniques dealing with these larger analytes are consequently discussed under the sialoglycopeptide section. The techniques and tools for general analysis of protein glycosylation have been reviewed previously.2,15–18 Here we will focus strictly on the sialylated component of the glycome/glycoproteome. It is stressed that the majority of the presented LC-MS based techniques are not only capable of analysing sialylated glycoproteins, but also their neutral counterparts; however, the techniques are irrespectively discussed with a sialic acid centric focus in this review. The bioinfomatics tools for the management and interpretation of LC-MS and tandem MS data and the use of glycosylation databases/repositories will not be covered here. Instead the reader is referred to other recent resources for detailed reviews.15,19–21
2. LC-MS analysis of sialoglycans
2.1 LC separation of sialoglycans
The hydrophilicity and small molecular mass/size are physicochemical features that allow relatively easy isolation of N- or O-linked glycans following release from their protein carriers in biological samples.3 In addition, the mammalian glycome has a somewhat limited number of analytes due to the enzymatic restrictions in the biosynthetic glycosylation machinery.22 Together, this means that the glycan mixtures released from proteins usually have much lower analyte complexities compared to equivalent peptide mixtures encountered in proteomics type experiments. The complexity is naturally reduced even further when the focus is restricted to encompass only the sialylated component of the N- and O-linked glycome. The main challenge in this context is, as such, usually not the number of sialoglycans in the mixture, but the close molecular relatedness that complicates their separation. For example, isobaric sialoglycans varying only in a sialyl linkage or in the location of a terminal sialic acid residue commonly occurs in biological samples. This emphasises the need for utilising LC techniques with high separation power for analytes with such small molecular discrepancies.Native (underivatised) sialoglycans are commonly separated using PGC, which avoids qualitative and quantitative distortions of the profile due to incomplete or biased chemical derivatisation or degradation. However, simple NaBH4 based reduction of the released sialoglycans to the alditol form is needed to yield a single peak for each eluting compound due to the α- and β-anomer formation of non-reduced glycans that separate well on PGC.30 PGC is arguably the LC method with the highest separation power of sialoglycans since its means of separation to resolve for example isobaric isomers is more orthogonal to the subsequent mass separation in the mass spectrometer than other LC separation techniques.17 This is evidenced by the separation by PGC of closely related structural variations of sialoglycans (see Fig. 1 for an example of a PGC separation of N-linked sialoglycans and Table 1 for an overview of LC based separation of sialoglycans and sialoglycopeptides). As such, PGC has the potential to separate: (i) sialoglycans with different monosaccharide compositions e.g. different number of Neu5Ac residues and/or Neu5Ac/Neu5Gc variants, (ii) sialoglycans with the same monosaccharide composition, but with different topologies e.g. Neu5Ac residues on the 3′ or 6′ arm of N-linked sialoglycans, and (iii) sialoglycans with the same monosaccharide composition and topology, but with different sialyl linkage types e.g. α2,3- or α2,6-linked Neu5Ac residues. Sialoglycans having a higher number of sialic acid residues and a higher number of antennas have a greater retention on PGC than less sialylated and less branched sialoglycans. In addition, α2,6-sialylated glycans have been shown to elute well before their α2,3-sialylated counterparts when applying a normal gradient of increasing acetonitrile concentration. As such, the retention order of the sialoglycans can assist in their structural characterisation.38 This is particularly useful for linkage-type isomers where molecular mass and tandem MS fragmentation often fail to confidently differentiate isobaric sialoglycans e.g. those isomers that differ only in the sialyl linkage type. The separation power of PGC-MS was recently demonstrated in a study where nearly 300 mouse serum N-glycan compounds from over 100 distinct monosaccharide compositions were separated.39 Together with MS/MS analysis this revealed a number of novel sialylated N-glycan structures as well as other modifications such as dehydration, O-acetylation and lactylation.
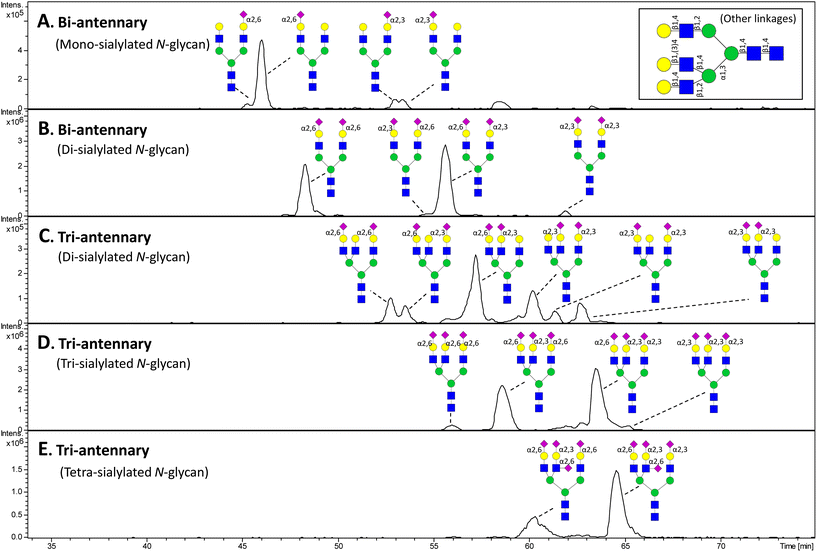 | ||
| Fig. 1 PGC LC-MS separation and detection of sialylated N-linked glycans released from bovine fetuin. The sialoglycans were reduced prior to analysis, but otherwise underivatised. Detection of the separated sialoglycans was performed using negative ion ESI IT MS. Structures have been assigned in the relevant extracted ion chromatograms for five m/z ratios corresponding to the major monosaccharide compositions for the reported N-linked sialoglycans from bovine fetuin.250,251 The high separation power of PGC for sialylated N-glycan isomers is showcased by its ability to separate sialoglycans with variations in their numbers of antennas/sialic acid residues and their linkages/branch points. Increasing number of sialic acid residues and antennas generate a stronger retention on PGC. In addition, glycans containing α2,6-linked sialic acid residues elute significantly before the α2,3-linked counterparts. | ||
| Chromatography | Separation of Neu5Ac composition isomers | Separation of sialo-topology isomers | Separation of sialo-linkage isomers | |
|---|---|---|---|---|
| a Permethylated or labelled glycans.b No general retention rule seem to apply; occasionally Neu5Ac on 3′ arm < Neu5Ac on 6′arm and occasionally Neu5Ac on 3′ arm < Neu5Ac on 6′arm.38c Weak separation of topology and/or linkage isomers was obtained using IP RPC of 2-AB labelled N-glycans.52,58d When IP conditions are used sialylated N-linked glycans containing a higher number of Neu5Ac residues are less retained on RPC.e This retention behaviour applies for ZIC HILIC separated sialoglycopeptides; other HILIC phases may retain sialoglycopeptides differently. | ||||
| Sialoglycans | PGC | Yes (higher # Neu5Ac → more retention) | Yes (structure specific retention)b | Yes (sialyl α2,6 < sialyl α2,3) |
| HILIC | Yes (higher # Neu5Ac → more retention) | Yes (Neu5Ac on 3′ arm < Neu5Ac on 6′ arm) | Yes (sialyl α2,3 < sialyl α2,6) | |
| RPCa | Yes (higher # Neu5Ac → more retention) | Weak separationc | Weak separationc | |
| Sialo-glycopeptides | RPC | Yes (higher # Neu5Ac → more retention)d | Weak (if any) separation | Weak (if any) separation |
| HILICe | Yes (higher # Neu5Ac → less retention)e | Weak (if any) separation | Weak (if any) separation | |
In one of the first glycomics applications using ZIC HILIC, neutral N-glycans labelled at their reducing end with 2-aminopyridine (PA) were well separated.46 Later, ZIC HLIC was demonstrated to be capable of separating 2-aminobenzoic acid (2-AA) derivatised N-linked sialoglycans according to their number of sialic acid residues.47 In addition, the separation of PA derivatised sialylated N-glycans including α2,3-/α2,6-Neu5Ac and β1,3-/β1,4-Gal linkage isomers were convincingly achieved using a ZIC HILIC phase.48 High separation capacity of ZIC HILIC was also illustrated in another study where a two-dimensional on-line LC approach consisting of HILIC and anion-exchange chromatography was sufficient to separate PA-labelled sialylated N-glycan topology isomers (i.e. Neu5Ac on 3′ or 6′ arm).49 Here, ZIC HILIC was shown to have a higher separation power for sialoglycan isomers than a neutral amide based (polar) stationary phase. α2,3-sialylated glycans have been shown to elute slightly before their α2,6-sialylated counterparts,50 but with less separation than on PGC (see Table 1 for overview of sialoglycan retention behavior for a variety of LC methods). New sub-2 μm particles of another neutral HILIC sorbent have improved the peak capacity when operated with ultra-high performance LC (UPLC).51 In a comparative study, HILIC was evaluated to have a higher separation power of labelled sialylated and neutral N-glycans than PGC and reversed phase chromatography (RPC).52 Amide HILIC columns are now available in chip-based formats for sensitive and easy analysis of sialoglycans.53,54
Although not LC based, other techniques can separate sialoglycans. Ion mobility spectrometry (IMS), which separates analytes based on molecular cross-section in the gas phase, is a promising separation technique for analytical glycoscience. Similarly to PGC, the separation using IMS is more orthogonal to the downstream mass based separation in the mass spectrometer compared to, for example HILIC, due to its structure based distinguishing properties. IMS can be utilised alone with MS based detection or in conjunction with other separation techniques.67–69 A few examples demonstrate that IMS is capable of separating sialylated N-glycan isomers including ones with different numbers of sialic acid residues and isobaric 3′ and 6′ arm sialic acid topological isomers.70,71 In addition, capillary electrophoresis (CE), also known as capillary zone electrophoresis, is a powerful separation technique that was shown to separate underivatised N-linked sialoglycans differing in the number of sialic acid residues.72 In other studies, CE combined with laser-induced fluorescence, or with MSn fragmentation in negative ion MS of N-linked glycans labelled with 8-aminopyrene-1,3,6-trisulfonic acid trisodium (APTS) or 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) was used to separate and identify sialylated and non-sialylated N-glycans.37,73 In addition, APTS labelled O-acetylated sialylated N-glycan isomers were recently separated using CE-MS proving that the method has high separation power for modified sialoglycans and is sufficiently gentle to retain the labile acetyl moieties during analysis.74 Finally, fluorophore-assisted carbohydrate electrophoresis is a less used separation technique that nonetheless is capable of separating ANTS labelled N-linked sialoglycans with different number and types of sialic acid residues, whilst it cannot resolve topology isomers of isobaric sialoglycans.75
2.2 MS detection of sialoglycans
The general use of MS techniques in glycomics has been covered elsewhere.18,76,77 Some MS aspects in sialic acid-focused glycomics were recently introduced78 and the reader is referred to this review as an alternative literature resource.The relatively low molecular mass of sialoglycans is well-suited for MS, where the technique generally shows its highest sensitivity (low 10−12 to 10−15 mol), resolution (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), dynamic range (several orders of magnitude) and mass accuracy (low or sub ppm). However, these optimal performance characteristics are heavily instrument dependent and are rarely achieved in a single experiment using a single instrument. The high speed of modern mass spectrometers (i.e. 5–50 tandem mass spectra per second) should be sufficient to cover multiple co-eluting compounds from even the most complex mixtures of sialoglycans; however, sialoglycan mixtures showing a large dynamic range are still challenging to profile and may leave some low-abundant sialylated glycans undetected in the mass spectrometer.
000), dynamic range (several orders of magnitude) and mass accuracy (low or sub ppm). However, these optimal performance characteristics are heavily instrument dependent and are rarely achieved in a single experiment using a single instrument. The high speed of modern mass spectrometers (i.e. 5–50 tandem mass spectra per second) should be sufficient to cover multiple co-eluting compounds from even the most complex mixtures of sialoglycans; however, sialoglycan mixtures showing a large dynamic range are still challenging to profile and may leave some low-abundant sialylated glycans undetected in the mass spectrometer.
2.2.1.a Ionisation. Where neutral glycans afford strong signals as [M + Na]+ ions in positive ion MALDI MS, signals for sialoglycans are usually stronger as [M − H]− in negative ion mode. Sialoglycoconjugates are inherently challenging to analyse in MALDI MS due to the instability of the α-glycosidic sialyl bond yielding potential in-source and post-source fragmentation by the loss of CO2 or the entire sialic acid.87 In addition, significant cation adduction to the negatively charged carboxyl group occurs in positive ion MALDI MS. Taken together, this means that intact sialoglycans are often observed at low signal strength in MALDI MS and that special considerations are required for their analysis. The matrix choice, sample preparation and MS settings are consequently of prime importance to facilitate a successful structural characterisation of sialoglycans using MALDI MS.
2.2.1.b Matrix choice. The common MALDI matrices for protein chemistry and proteomics including 2,5-dihydroxybenzoic acid (DHB), α-cyano-4-hydroxycinnamic acid (CHCA) and 2-(4′-hydroxyphenylazo)benzoic acid have repeatedly been shown to be unsuitable for the MALDI MS based analysis of sialoglycans due to significant losses of sialic acids during ionisation under normal acquisition settings. Instead, cooler matrices with lower energy transfer to the analyte are typically favoured such as 6-aza-2-thiothymine (ATT),88 2,6-dihydroxyacetophenone89 and 2,4,6-trihydroxyacetophenone (THAP) in the presence of low concentrations of ammonium citrate.90 In particular THAP yielded no evident in-source fragmentation of sialoglycans in negative ion MALDI MS with linear mode acquisition while retaining the high sensitivity of the detection technique in the low femtomolar range.88 Another systematic study reported that D-arabinosazone was superior to other matrices in negative ion MALDI MS by yielding higher signal strength and lower salt adduct formation than more common matrices.91 In addition, spermine minimised the adduct formation of sialoglycans when used as a co-matrix with DHB.92 Furthermore, modified CHCA matrices have proved superior for the analysis of sialoglycans: chlorinated CHCA (Cl-CHCA) was shown to minimise desialylation of N- and O-linked sialoglycans by eliminating MS induced in-source and metastable decay in negative ion MALDI MS93 and the 1,1,3,3-tetramethylguanidium (TMG) salt of CHCA generated preferential ionisation of sialoglycans.94
2.2.1.c Instrument settings in MALDI MS. In addition to the in-source (or ‘prompt’) fragmentation, the sialoglycans can also be desialylated post source in the ion transfer or downstream in the mass analyzer, a mechanism termed post-source decay. For example, desialylation occurring post source is normally very visible for MALDI time-of-flight (TOF) MS performed in reflectron mode with the extraction delay functionality enabled.12,40 This can largely be avoided by performing the acquisition in linear mode with limited, or even without, extraction delay of the generated ions, but at a significant expense of signal resolution.95 Another example is the pulsed application of cooling gas in MALDI fourier transform-ion cyclotron resonance (FT-ICR) MS, which together with a low extraction voltage, promises to minimise the loss of sialic acid residues from sialoglycans.40 In comparison, significant losses of sialic acid residues occur in a typical vacuum MALDI source without cooling of the sialoglycan ions.79 Hence, abundant losses of sialic acids seem avoidable by using intermediate pressure MALDI sources where the sialoglycans are allowed to dissipate excess energy prior to the mass analysis. This, in turn, opens up the use of ‘hotter’ and more user-friendly matrices facilitating easier sample preparation and data acquisition for MALDI MS based analysis of sialoglycoconjugates.96–98
2.2.1.d Quantitation. Until recently, quantitative profiling of sialylated and neutral glycans was mostly performed using normal phase chromatography (NPC) based separation and fluorescence detection of glycans labelled at the reducing end e.g. with PA, 2-AB or 2-AA;99,100 but this approach lacks speed and the capacity to resolve some structural configurations such as some fucosylated structures from their non-fucosylated counterpart. Recent advances in NPC, however, have increased its peak capacity and the speed of acquisition.51 Due to the higher speed and sensitivity, MALDI MS based quantitation has been investigated as an alternative to NPC for quantifying sialoglycans. It was found that MALDI MS signal intensities can be used as accurate and reproducible quantitative measures of the relative N-glycan distribution when populations consisting only of sialylated or neutral glycans are analysed separately in their native or derivatised forms, Fig. 2.88,101–104 In contrast, when mixtures of sialylated and non-sialylated glycans are analysed together, significant detection biases for or against sialoglycans are observed depending on the MS polarity mode.2,11,102,103,105,106 This limits the use of MALDI MS for direct accurate quantitation of biological mixtures, which usually comprise sialo and asialo N- and O-linked glycans. To obtain equal ionisation responses for sialylated and neutral glycans it has been suggested to either neutralise the charge on the carboxyl group of the sialic acid residues by derivatisation or to modify the MALDI matrix. For the charge neutralisation, the quantitative detection biases were overcome by simple methyl esterification107–109 or amidation106 (see Part I of the review3 for more information). This permanent charge neutralisation yielded very accurate relative quantitation of sialylated and neutral reducing-end labelled N-glycans when analysed in positive ion MALDI TOF MS in good agreement with quantitation profiles obtained by conventional NPC separation and fluorescence detection.109,110 For the matrix modification, spiking the matrix with trace amounts of either H2SO4 or alkylsulfonates yielded equal in-spectrum ionisation responses in negative ion MALDI MS of neutral glycans (observed as [M + H2SO4]− or [M + RSO3]− ions) and sialoglycans (observed as [M − H]− ions).111,112 Another group introduced a light and heavy tag in the reaction step for the reducing end labelling with 2-AA to yield accurate glycoform quantitation of the glycans between different samples (but not necessarily between the glycans within a given sample) using negative ion MALDI TOF MS.113
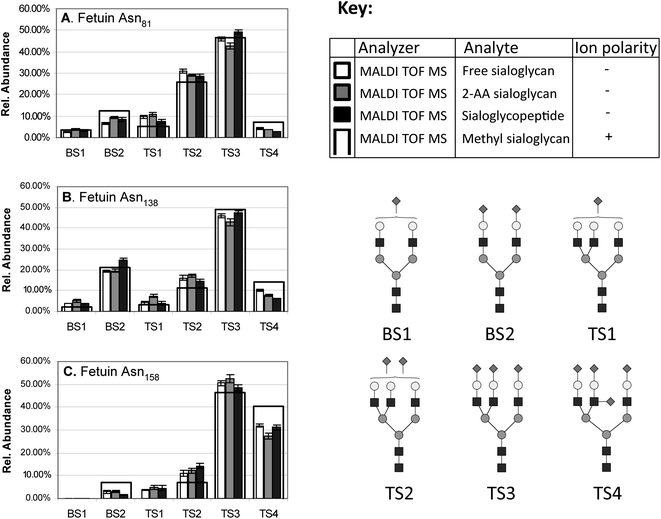 | ||
| Fig. 2 Site-specific relative quantitation of the six sialylated N-linked glycan monosaccharide compositions occupying the three N-glycosylation sites of bovine fetuin i.e. (A) Asn81, (B) Asn138 and (C) Asn158. The relative quantitation is based on MALDI TOF MS signal intensities of free non-reduced sialoglycans, 2-AA labelled sialoglycans and tryptic sialoglycopeptides were all recorded in negative ion linear mode. These profiles are compared to a reference profile of methylated (stabilised) fetuin sialoglycans (black outline, see key for explanation). The figure is modified with permission from Thaysen-Andersen et al. Anal. Chem., 2009, 81, 3933–3943,102 which also contains more information about the specific experiment. | ||
2.2.2.a Ionisation. Non-reduced or reduced native sialoglycans are commonly analysed in negative ion ESI MS because of the higher signal response in this ion polarity in which MALDI MS shows lower performance. On the other hand, permethylated sialoglycans produce stronger signal responses in positive ion ESI MS compared to their underivatised equivalents.114 Compared to MALDI, ESI inherently generates cooler ions and limited ionisation induced fragmentation is consequently observed when gentle desolvation settings of the source are used.17,77 Here deprotonation is the dominating ion formation rather than ionisation by anion adduction. Unlike sialoglycans, neutral glycans usually appear in negative ion ESI MS as the anion adducts when doped with anions such as chloride.115 However, neutral glycans can also be ionised in negative ion mode as [M − H]− ions without anion doping. As described below, another advantage of negative ion ESI MS is that sialylated and neutral native glycans ionise with more similar ionisation efficiencies, which is beneficial for quantitation purposes. In addition, negative ion MS usually gives more informative CID fragmentation spectra when native reduced glycans are analysed by tandem MS. This has paved the way for detailed characterisation of many sialylated glycans derived from both N- and O-linked sialoglycoproteins.116–119 Structural information obtained includes monosaccharide compositions, topologies, branches and linkage types; however, extraction of parts of such structural information is often dependent on the simultaneous use of PGC retention time information and parallel exoglycosidase treatment.120
2.2.2.b Quantitation. The same ionisation biases of neutral and sialylated glycans observed in MALDI may apply for ESI, but such ionisation differences, if any, have been reported to be less prominent in ESI MS.11,34,40,121 Some researchers have actually observed no significant ionisation differences between native sialo- and asialoglycans when analysed in negative ion ESI MS when evaluated quantitatively against the glycan profile obtained using conventional NPC analysis of the same sample.122 In support, signal intensities were found to be an accurate measure of the quantitative amounts of sialylated and neutral reduced N-glycans from human prostate specific antigen when recorded in negative ion ESI MS on an ion trap (IT) MS.123 However, in ESI MS, the sialoglycans are observed with different ion charge state distributions than neutral glycans meaning that all observed charge states need to be considered to obtain accurate quantitation.11 In a systematic study using PGC ESI MS it was concluded that solvent pH has marginal effect on the ionisation efficiency of sialoglycans in positive and negative ion ESI MS, but that the content of the organic solvent strongly influences the signal strength.34 The same researchers concluded that positive ion ESI MS appears preferable when neutral and sialylated glycans are quantified in the same sample. Others have calculated the difference in signal response factors of some sialylated and non-sialylated glycans released from mucins in negative ion ESI MS such that the biased detection of sialoglycans is able to be compensated for.124 The permanent charge neutralisation techniques used for derivatisation of sialoglycans, including methyl esterification, amidation and permethylation as described previously,3 are also compatible with downstream ESI MS based analysis. As an example, permethylation enabled accurate quantitation of mixtures of neutral and sialylated N-glycans derived from specific glycoproteins by MS driven glycoprofiling based on comparison with profiles obtained using NPC analysis of 2-AB labelled N-glycans derived from the same glycoproteins.125 Detection biases for or against sialoglycan ion intensities in ESI and MALDI MS becomes less important when comparative studies are undertaken since any suppression or underrepresentation of each glycan species would occur in both samples. The literature harbours plenty of examples of such comparative studies where the accuracy of the individual relative amounts of each component within a sample is of lower importance than the relative differences between samples. As an example, this was illustrated in a study where the cytokines interleukin-1β and tumor necrosis factor-α were shown to quantitatively induce a shift towards expression of glycoproteins carrying α2,3-linked sialic acid containing glycans in primary human chondrocytes compared to the expression of this linkage type before cytokine treatment.126 Direct quantitative comparison of released sialoglycans from two sample origins may also be quantified using a recently introduced glyco tandem mass tag, which showed accurate quantification over a broad dynamic range.127
2.2.3.a CID for tandem MS and multistage MSn. Collision induced dissociation (CID) is by far the most used fragmentation technique for biomolecules in modern MS. CID is also routinely used to fragment sialylated and neutral N- and O-linked glycans. The general monosaccharide compositions of sialoglycans may be elucidated and confirmed by the presence of abundant B/C- and Y/Z-type ions generated from glycosidic bond cleavages using lower energy CID tandem MS fragmentation which is feasible on various mass spectrometers including triple quadrupoles (Q), IT, Q-TOF and FT-ICR analyzers. Native sialylated glycans tend to produce an abundant B1-ion (e.g. m/z 290 for Neu5Ac and m/z 306 for Neu5Gc containing glycans, respectively in negative ion MS) reducing the abundance of the informative C-type ions.114 Higher energy CID fragmentation, which can be obtained on MALDI TOF-TOF instrumentation yields more cross-ring cleavages such as X-type ions of neutral and sialylated glycans.134 Fragmentation of sialylated and neutral N-glycans labelled with 2-AA generates mostly Y-type ions and some A-type cross-ring cleavages.135 Again it is stressed that the fragmentation pattern and, hence, the structural information obtained by tandem MS varies depending on the charge state and the polarity of the molecular precursor.
Cross-ring cleavages in the forms of An- and Xn-ions are often needed to interpret the fine structure of native sialoglycans. In contrast, some linkage and topology information can be extracted from so-called ‘scars’ generated from glycosidic cleavages of permethylated sialoglycans.76 Such information is based on the appearance of unmodified hydroxyl groups formed during tandem MS of the permethylated sialoglycans indicating which glycosidic bonds have been broken and which hydroxyl groups were originally free. The arm-specific position of sialic acid residues on native multi-antennary N-glycans has been obtained using negative ion ESI tandem MS on Q-TOF mass analyzers.136 For this type of analysis the D-ion, which is defined as the fragment ion formed by the loss of the 3′ antenna together with the N-glycan chitobiose core, is very informative.14 In addition, it has been shown that the two common sialic acid linkages (α2,3- or α2,6-) can be differentiated in positive ion ESI MS using CID fragmentation in combination with transition metal–ligand derivatisation.137 For permethylated sialoglycans, the formation of specific A-type ions in positive ion ESI MS was shown to allow differentiation of α2,3- or α2,6-sialyl linkage isomers.138–140 Cross-ring cleavages that provided valuable linkage information of the structure of permethylated sialoglycans were also observed in positive ion MALDI TOF-TOF MS using higher energy CID fragmentation.141
Negative ion multistage MSn where the molecular precursor is undergoing several rounds of isolation, fragmentation and mass separation/analysis has been shown to be capable of distinguishing closely related PA-labelled N-linked sialoglycans using a linear IT-TOF mass analyzer.142,143 The resulting fragment mass spectra was sufficiently reproducible and unique to enable differentiation of the topological isomers (e.g. specific fragments could be used to distinguish between N-glycans with sialic acid residues on either 3′ or 6′ arm) as well as the sialyl linkage type isomers (e.g. distinguish between structures carrying α2,3- or α2,6-Neu5Ac) by spectral matching. In addition, the C4- and D-type ions in MS3 enhanced the confidence in the assignment. The identification of glycans by MSn spectral matching had been suggested previously.144 The need to perform multistage MSn to structurally analyse sialoglycans by first removing the sialic acid(s) in the first round of fragmentation was stressed in a recent publication.33 Here the on-line MSn analysis was enabled using nano-flow PGC MS with alkaline mobile phases in negative ion mode for the sensitive detection of the sialoglycans. In addition, sialic acid linkage determination on O-linked sialoglycans using negative ion ESI MSn on IT instrumentation was recently reported.145 Furthermore, polarity switching combined with MSn was found to be beneficial for characterising 2-AB labelled sialylated and neutral N-glycans separated by RPC.146 Multistage MSn was also used to fragment and characterise charge neutralised free oligosialic acids using negative ion ESI MS.9 In the same study, MALDI MSn proved capable of characterising the larger polysialic acids. Here, laser induced dissociation produced a range of different fragments including novel cross-ring cleavages (e.g.3,5L fragment ions), more classical ring fragments (e.g.0,2A- and 0,4A-type ions) and common fragment ions generated by glycosidic bond cleavages.9 Different pyrene labels including pyrene butanoic acid hydrazide and 1-pyrenyl diazomethane (PDAM) derivatives have proved useful for the characterisation of sialoglycans in negative ion MALDI MSn by yielding A-, D- and Y-ions.147,148 Taken together, multistage MSn is a powerful approach for deep characterisation of sialoglycans; however, considering the much increased time and sample consumption of each additional tandem MS stage, multistage MSn is probably most useful for abundant sialoglycan structures. Hence, for global glycome profiling purposes it is generally desirable to obtain the structural information in a single stage of tandem MS.77
2.2.3.b Other fragmentation techniques. Fragmentation techniques other than CID have increasingly been used for native and derivatised sialoglycans. Similar to CID, infrared multiphoton dissociation (IRMPD) facilitates dissociation by collisional heating and, hence, mostly generates cleavages of the most labile bonds, the glycosidic bonds.77
Activated electron dissociation (ExD) has recently received some attention in glycomics because cross ring cleavages of the monosaccharides of the glycans are being generated upon activation using ExD. This produces fragment ions which complement the fragments obtained by glycosidic cleavages. However, only limited work has been presented for sialoglycans. In recent studies, electron capture dissociation (ECD)149 and electron transfer dissociation (ETD)77 both produced glycan fragment ions that complemented the ions obtained from CID type fragmentation events. In addition, electron detachment dissociation (EDD) of sialoglycans was shown to be efficient for sialoglycan characterisation by yielding complementary structural information to that obtained from IRMPD by the formation of additional A-type cross-ring cleavages, Fig. 3.150,151 Even better performance of EDD fragmentation of sialoglycans was obtained with chloride anion attachment.152
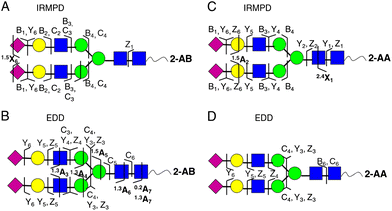 | ||
| Fig. 3 Complementary fragmentation of 2-AA- (A and B) and 2-AB- (C and D) labelled N-linked sialoglycans derived from human transferrin using IRMPD (A and C) and EDD MS/MS (B and D). The resulting fragment ions are indicated on the structures. Figure adapted with permission.151 | ||
Higher-energy C-trap dissociation (HCD) and pulsed-Q-dissociation (PQD) are alternative fragmentation techniques to CID on the LTQ-Orbitrap platform.153,154 The complementarity of HCD and PQD to CID was shown for neutral linear and cyclic glycans,155 but the performance of these fragmentation techniques still needs to be established for sialoglycans.
Post-source decay fragmentation in MALDI TOF MS was also shown to be capable of differentiating α2,3- and α2,6-linked sialic acids on sialyllactose and sialyl-N-acetyllactosamines.156 Finally, laser-induced photofragmentation, which was shown to produce diagnostic ions for specifying the sialyl linkages of N-linked sialoglycans represents another exciting fragmentation technique for native and modified sialoglycans.157
3. LC-MS analysis of sialoglycopeptides
Sialylated glycopeptides, which is usually generated by hydrolysis of sialoglycoproteins by specific or unspecific proteases (see more in Part I of the reviews paragraph 3.2.13), can vary dramatically in size and charge. Where N-linked sialoglycopeptides typically have a single, but larger, N-linked sialoglycan per proteolytically generated peptide, mucin type O-linked sialoglycopeptides may contain multiple shorter O-sialoglycans in clusters inaccessible to proteases. To date, a universal workflow capable of analysing the full range of sialylated glycopeptides that can be encountered in nature remains undescribed. Custom made experimental designs tailored to the specific research question being investigated are consequently still required in glycoproteomics to yield information-rich data. This means that relative few glycoproteins has been site-specifically glycoprofiled (list of profiled mammalian N-glycoproteins can be found in supplementary data in a recent publication158). In contrast to released sialoglycans, the negative charge provided by the sialic acid on the sialoglycopeptides is not a unique feature since it is buffered by negative and positive charges from the acidic and basic amino acid residues of the attached polypeptide chain. Nonetheless, as described in the first of these two reviews,3 efficient enrichment tools are available to isolate the sialoglycopeptides from the neutral subset of the glycoproteome (or glycopeptidome) as well as from the non-glycosylated component for the enhanced analysis of sialoglycopeptides using LC-MS techniques.3.1 LC separation of sialoglycopeptides
3.2 MS detection of sialoglycopeptides
3.2.1.a Ionisation. Due to the lower stoichiometry and the general hydrophilicity, sialoglycopeptides tend to have lower ionisation strength in MALDI MS compared to nonglycosylated peptides. Sialylated glycopeptides have mostly been analysed in positive ion MALDI MS where the detection is more sensitive.175–178 However, negative ion MALDI MS was shown to be a viable alternative to positive ion mode because of less discrimination in ionisation between neutral and sialoglycopeptides and more informative fragmentation of the sialoglycopeptides using tandem MS.179 In another study, on-plate derivatisation of the carboxyl group of the C-terminus as well as the acidic amino acid side chains and the sialic acid residues of the sialoglycopeptides with PDAM increased the intensities of sialylated and neutral glycopeptides relative to the non-glycosylated peptides, increased the stability of the sialoglycopeptides, and allowed their characterisation using negative ion MALDI MSn.180 The same authors also showed that the linkage of α2,3- and α2,6-Neu5Ac containing glycopeptides derivatised with PDAM could be distinguished using negative ion MALDI MS.5,148
3.2.1.b Matrix choice. As for sialoglycans, the commonly used DHB and CHCA matrices are unsuitable for sialoglycopeptides due to significant losses of sialic acids during fragmentation under typical MALDI acquisition settings (i.e. reflector mode with delayed extraction enabled). Instead, cooler matrices are typically favoured. Both Cl-CHCA93 and THAP88,102 are suitable matrices for negative ion MALDI MS of underivatised sialoglycopeptides with no ionisation-induced fragmentation observed, even with acquisition in reflector mode with delayed extraction enabled. Derivatised and thereby stabilised sialoglycopeptides e.g. with PDAM, can be analysed without sialic acid loss, even using the ‘hotter’ matrices including the popular DHB matrix.148
3.2.1.c Instrument settings. As is the case for sialoglycans, sialoglycopeptides can lose their sialic acids post source in the ion transfer or downstream in the mass analyzer. Post source desialylation is normally detectable for MALDI TOF MS performed in reflectron mode with extraction delay. This can largely be avoided by performing the acquisition in linear mode with limited or even without extraction delay, but results in lower signal resolution. Another example of minimising the loss of sialic acids from sialoglycopeptides is the pulsed application of cooling gas in MALDI FT-ICR MS together with a low extraction voltage.181 In comparison, it was shown that sialoglycopeptides formed in a typical vacuum MALDI source suffered from significant losses of sialic acid residues due to the lack of cooling of the sialoglycopeptide ions.
3.2.1.d Quantitation. Relative quantitation of sialoglycopeptides by MALDI MS signal intensities has been shown to be accurate when the sample preparation and MS settings are carefully considered.102 However, sialylated glycopeptide mass signals are weaker than the signals arising from their non-sialylated counterparts irrespective of ion polarity, limiting the use of signal intensities for relative quantitation when sialo- and asialoglycopeptides are analysed together.86,182 Recently, it was shown that positive ion MALDI FT-ICR MS discriminates less against sialoglycopeptide ionisation than positive ion MALDI TOF MS; when analysed together by MALDI FT-ICR MS the sialylated glycopeptides and the asialylated counterparts yielded essentially the same MS signal responses.181
3.2.2.a Ionisation. ESI MS of sialoglycopeptides has mostly been carried out in positive ion mode. Although sialic acid residues are negatively charged, the charge state of sialylated glycopeptides, containing one or more sialic acid residues, is surprisingly higher than the corresponding deglycosylated peptides in positive ion mode (unpublished observations and Lendal et al., in preparation). The extra proton(s) may tentatively be assigned to the nitrogen atoms of the sialic acid residues or the fact that sialoglycopeptides have a higher mass, but more investigation is needed to confirm this. In a study comparing ionisation efficiencies, HILIC separated sialoglycopeptides were analysed in positive and negative ion MSn.166 Here positive ion mode was preferred since information on the sialoglycan structure was obtained already at the MS2 stage by sequential glycosidic bond fragmentations (Yn-type ions).166 Information on the peptide structure was obtained at the MS3 stage through formation of polypeptide backbone fragment ions including bn-type ions. In other studies, data acquisition combining positive and negative ion polarities with different fragmentation techniques was found to be very useful for characterising N- and O-linked sialoglycopeptides.183,184
3.2.2.b Quantitation. ESI MS based relative quantitation of sialoglycopeptides using MS signal intensities has been shown to be accurate by careful consideration and optimisation of the sample preparation and MS settings.102 It needs to be emphasised that the intensities of all charge states of each sialylated glycopeptide need to be included in the quantitation since the glycopeptides isomers ionises in slightly different charge state distributions.118 Positive ion ESI MS intensities were recently shown to discriminate somewhat against sialoglycopeptides when analysed together with the asialylated glycopeptide counterpart.181 Importantly, this study also highlighted that both the neutral and negatively charged sialylated N-glycopeptide ions are dramatically under-represented compared to the non-glycosylated and deglycosylated counterparts when equimolar amounts are analysed in positive ion ESI and MALDI MS. These important findings should be considered when glycosylation site occupancy is evaluated by MS signal strength.
3.2.3.a CID for tandem MS and multistage MSn. The most popular fragmentation technique for analysis of sialylated glycopeptides is by far CID with acquisition in positive ion polarity. CID fragmentation of sialoglycopeptides is dominated by cleavages of the glycosidic linkages of the peptide bound sialoglycans generating B- and Y-ions.190–194 In particular, the sialic acid linkage to the penultimate residue is cleaved with high efficiency leading to a strong B1 ion at m/z 290 (negative ion mode) or m/z 292 (and m/z 274, B1-H2O) (positive ion mode) and the corresponding Y-ions.195 Extracted ion chromatograms for these B1 ions and some oxonium ions (e.g. m/z 366, HexHexNAc in positive ion mode) are helpful in localising sialoglycopeptide tandem MS CID spectra in the LC-MS data.161 CID fragmentation is usually sufficient to verify the monosaccharide composition of the sialoglycopeptides when combined with the accurate molecular mass of the precursor.196 Dedicated software has been developed to aid the determination of glycopeptide composition from tandem MS data.197 However, the identification of the peptide carrier and the site-specific attachment is usually difficult due to low-intensity peptide fragment ions in CID,198 but can be obtained either through MSn CID approaches or by alternative fragmentation techniques including HCD199 or ExD189 as described below. For the former, positive and negative ion ESI linear IT-TOF MSn has been employed for the analysis of sialylated N-linked glycopeptides with rich structural information on the attached sialoglycans being generated.184,200 This led to the suggestion that the structural assignment of sialylated N-glycans on glycopeptides can be determined by MSn spectral matching.184 As in the analysis of released glycans, rearrangement during the tandem MS process is known to happen particularly for glycopeptides that ionise by protonation, but reports have mostly described deoxyhexose rearrangements and not rearrangements of sialic acid residues.131,132
3.2.3.b HCD, ExD and other fragmentation techniques. Realising the limitations of CID fragmentation in determining the complete sialoglycopeptide structure, glycoscientists have increasingly explored the possibilities for using alternative fragmentation techniques for the characterisation of sialoglycopeptides. Infrared multiphoton dissociation (IRMPD) facilitates dissociation by collisional heating and hence generates cleavage of the most labile bonds, the glycosidic bonds. IRMPD is consequently rather similar to CID in its fragmentation behaviour,201 but with additional polypeptide backbone fragmentation being observed.202
Activated electron dissociation (ExD) methods have become popular in MS of peptides, but limited work has been presented for sialoglycopeptides. Electron capture dissociation (ECD) was shown to produce ions that complement the ions generated from CID type fragmentation events.183,203,204 Proof-of-concept studies have also been presented for the related fragmentation technique electron transfer dissociation (ETD). Here, multi-sialylated glycopeptides were seemingly less efficiently fragmented compared to ETD of neutral glycopeptides, but orthogonal information on the sialoglycopeptide structure could still be obtained when dedicated bioinformatic tools were used to interpret the obtained data.169,205,206 Nonetheless, most researchers carry out ETD or ECD on neutral or desialylated glycans.207,208 Neutral loss of the sialic acids and general charge reduction were observed when sialylated N- and O-linked glycopeptides were analysed using ETD MS195,209. An approach has been presented where ETD and ECD were utilised together with CID fragmentation to characterise formerly sialylated N- and O-linked glycopeptides following enrichment based on sialic acid capture and release of the sialic acids by acid hydrolysis prior to MS detection; however, in this approach no information on the conjugated sialoglycan is obtained.210–212 Finally, fragmentation techniques using negative ion polarity including electron detachment dissociation (EDD) and negative ETD (nETD) may very well be useful for the analysis of sialoglycopeptides, but these alternative fragmentation techniques need more investigation.189
The performance of CID combined with either HCD or ETD as parallel fragmentation techniques has been compared in a study where the neutral N-glycoproteome of Campylobacter jejuni was investigated.213 The combination CID/HCD enabled identification of the glycan structure and peptide backbone whereas CID/ETD enabled elucidation of the glycosylation site. In another study, 88 previously uncharacterised neutral N-glycopeptides were identified from hen egg glycoprotein mixtures using HCD and accurate molecular mass, but no sialylated glycopeptides were reported.214 Recently, HCD and ETD were utilised in combination to identify a high number of neutral and sialylated N-linked glycopeptides of the secretome of human endothelial cells.215 In this study alternating HCD-ETD detected predominantly high-intensity, multiply charged N-glycopeptides. HCD-product-dependent ETD preferentially selected complex/hybrid N-glycopeptide precursors for fragmentation. The benefits of using HCD fragmentation for N-linked sialoglycopeptides includes the presence of distinct Y1 ions (peptide + GlcNAc), which can be used for further MS3 based identification, abundant oxonium ions and B-type ions (e.g. m/z 292 from Neu5Ac and m/z 657 from sialyl LacNAc) and the high mass accuracy, resolution and extended m/z range offered by the Orbitrap.199 The combined use of CID, HCD and ETD on an Orbitrap instrumentation to characterise intact glycopeptides originating from the partly sialylated rat N-glycoproteome enabled identification of a total of 863 N-linked glycopeptides from 161 rat brain glycoproteins (Parker et al., 2013, submitted). This deep N-glycoproteome analysis was aided by a parallel characterisation of the released N-glycome and sophisticated bioinformatic tools to identify and verify the glycopeptides as intact molecules. Moreover, a total of 569 unique N-linked intact sialylated glycopeptides from 190 unique peptide sequences were identified from 98 proteins in human plasma (S. Lendal et al., in preparation). The identification of intact sialylated glycopeptides was performed using high accuracy mass spectrometry combined with computational tools.
3.3 LC-MS of sialoglycoproteins
Limited data has been presented for the LC-MS based analysis of intact sialoglycoproteins. However, this field is maturing quickly, driven by the well recognised analytical benefits of detecting the intact biomolecules instead of the post analysis piecing together the structural information in bottom up approaches.For increased LC resolution of sialoglycoforms, physicochemical parameters other than hydrophobicity have been used for separation. Anion exchange chromatography (AEC) is a rational choice to separate the negatively charged sialoglycoproteins, although the negative charges from acidic amino acid residues in the polypeptide backbone can dilute the charge difference between the sialoglycoforms to a certain extent. Nonetheless, AEC was able to differentiate human kallikrein-6 sialylated isoforms comprising different numbers of sialic acid residues.218,219
In addition to LC based separations, CE has proven very powerful in separating intact sialoglycoproteins. Glycoproteins with different numbers of sialic acid residues can routinely be separated using CE with either UV or molecular mass detection by ESI MS.117,220–225 Examples include CE-MS based separation and detection of glycoforms of intact bovine α1-acid-glycoprotein (AGP) and fetuin that differed in the number of sialic acid residues.72 Glycoform separation was also shown for recombinant human EPO and transferrin using CE-UV or CE-MS, but in neither of these cases could the branching and linkage isoforms of the attached sialoglycans be resolved.220,221 Another very useful alternative for separating differently sialylated glycoproteins is two dimensional (2D) gel electrophoresis where the isoelectric focusing (IEF) in the first dimension is capable of separating sialoglycoforms.36,226 The higher the number of sialic acids residues, the more the migration of the sialoglycoform to the acidic end of the pH region of the IEF strip. Using high resolution IEF strips, sialoglycoforms differing by a single sialic acid residue can usually be resolved. One of the inherent limitations of this approach, however, is that membrane sialoglycoproteins are under-represented due to their low solubility in the detergents used for the IEF dimension.16 Recently, a systematic study established rules for roughly predicting the pI of sialoglycoproteins separated by 2D gel electrophoresis based on their sialic acid content.227
4. Conclusion
Analytical glycoscience including the analysis of N- and O-linked glycoprotein sialylation has benefitted immensely from the advancement of LC-MS technologies in recent decades. Many of these advancements have been driven by technology improvements within the larger proteomics research field. Due to the unique analytical challenges encountered in structural glycobiology, many of these technological improvements have only become useful for researchers in glycoscience after tailored alterations of the workflows and tedious method optimisation. This is in particular true for the analysis of glycoprotein sialylation where the lability of the sialic acid residues together with their negative charge, linkage and modification heterogeneity, demands dedicated workflows. Methods for sophisticated analyte manipulation where the sialoglycoconjugates are rendered more “LC-MS friendly” by chemical derivatisation prior to the LC-MS analysis have also played a significant part in the successful analytical achievements in recent years.Although it still is commonly accepted that no analytical technique can yield the complete structural information regarding glycoprotein sialylation in a single analysis from a typical biological sample, LC-MS is gradually proving to be an extremely powerful approach to define the structure of sialoglycans and sialoglycopeptides at biologically relevant levels and complexities. Here it is important to stress that the biological function of the sialic acid and the protein glycosylation as a whole often can be learned from less-than-complete structural information of the sialylated N- or O-linked glycoprotein. In many known (and unknown) cases, however, the glycobiological function can be regulated by changes at the fine structural level such that the detailed and exact structure of the sialoglycoproteins is required to understand the underlying biology.
Although this review has focused on the analysis of the ubiquitous terminal modification of sialylation on N- and O-linked glycoproteins, it should be noted that our understanding of system glycobiology cannot be expected to be achieved by concentrating only on the sialylated protein component of the glycome/glycoproteome. The neutral glycome/glycoproteome equally needs to be specified from the same sample to define the entire glycosylation phenotype and to link this phenotype to biological functions. The spatial and temporal relationship with sialylated lipids is also important to consider particularly when cellular or system-wide processes are investigated. However, at this stage, holistic efforts are rarely undertaken due to the lack of such broad analytical knowledge and technology in a single laboratory.
At present, the main limitation of the available analytical techniques for determining glycoprotein sialylation includes the lack of automation of most experimental workflows. This obstacle severely limits the detailed structural characterisation of glycoprotein sialylation on the system-wide (cellular/organism) scale. Integration of bioinformatic tools and glycan repositories to aid the data handling and interpretation are on-going initiatives that are expected to facilitate greater coverage of the sialylated glycoproteome within the next few years by improving the throughput of data analysis. Spectral matching to libraries of tandem MS CID spectra of well-characterised sialylated N- and O-linked glycans is being pursued and will reduce the dependence on manual data interpretation, which, at present, is limited to relatively few expert research groups. The requirement for expertise is even more pronounced for the interpretation of fragment MS spectra of sialoglycopeptides. Since tandem mass spectral matching approaches are presently unrealistic because of the astronomical number of possible sialoglycopeptides that can be encountered in a proteolytical digest of a biological sample (number of glycosylated N- and O-linked peptides in the proteome × number of N- and O-linked sialoglycans in the glycome), bioinformatic tools and workflows must aim to assist the identification by partial de novo characterisation. This, in turn, relies strongly on the availability of well-understood fragmentation techniques including CID, HCD and ExD to allow acquisition of complementary structural information on the glycan structure, the peptide carrier as well as information on the sites of attachment.
LC-MS has proven capable of yielding accurate quantitative evaluation of sialylated and neutral glycans/glycopeptides comparable to the accuracy obtained using conventional UV-HPLC based detection techniques. However, accurate quantitation is highly dependent on the use of appropriate experimental designs since significant biases towards the derivatisation/ionisation/detection of some structures over others can be easily introduced during sample preparation and analyte detection. Recent advancements in the synthesis of sialoglycopeptides and glycoproteins using Fmoc solid-phase peptide synthesis169,246,247 may provide standards that will facilitate better understanding of fragmentation pathways and accurate definition of ionisation and detection efficiencies to yield more confident characterisation and quantitation of glycoprotein sialylation at the site-specific glycopeptide level.181
Another milestone to reach is the development and the further maturation and implementation of top-down MS technologies for the routine analysis of intact sialoglycoproteins. At present, little analytical work on intact sialoglycoproteins has been published, but the field is maturing rapidly.248 In addition to increasing the throughput of analysis by reducing the need for the extensive sample handling and processing required for bottom-up approaches, top-down approaches would allow researchers to study the intra-molecular relationships between sialoglycans on multiple glycosylation sites and other post-translational modifications to the intact polypeptide backbone. This would facilitate a more accurate characterisation of the individual “glycoforms” (more recently termed the “proteoform”) than present-day bottom-up approaches.249 In the meantime migratory separation techniques such as 2D gels remain a helpful tool to visualise and separate sialylation on the intact glycoprotein level, but, needless to mention, at low resolution.226
In conclusion, remarkable analytical advances in the LC-MS technologies have been accomplished in recent decades enabling increased depth, sensitivity and accuracy in the characterisation of glycoprotein sialylation in biologically relevant samples. However, there are still unmet needs in structural glycobiology. Improvements are consequently needed at all analyte levels from sample preparation through LC-MS acquisition to data analysis and interpretation. The combined efforts will allow us to dig deeper into the barely explored sialylated glycoproteome to address the functional relevance of sialic acid residues attached to specific glycoproteins.
Abbreviations
| 2-AA | 2-Aminobenzoic acid |
| 2-AB | 2-Aminobenzamide |
| 2D | Two-dimensional |
| AEC | Anion exchange chromatography |
| AGP | α1-Acid-glycoprotein |
| ANTS | 8-Aminonaphthalene-1,3,6-trisulfonic acid |
| APTS | 8-Aminopyrene-1,3,6-trisulfonic acid trisodium |
| ATT | 6-Aza-2-thiothymine |
| CE | Capillary electrophoresis |
| CHCA | α-Cyano-4-hydroxycinnamic acid |
| CID | Collision induced dissociation |
| Cl-CHCA | Chlorinated CHCA |
| CMP | Cytidine 5-monophosphate |
| DHB | 2,5-Dihydroxybenzoic acid |
| ECD | Electron capture dissociation |
| EDD | Electron detachment dissociation |
| ELISA | Enzyme-linked immunosorbent assay |
| EPO | Erythropoietin |
| ESI | Electrospray ionisation |
| ETD | Electron transfer dissociation |
| ExD | Activated electron dissociation |
| FT-ICR | Fourier transform-ion cyclotron resonance |
| HCD | Higher-energy C-trap dissociation |
| HILIC | Hydrophilic interaction liquid chromatography |
| HPAEC | High pH anion exchange chromatography |
| IMS | Ion mobility spectrometry |
| IP | Ion pairing |
| IRMPD | Infrared multiphoton dissociation |
| Kdo | 3-Deoxy-manno-octulosonic acid |
| IEF | Isoelectric focusing |
| IT | Ion trap |
| LC | Liquid chromatography |
| MALDI | Matrix assisted laser desorption ionisation |
| MS | Mass spectrometry |
| NCAM | Neural cell adhesion molecule |
| nETD | Negative ETD |
| Neu5Ac | N-Acetylneuraminic acid |
| Neu5Gc | N-Glycolylneuraminic acid |
| NPC | Normal phase chromatography |
| PA | 2-Aminopyridine |
| PDAM | 1-Pyrenyl diazomethane |
| PGC | Porous graphitized carbon |
| PQD | Pulsed-Q-dissociation |
| Q | Quadrupole |
| RPC | Reversed phase chromatography |
| SNA | Sambucus nigra agglutinin |
| TFA | Trifluoroacetic acid |
| THAP | 2,4,6-Trihydroxyacetophenone |
| TOF | Time-of-flight |
| UPLC | Ultra-high performance liquid chromatography |
| ZIC | Zwitter-ionic |
Acknowledgements
We thank Dr Daniel Kolarich for critical reading of the manuscript. M.T.-A. was funded by the Australian Research Council by a Super Science Fellowship. GP was supported by the Danish Medical Science Research Council (Grant no. 11-107551). MRL was supported by the Lundbeck foundation (Junior Group Leader Fellowship).References
- J. L. Benesch and B. T. Ruotolo, Curr. Opin. Struct. Biol., 2011, 21, 641–649 CrossRef CAS PubMed.
- M. Wuhrer, A. M. Deelder and C. H. Hokke, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 825, 124–133 CrossRef CAS PubMed.
- M. Thaysen-Andersen, M. R. Larsen, N. H. Packer and G. Palmisano, RSC Adv., 2013 10.1039/C3RA42960A.
- A. G. Marshall and C. L. Hendrickson, Annu. Rev. Anal. Chem., 2008, 1, 579–599 CrossRef CAS PubMed.
- J. P. Kamerling and G. J. Gerwig, Methods Mol. Biol., 2006, 347, 69–91 CAS.
- S. P. Galuska, H. Geyer, B. Weinhold, M. Kontou, R. C. Rohrich, U. Bernard, R. Gerardy-Schahn, W. Reutter, A. Munster-Kuhnel and R. Geyer, Anal. Chem., 2010, 82, 4591–4598 CrossRef CAS PubMed.
- S. G. Penn, M. T. Cancilla and C. B. Lebrilla, Int. J. Mass Spectrom., 2000, 195, 259–269 CrossRef.
- S. P. Galuska, R. Geyer, M. Muhlenhoff and H. Geyer, Anal. Chem., 2007, 79, 7161–7169 CrossRef CAS PubMed.
- S. P. Galuska, H. Geyer, C. Bleckmann, R. C. Rohrich, K. Maass, A. K. Bergfeld, M. Muhlenhoff and R. Geyer, Anal. Chem., 2010, 82, 2059–2066 CrossRef CAS PubMed.
- Y. Wada, A. Dell, S. M. Haslam, B. Tissot, K. Canis, P. Azadi, M. Backstrom, C. E. Costello, G. C. Hansson, Y. Hiki, M. Ishihara, H. Ito, K. Kakehi, N. Karlsson, C. E. Hayes, K. Kato, N. Kawasaki, K. H. Khoo, K. Kobayashi, D. Kolarich, A. Kondo, C. Lebrilla, M. Nakano, H. Narimatsu, J. Novak, M. V. Novotny, E. Ohno, N. H. Packer, E. Palaima, M. B. Renfrow, M. Tajiri, K. A. Thomsson, H. Yagi, S. Y. Yu and N. Taniguchi, Mol. Cell. Proteomics, 2010, 9, 719–727 CAS.
- Y. Wada, P. Azadi, C. E. Costello, A. Dell, R. A. Dwek, H. Geyer, R. Geyer, K. Kakehi, N. G. Karlsson, K. Kato, N. Kawasaki, K. H. Khoo, S. Kim, A. Kondo, E. Lattova, Y. Mechref, E. Miyoshi, K. Nakamura, H. Narimatsu, M. V. Novotny, N. H. Packer, H. Perreault, J. Peter-Katalinic, G. Pohlentz, V. N. Reinhold, P. M. Rudd, A. Suzuki and N. Taniguchi, Glycobiology, 2007, 17, 411–422 CrossRef CAS PubMed.
- L. R. Ruhaak, G. Zauner, C. Huhn, C. Bruggink, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2010, 397, 3457–3481 CrossRef CAS PubMed.
- J. S. Rohrer, G. A. Cooper and R. R. Townsend, Anal. Biochem., 1993, 212, 7–16 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 1999, 18, 349–450 CrossRef CAS PubMed.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6, 713–723 CrossRef CAS PubMed.
- H. Geyer and R. Geyer, Biochim. Biophys. Acta, 2006, 1764, 1853–1869 CrossRef CAS PubMed.
- M. Pabst and F. Altmann, Proteomics, 2011, 11, 631–643 CrossRef CAS PubMed.
- M. Wuhrer, Glycoconjugate J., 2013, 30, 11–22 CrossRef CAS PubMed.
- N. V. Artemenko, A. G. McDonald, G. P. Davey and P. M. Rudd, Methods Mol. Biol., 2012, 899, 325–350 CAS.
- T. Lutteke, Beilstein J. Org. Chem., 2012, 8, 915–929 CrossRef PubMed.
- M. Frank and S. Schloissnig, Cell. Mol. Life Sci., 2010, 67, 2749–2772 CrossRef CAS PubMed.
- A. Helenius and M. Aebi, Annu. Rev. Biochem., 2004, 73, 1019–1049 CrossRef CAS PubMed.
- L. R. Ruhaak, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2009, 394, 163–174 CrossRef CAS PubMed.
- M. Davies, K. D. Smith, A. M. Harbin and E. F. Hounsell, J. Chromatogr., 1992, 609, 125–131 CrossRef CAS PubMed.
- J. Q. Fan, A. Kondo, I. Kato and Y. C. Lee, Anal. Biochem., 1994, 219, 224–229 CrossRef CAS PubMed.
- N. G. Karlsson, N. L. Wilson, H. J. Wirth, P. Dawes, H. Joshi and N. H. Packer, Rapid Commun. Mass Spectrom., 2004, 18, 2282–2292 CrossRef CAS PubMed.
- W. Ni, J. Bones and B. L. Karger, Anal. Chem., 2013, 85, 3127–3135 CrossRef CAS PubMed.
- J. H. Knox and P. Ross, Adv. Chromatogr., 1997, 37, 73–119 CAS.
- L. Pereira, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 1687–1731 CrossRef CAS.
- P. H. Jensen, N. G. Karlsson, D. Kolarich and N. H. Packer, Nat. Protoc., 2012, 7, 1299–1310 CrossRef CAS PubMed.
- A. V. Everest-Dass, D. Jin, M. Thaysen-Andersen, H. Nevalainen, D. Kolarich and N. H. Packer, Glycobiology, 2012, 22, 1465–1479 CrossRef CAS PubMed.
- S. Itoh, N. Kawasaki, M. Ohta and T. Hayakawa, J. Chromatogr., A, 2002, 978, 141–152 CrossRef CAS PubMed.
- K. A. Thomsson, M. Backstrom, J. M. Holmen Larsson, G. C. Hansson and H. Karlsson, Anal. Chem., 2010, 82, 1470–1477 CrossRef CAS PubMed.
- M. Pabst and F. Altmann, Anal. Chem., 2008, 80, 7534–7542 CrossRef CAS PubMed.
- M. Pabst, J. Grass, R. Fischl, R. Leonard, C. Jin, G. Hinterkorner, N. Borth and F. Altmann, Anal. Chem., 2010, 82, 9782–9788 CrossRef CAS PubMed.
- Y. Mechref and M. V. Novotny, Chem. Rev., 2002, 102, 321–369 CrossRef CAS PubMed.
- F. T. Chen and R. A. Evangelista, Electrophoresis, 1998, 19, 2639–2644 CrossRef CAS PubMed.
- M. Pabst, J. S. Bondili, J. Stadlmann, L. Mach and F. Altmann, Anal. Chem., 2007, 79, 5051–5057 CrossRef CAS PubMed.
- S. Hua, H. N. Jeong, L. M. Dimapasoc, I. Kang, C. Han, J. S. Choi, C. B. Lebrilla and H. J. An, Anal. Chem., 2013, 85, 4636–4643 CrossRef CAS PubMed.
- M. Wuhrer, A. R. de Boer and A. M. Deelder, Mass Spectrom. Rev., 2009, 28, 192–206 CrossRef CAS PubMed.
- G. Zauner, A. M. Deelder and M. Wuhrer, Electrophoresis, 2011, 32, 3456–3466 CrossRef CAS PubMed.
- M. Thaysen-Andersen, K. Engholm-Keller and P. Roepstorff, in Hydrophilic interaction liquid chromatography (HILIC) and advanced applications, ed. P. G. Wang and W. He, CRC Press, Boca Raton, 2011, pp. 551–575 Search PubMed.
- Z. Hao, B. Xiao and N. Weng, J. Sep. Sci., 2008, 31, 1449–1464 CrossRef CAS PubMed.
- P. Hemstrom and K. Irgum, J. Sep. Sci., 2006, 29, 1784–1821 CrossRef PubMed.
- A. J. Alpert, Anal. Chem., 2008, 80, 62–76 CrossRef CAS PubMed.
- Y. Takegawa, K. Deguchi, T. Keira, H. Ito, H. Nakagawa and S. Nishimura, J. Chromatogr., A, 2006, 1113, 177–181 CrossRef CAS PubMed.
- Y. Yamakoshi, F. Yamakoshi, J. C. Hu and J. P. Simmer, Eur. J. Oral Sci., 2011, 119(suppl. 1), 234–240 CrossRef PubMed.
- Y. Takegawa, K. Deguchi, H. Ito, T. Keira, H. Nakagawa and S. Nishimura, J. Sep. Sci., 2006, 29, 2533–2540 CrossRef CAS PubMed.
- K. Deguchi, T. Keira, K. Yamada, H. Ito, Y. Takegawa, H. Nakagawa and S. Nishimura, J. Chromatogr., A, 2008, 1189, 169–174 CrossRef CAS PubMed.
- D. Kolarich, P. L. Turecek, A. Weber, A. Mitterer, M. Graninger, P. Matthiessen, G. A. Nicolaes, F. Altmann and H. P. Schwarz, Transfusion, 2006, 46, 1959–1977 CrossRef CAS PubMed.
- J. Ahn, J. Bones, Y. Q. Yu, P. M. Rudd and M. Gilar, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2010, 878, 403–408 CrossRef CAS PubMed.
- M. Melmer, T. Stangler, A. Premstaller and W. Lindner, J. Chromatogr., A, 2011, 1218, 118–123 CrossRef CAS PubMed.
- G. O. Staples, M. J. Bowman, C. E. Costello, A. M. Hitchcock, J. M. Lau, N. Leymarie, C. Miller, H. Naimy, X. Shi and J. Zaia, Proteomics, 2009, 9, 686–695 CrossRef CAS PubMed.
- G. O. Staples, H. Naimy, H. Yin, K. Kileen, K. Kraiczek, C. E. Costello and J. Zaia, Anal. Chem., 2010, 82, 516–522 CrossRef CAS PubMed.
- Y. Takegawa, K. Deguchi, S. Ito, S. Yoshioka, H. Nakagawa and S. Nishimura, Anal. Chem., 2005, 77, 2097–2106 CrossRef CAS PubMed.
- J. Delaney and P. Vouros, Rapid Commun. Mass Spectrom., 2001, 15, 325–334 CrossRef CAS PubMed.
- X. Chen and G. C. Flynn, Anal. Biochem., 2007, 370, 147–161 CrossRef CAS PubMed.
- L. A. Gennaro, D. J. Harvey and P. Vouros, Rapid Commun. Mass Spectrom., 2003, 17, 1528–1534 CrossRef CAS PubMed.
- R. Zeleny, D. Kolarich, R. Strasser and F. Altmann, Planta, 2006, 224, 222–227 CrossRef CAS PubMed.
- J. L. Behan and K. D. Smith, Biomed. Chromatogr., 2011, 25, 39–46 CrossRef CAS PubMed.
- D. C. Hurum and J. S. Rohrer, J. Dairy Sci., 2012, 95, 1152–1161 CrossRef CAS PubMed.
- R. R. Townsend, M. R. Hardy, D. A. Cumming, J. P. Carver and B. Bendiak, Anal. Biochem., 1989, 182, 1–8 CrossRef CAS PubMed.
- M. von Der Ohe, S. F. Wheeler, M. Wuhrer, D. J. Harvey, S. Liedtke, M. Muhlenhoff, R. Gerardy-Schahn, H. Geyer, R. A. Dwek, R. Geyer, D. R. Wing and M. Schachner, Glycobiology, 2002, 12, 47–63 CrossRef CAS PubMed.
- J. R. Barr, K. R. Anumula, M. B. Vettese, P. B. Taylor and S. A. Carr, Anal. Biochem., 1991, 192, 181–192 CrossRef CAS PubMed.
- J. Bones, N. McLoughlin, M. Hilliard, K. Wynne, B. L. Karger and P. M. Rudd, Anal. Chem., 2011, 83, 4154–4162 CrossRef CAS PubMed.
- N. Takahashi, J. Chromatogr., A, 1996, 720, 217–225 CrossRef CAS PubMed.
- W. Winkler, W. Huber, R. Vlasak and G. Allmaier, Rapid Commun. Mass Spectrom., 2011, 25, 3235–3244 CrossRef CAS PubMed.
- Y. Yamaguchi, W. Nishima, S. Re and Y. Sugita, Rapid Commun. Mass Spectrom., 2012, 26, 2877–2884 CrossRef CAS PubMed.
- M. M. Gaye, S. J. Valentine, Y. Hu, N. Mirjankar, Z. T. Hammoud, Y. Mechref, B. K. Lavine and D. E. Clemmer, J. Proteome Res., 2012, 11, 6102–6110 CAS.
- D. Isailovic, R. T. Kurulugama, M. D. Plasencia, S. T. Stokes, Z. Kyselova, R. Goldman, Y. Mechref, M. V. Novotny and D. E. Clemmer, J. Proteome Res., 2008, 7, 1109–1117 CrossRef CAS PubMed.
- Y. Mechref, Electrophoresis, 2011, 32, 3467–3481 CrossRef CAS PubMed.
- E. Balaguer and C. Neususs, Anal. Chem., 2006, 78, 5384–5393 CrossRef CAS PubMed.
- L. A. Gennaro, J. Delaney, P. Vouros, D. J. Harvey and B. Domon, Rapid Commun. Mass Spectrom., 2002, 16, 192–200 CrossRef CAS PubMed.
- R. G. Jayo, J. Li and D. D. Chen, Anal. Chem., 2012, 84, 8756–8762 CrossRef CAS PubMed.
- O. Quintero, R. Montesino and J. A. Cremata, Anal. Biochem., 1998, 256, 23–32 CrossRef CAS PubMed.
- J. Zaia, OMICS, 2010, 14, 401–418 CrossRef CAS PubMed.
- N. Leymarie and J. Zaia, Anal. Chem., 2012, 84, 3040–3048 CrossRef CAS PubMed.
- H. Nie, Y. Li and X. L. Sun, J. Proteomics, 2012, 75, 3098–3112 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2006, 25, 595–662 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2008, 27, 125–201 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2009, 28, 273–361 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2011, 30, 1–100 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2012, 31, 183–311 CrossRef CAS PubMed.
- P. Domann, D. I. Spencer and D. J. Harvey, Rapid Commun. Mass Spectrom., 2012, 26, 469–479 CrossRef CAS PubMed.
- J. Colangelo and R. Orlando, Anal. Chem., 1999, 71, 1479–1482 CrossRef CAS PubMed.
- C. W. Sutton, J. A. O'Neill and J. S. Cottrell, Anal. Biochem., 1994, 218, 34–46 CrossRef CAS PubMed.
- A. K. Powell and D. J. Harvey, Rapid Commun. Mass Spectrom., 1996, 10, 1027–1032 CrossRef CAS PubMed.
- D. I. Papac, A. Wong and A. J. Jones, Anal. Chem., 1996, 68, 3215–3223 CrossRef CAS PubMed.
- J. J. Pitt and J. J. Gorman, Rapid Commun. Mass Spectrom., 1996, 10, 1786–1788 CrossRef CAS.
- D. Sagi, P. Kienz, J. Denecke, T. Marquardt and J. Peter-Katalinic, Proteomics, 2005, 5, 2689–2701 CrossRef CAS PubMed.
- S. F. Wheeler and D. J. Harvey, Anal. Chem., 2000, 72, 5027–5039 CrossRef CAS PubMed.
- Y. Mechref and M. V. Novotny, J. Am. Soc. Mass Spectrom., 1998, 9, 1293–1302 CrossRef CAS PubMed.
- M. H. Selman, M. Hoffmann, G. Zauner, L. A. McDonnell, C. I. Balog, E. Rapp, A. M. Deelder and M. Wuhrer, Proteomics, 2012, 12, 1337–1348 CrossRef CAS PubMed.
- Y. Fukuyama, S. Nakaya, Y. Yamazaki and K. Tanaka, Anal. Chem., 2008, 80, 2171–2179 CrossRef CAS PubMed.
- L. R. Ruhaak, C. Huhn, W. J. Waterreus, A. R. de Boer, C. Neususs, C. H. Hokke, A. M. Deelder and M. Wuhrer, Anal. Chem., 2008, 80, 6119–6126 CrossRef CAS PubMed.
- M. H. Selman, L. A. McDonnell, M. Palmblad, L. R. Ruhaak, A. M. Deelder and M. Wuhrer, Anal. Chem., 2010, 82, 1073–1081 CrossRef CAS PubMed.
- E. Ito, A. Tominaga, H. Waki, K. Miseki, A. Tomioka, K. Nakajima, K. Kakehi, M. Suzuki, N. Taniguchi and A. Suzuki, Neurochem. Res., 2012, 37, 1315–1324 CrossRef CAS PubMed.
- P. B. O'Connor, E. Mirgorodskaya and C. E. Costello, J. Am. Soc. Mass Spectrom., 2002, 13, 402–407 CrossRef.
- M. Nakano, K. Kakehi, M. H. Tsai and Y. C. Lee, Glycobiology, 2004, 14, 431–441 CrossRef CAS PubMed.
- P. J. Domann, A. C. Pardos-Pardos, D. L. Fernandes, D. I. Spencer, C. M. Radcliffe, L. Royle, R. A. Dwek and P. M. Rudd, Proteomics, 2007, 7(suppl. 1), 70–76 CrossRef PubMed.
- D. J. Harvey, Rapid Commun. Mass Spectrom., 1993, 7, 614–619 CrossRef CAS PubMed.
- M. Thaysen-Andersen, S. Mysling and P. Hojrup, Anal. Chem., 2009, 81, 3933–3943 CrossRef CAS PubMed.
- G. C. Gil, Y. G. Kim and B. G. Kim, Anal. Biochem., 2008, 379, 45–59 CrossRef CAS PubMed.
- B. J. Harmon, X. Gu and D. I. Wang, Anal. Chem., 1996, 68, 1465–1473 CrossRef CAS PubMed.
- T. J. Naven and D. J. Harvey, Rapid Commun. Mass Spectrom., 1996, 10, 1361–1366 CrossRef CAS PubMed.
- M. Toyoda, H. Ito, Y. K. Matsuno, H. Narimatsu and A. Kameyama, Anal. Chem., 2008, 80, 5211–5218 CrossRef CAS PubMed.
- Y. Miura, Y. Shinohara, J. Furukawa, N. Nagahori and S. Nishimura, Chemistry, 2007, 13, 4797–4804 CrossRef CAS PubMed.
- B. Kuster, A. P. Hunter, S. F. Wheeler, R. A. Dwek and D. J. Harvey, Electrophoresis, 1998, 19, 1950–1959 CrossRef CAS PubMed.
- K. S. Jang, Y. G. Kim, G. C. Gil, S. H. Park and B. G. Kim, Anal. Biochem., 2009, 386, 228–236 CrossRef CAS PubMed.
- G. C. Gil, B. Iliff, R. Cerny, W. H. Velander and K. E. Van Cott, Anal. Chem., 2010, 82, 6613–6620 CrossRef CAS PubMed.
- A. W. Wong, M. T. Cancilla, L. R. Voss and C. B. Lebrilla, Anal. Chem., 1999, 71, 205–211 CrossRef CAS PubMed.
- A. W. Wong, H. Wang and C. B. Lebrilla, Anal. Chem., 2000, 72, 1419–1425 CrossRef CAS PubMed.
- S. Tep, M. Hincapie and W. S. Hancock, Carbohydr. Res., 2012, 347, 121–129 CrossRef CAS PubMed.
- J. Zaia, Mass Spectrom. Rev., 2004, 23, 161–227 CrossRef CAS PubMed.
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2005, 16, 622–630 CrossRef CAS PubMed.
- A. Lee, J. M. Chick, D. Kolarich, P. A. Haynes, G. R. Robertson, M. Tsoli, L. Jankova, S. J. Clarke, N. H. Packer and M. S. Baker, Mol. Cell. Proteomics, 2011, 10, M900538MCP900200 Search PubMed.
- Z. Sumer-Bayraktar, T. Nguyen-Khuong, R. Jayo, D. D. Chen, S. Ali, N. H. Packer and M. Thaysen-Andersen, Proteomics, 2012, 12, 3315–3327 CrossRef CAS PubMed.
- N. Deshpande, P. H. Jensen, N. H. Packer and D. Kolarich, J. Proteome Res., 2010, 9, 1063–1075 CrossRef CAS PubMed.
- Z. Sumer-Bayraktar, D. Kolarich, M. P. Campbell, S. Ali, N. H. Packer and M. Thaysen-Andersen, Mol. Cell. Proteomics, 2011, 10, M111 009100 Search PubMed.
- D. Kolarich, A. Weber, P. L. Turecek, H. P. Schwarz and F. Altmann, Proteomics, 2006, 6, 3369–3380 CrossRef CAS PubMed.
- S. Itoh, N. Kawasaki, N. Hashii, A. Harazono, Y. Matsuishi, T. Hayakawa and T. Kawanishi, J. Chromatogr., A, 2006, 1103, 296–306 CrossRef CAS PubMed.
- J. Stadlmann, M. Pabst, D. Kolarich, R. Kunert and F. Altmann, Proteomics, 2008, 8, 2858–2871 CrossRef CAS PubMed.
- N. Leymarie, P. J. Griffin, K. Jonscher, D. Kolarich, R. Orlando, M. McComb, J. Zaia, J. Aguilan, W. R. Alley, F. Altmann, L. E. Ball, L. Basumallick, C. R. Bazemore-Walker, H. Behnken, M. A. Blank, K. J. Brown, S. C. Bunz, C. W. Cairo, J. F. Cipollo, R. Daneshfar, H. Desaire, R. R. Drake, E. P. Go, R. Goldman, C. Gruber, A. Halim, Y. Hathout, P. J. Hensbergen, D. M. Horn, D. Hurum, W. Jabs, G. Larson, M. Ly, B. F. Mann, K. Marx, Y. Mechref, B. Meyer, U. Moginger, C. Neususs, J. Nilsson, M. V. Novotny, J. O. Nyalwidhe, N. H. Packer, P. Pompach, B. Reiz, A. Resemann, J. S. Rohrer, A. Ruthenbeck, M. Sanda, J. M. Schulz, U. Schweiger-Hufnagel, C. Sihlbom, E. Song, G. O. Staples, D. Suckau, H. Tang, M. Thaysen-Andersen, R. I. Viner, Y. An, L. Valmu, Y. Wada, M. Watson, M. Windwarder, R. Whittal, M. Wuhrer, Y. Zhu and C. Zou, Mol. Cell. Proteomics, 2013 DOI:10.1074/mcp.M113.030643.
- F. J. Olson, M. Backstrom, H. Karlsson, J. Burchell and G. C. Hansson, Glycobiology, 2005, 15, 177–191 CrossRef CAS PubMed.
- N. Viseux, X. Hronowski, J. Delaney and B. Domon, Anal. Chem., 2001, 73, 4755–4762 CrossRef CAS PubMed.
- M. Pabst, S. Q. Wu, J. Grass, A. Kolb, C. Chiari, H. Viernstein, F. M. Unger, F. Altmann and S. Toegel, Carbohydr. Res., 2010, 345, 1389–1393 CrossRef CAS PubMed.
- H. Hahne, P. Neubert, K. Kuhn, C. Etienne, R. Bomgarden, J. C. Rogers and B. Kuster, Anal. Chem., 2012, 84, 3716–3724 CrossRef CAS PubMed.
- M. Wuhrer and A. M. Deelder, Rapid Commun. Mass Spectrom., 2006, 20, 943–951 CrossRef CAS PubMed.
- D. J. Harvey, T. S. Mattu, M. R. Wormald, L. Royle, R. A. Dwek and P. M. Rudd, Anal. Chem., 2002, 74, 734–740 CrossRef CAS PubMed.
- J. L. Seymour, C. E. Costello and J. Zaia, J. Am. Soc. Mass Spectrom., 2006, 17, 844–854 CrossRef CAS PubMed.
- M. Wuhrer, A. M. Deelder and Y. E. van der Burgt, Mass Spectrom. Rev., 2011, 30, 664–680 CrossRef CAS PubMed.
- M. Wuhrer, C. A. Koeleman and A. M. Deelder, Anal. Chem., 2009, 81, 4422–4432 CrossRef CAS PubMed.
- M. Wuhrer, C. A. Koeleman, C. H. Hokke and A. M. Deelder, Rapid Commun. Mass Spectrom., 2006, 20, 1747–1754 CrossRef CAS PubMed.
- U. Lewandrowski, A. Resemann and A. Sickmann, Anal. Chem., 2005, 77, 3274–3283 CrossRef CAS PubMed.
- D. J. Harvey, J. Mass Spectrom., 2005, 40, 642–653 CrossRef CAS PubMed.
- D. Sagi, J. Peter-Katalinic, H. S. Conradt and M. Nimtz, J. Am. Soc. Mass Spectrom., 2002, 13, 1138–1148 CrossRef CAS PubMed.
- M. D. Leavell and J. A. Leary, J. Am. Soc. Mass Spectrom., 2001, 12, 528–536 CrossRef CAS PubMed.
- J. Lemoine, G. Strecker, Y. Leroy, B. Fournet and G. Ricart, Carbohydr. Res., 1991, 221, 209–217 CrossRef CAS PubMed.
- Y. Mechref and M. V. Novotny, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 841, 65–78 CrossRef CAS PubMed.
- Y. Mechref, P. Kang and M. V. Novotny, Rapid Commun. Mass Spectrom., 2006, 20, 1381–1389 CrossRef CAS PubMed.
- W. Morelle, V. Faid and J. C. Michalski, Rapid Commun. Mass Spectrom., 2004, 18, 2451–2464 CrossRef CAS PubMed.
- K. Deguchi, Y. Takegawa, H. Ito, N. Miura, S. Yoshioka, S. Nagai, H. Nakagawa and S. Nishimura, Rapid Commun. Mass Spectrom., 2006, 20, 412–418 CrossRef CAS PubMed.
- H. Ito, K. Yamada, K. Deguchi, H. Nakagawa and S. Nishimura, Rapid Commun. Mass Spectrom., 2007, 21, 212–218 CrossRef CAS PubMed.
- A. Kameyama, N. Kikuchi, S. Nakaya, H. Ito, T. Sato, T. Shikanai, Y. Takahashi, K. Takahashi and H. Narimatsu, Anal. Chem., 2005, 77, 4719–4725 CrossRef CAS PubMed.
- E. Casal, R. Lebron-Aguilar, F. J. Moreno, N. Corzo and J. E. Quintanilla-Lopez, Rapid Commun. Mass Spectrom., 2010, 24, 885–893 CrossRef CAS PubMed.
- X. Chen and G. C. Flynn, J. Am. Soc. Mass Spectrom., 2009, 20, 1821–1833 CrossRef CAS PubMed.
- J. Amano, D. Sugahara, K. Osumi and K. Tanaka, Glycobiology, 2009, 19, 592–600 CrossRef CAS PubMed.
- T. Nishikaze, T. Nakamura, H. Jinmei and J. Amano, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1419–1428 CrossRef CAS PubMed.
- C. Zhao, B. Xie, S. Y. Chan, C. E. Costello and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2008, 19, 138–150 CrossRef CAS PubMed.
- J. T. Adamson and K. Hakansson, J. Am. Soc. Mass Spectrom., 2007, 18, 2162–2172 CrossRef CAS PubMed.
- W. Zhou and K. Hakansson, Electrophoresis, 2011, 32, 3526–3535 CrossRef CAS PubMed.
- J. R. Kornacki, J. T. Adamson and K. Hakansson, J. Am. Soc. Mass Spectrom., 2012, 23, 2031–2042 CrossRef CAS PubMed.
- J. V. Olsen, B. Macek, O. Lange, A. Makarov, S. Horning and M. Mann, Nat. Methods, 2007, 4, 709–712 CrossRef CAS PubMed.
- T. Guo, C. S. Gan, H. Zhang, Y. Zhu, O. L. Kon and S. K. Sze, J. Proteome Res., 2008, 7, 4831–4840 CrossRef CAS PubMed.
- C. Przybylski and V. Bonnet, Rapid Commun. Mass Spectrom., 2013, 27, 75–87 CrossRef CAS PubMed.
- T. Yamagaki and H. Nakanishi, Glycoconjugate J., 1999, 16, 385–389 CrossRef CAS PubMed.
- A. Devakumar, Y. Mechref, P. Kang, M. V. Novotny and J. P. Reilly, Rapid Commun. Mass Spectrom., 2007, 21, 1452–1460 CrossRef CAS PubMed.
- M. Thaysen-Andersen and N. H. Packer, Glycobiology, 2012, 22, 1440–1452 CrossRef CAS PubMed.
- M. Ohta, N. Kawasaki, S. Itoh and T. Hayakawa, Biologicals, 2002, 30, 235–244 CrossRef CAS PubMed.
- M. Ohta, N. Kawasaki, S. Hyuga, M. Hyuga and T. Hayakawa, J. Chromatogr., A, 2001, 910, 1–11 CrossRef CAS PubMed.
- Y. Takegawa, H. Ito, T. Keira, K. Deguchi, H. Nakagawa and S. Nishimura, J. Sep. Sci., 2008, 31, 1585–1593 CrossRef CAS PubMed.
- J. Zhang and D. I. Wang, J. Chromatogr., B: Biomed. Sci. Appl., 1998, 712, 73–82 CrossRef CAS.
- W. Ding, H. Nothaft, C. M. Szymanski and J. Kelly, Mol. Cell. Proteomics, 2009, 8, 2170–2185 CAS.
- S. Mysling, G. Palmisano, P. Hojrup and M. Thaysen-Andersen, Anal. Chem., 2010, 82, 5598–5609 CrossRef CAS PubMed.
- W. Ding, J. J. Hill and J. Kelly, Anal. Chem., 2007, 79, 8891–8899 CrossRef CAS PubMed.
- O. Hernandez-Hernandez, R. Lebron-Aguilar, J. E. Quintanilla-Lopez, M. L. Sanz and F. J. Moreno, Proteomics, 2010, 10, 3699–3711 CrossRef CAS PubMed.
- M. J. Davies, K. D. Smith, R. A. Carruthers, W. Chai, A. M. Lawson and E. F. Hounsell, J. Chromatogr., 1993, 646, 317–326 CrossRef CAS PubMed.
- J. S. Rohrer, J. Chromatogr., A, 1994, 667, 75–83 CrossRef CAS PubMed.
- M. Thaysen-Andersen, B. L. Wilkinson, R. J. Payne and N. H. Packer, Electrophoresis, 2011, 32, 3536–3545 CrossRef CAS PubMed.
- K. Lohrig, A. Sickmann and U. Lewandrowski, Methods Mol. Biol., 2011, 753, 299–308 CAS.
- U. Lewandrowski, R. P. Zahedi, J. Moebius, U. Walter and A. Sickmann, Mol. Cell. Proteomics, 2007, 6, 1933–1941 CAS.
- S. I. Snovida, V. C. Chen, O. Krokhin and H. Perreault, Anal. Chem., 2006, 78, 6556–6563 CrossRef CAS PubMed.
- R. Qiu and F. E. Regnier, Anal. Chem., 2005, 77, 2802–2809 CrossRef CAS PubMed.
- R. Qiu and F. E. Regnier, Anal. Chem., 2005, 77, 7225–7231 CrossRef CAS PubMed.
- Z. Valnickova, M. Thaysen-Andersen, P. Hojrup, T. Christensen, K. W. Sanggaard, T. Kristensen and J. J. Enghild, BMC Biochem., 2009, 10, 13 CrossRef PubMed.
- L. Dissing-Olesen, M. Thaysen-Andersen, M. Meldgaard, P. Hojrup and B. Finsen, J. Pharmacol. Exp. Ther., 2008, 326, 338–347 CrossRef CAS PubMed.
- M. Thaysen-Andersen, I. B. Thogersen, U. Lademann, H. Offenberg, A. M. Giessing, J. J. Enghild, H. J. Nielsen, N. Brunner and P. Hojrup, Biochim. Biophys. Acta, 2008, 1784, 455–463 CrossRef CAS PubMed.
- M. Thaysen-Andersen, I. B. Thogersen, H. J. Nielsen, U. Lademann, N. Brunner, J. J. Enghild and P. Hojrup, Mol. Cell. Proteomics, 2007, 6, 638–647 CAS.
- C. C. Nwosu, J. S. Strum, H. J. An and C. B. Lebrilla, Anal. Chem., 2010, 82, 9654–9662 CrossRef CAS PubMed.
- J. Amano, T. Nishikaze, F. Tougasaki, H. Jinmei, I. Sugimoto, S. Sugawara, M. Fujita, K. Osumi and M. Mizuno, Anal. Chem., 2010, 82, 8738–8743 CrossRef CAS PubMed.
- K. Stavenhagen, H. Hinneburg, M. Thaysen-Andersen, L. Hartmann, D. Varon Silva, J. Fuchser, S. Kaspar, E. Rapp, P. H. Seeberger and D. Kolarich, J. Mass Spectrom., 2013, 48, 627–639 CrossRef CAS PubMed.
- Y. Zhang, E. P. Go and H. Desaire, Anal. Chem., 2008, 80, 3144–3158 CrossRef CAS PubMed.
- K. Deguchi, H. Ito, T. Baba, A. Hirabayashi, H. Nakagawa, M. Fumoto, H. Hinou and S. Nishimura, Rapid Commun. Mass Spectrom., 2007, 21, 691–698 CrossRef CAS PubMed.
- H. Ito, Y. Takegawa, K. Deguchi, S. Nagai, H. Nakagawa, Y. Shinohara and S. Nishimura, Rapid Commun. Mass Spectrom., 2006, 20, 3557–3565 CrossRef CAS PubMed.
- M. Wuhrer, M. I. Catalina, A. M. Deelder and C. H. Hokke, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 849, 115–128 CrossRef CAS PubMed.
- U. M. Demelbauer, M. Zehl, A. Plematl, G. Allmaier and A. Rizzi, Rapid Commun. Mass Spectrom., 2004, 18, 1575–1582 CrossRef CAS PubMed.
- G. Zauner, R. P. Kozak, R. A. Gardner, D. L. Fernandes, A. M. Deelder and M. Wuhrer, Biol. Chem., 2012, 393, 687–708 CrossRef CAS PubMed.
- Y. Mechref, Curr Protoc Protein Sci, 2012, ch. 12, Unit 12 11pp. 1–11 Search PubMed.
- E. D. Dodds, Mass Spectrom. Rev., 2012, 31, 666–682 CrossRef CAS PubMed.
- M. A. Ritchie, A. C. Gill, M. J. Deery and K. Lilley, J. Am. Soc. Mass Spectrom., 2002, 13, 1065–1077 CrossRef CAS PubMed.
- F. Wang, A. Nakouzi, R. H. Angeletti and A. Casadevall, Anal. Biochem., 2003, 314, 266–280 CrossRef CAS PubMed.
- Y. Satomi, Y. Shimonishi and T. Takao, FEBS Lett., 2004, 576, 51–56 CrossRef CAS PubMed.
- Y. Satomi, Y. Shimonishi, T. Hase and T. Takao, Rapid Commun. Mass Spectrom., 2004, 18, 2983–2988 CrossRef CAS PubMed.
- A. Kondo, M. Thaysen-Andersen, K. Hjerno and O. N. Jensen, J. Sep. Sci., 2010, 33, 891–902 CrossRef CAS PubMed.
- Z. Darula and K. F. Medzihradszky, Mol. Cell. Proteomics, 2009, 8, 2515–2526 CAS.
- J. W. Froehlich, M. Barboza, C. Chu, L. A. Lerno Jr, B. H. Clowers, A. M. Zivkovic, J. B. German and C. B. Lebrilla, Anal. Chem., 2011, 83, 5541–5547 CrossRef CAS PubMed.
- C. L. Woodin, D. Hua, M. Maxon, K. R. Rebecchi, E. P. Go and H. Desaire, Anal. Chem., 2012, 84, 4821–4829 CrossRef CAS PubMed.
- C. Zhang, A. Doherty-Kirby, R. Huystee Rv and G. Lajoie, Phytochemistry, 2004, 65, 1575–1588 CrossRef CAS PubMed.
- Z. M. Segu and Y. Mechref, Rapid Commun. Mass Spectrom., 2010, 24, 1217–1225 CrossRef CAS PubMed.
- K. Deguchi, H. Ito, Y. Takegawa, N. Shinji, H. Nakagawa and S. Nishimura, Rapid Commun. Mass Spectrom., 2006, 20, 741–746 CrossRef CAS PubMed.
- K. Hakansson, M. J. Chalmers, J. P. Quinn, M. A. McFarland, C. L. Hendrickson and A. G. Marshall, Anal. Chem., 2003, 75, 3256–3262 CrossRef CAS PubMed.
- R. R. Seipert, E. D. Dodds, B. H. Clowers, S. M. Beecroft, J. B. German and C. B. Lebrilla, Anal. Chem., 2008, 80, 3684–3692 CrossRef CAS PubMed.
- F. Kjeldsen, K. F. Haselmann, B. A. Budnik, E. S. Sorensen and R. A. Zubarev, Anal. Chem., 2003, 75, 2355–2361 CrossRef CAS PubMed.
- K. F. Haselmann, B. A. Budnik, J. V. Olsen, M. L. Nielsen, C. A. Reis, H. Clausen, A. H. Johnsen and R. A. Zubarev, Anal. Chem., 2001, 73, 2998–3005 CrossRef CAS PubMed.
- D. Wang, M. Hincapie, T. Rejtar and B. L. Karger, Anal. Chem., 2011, 83, 2029–2037 CrossRef CAS PubMed.
- Z. Zhu, D. Hua, D. F. Clark, E. P. Go and H. Desaire, Anal. Chem., 2013, 85, 5023–5032 CrossRef CAS PubMed.
- K. Takahashi, S. B. Wall, H. Suzuki, A. D. T. Smith, S. Hall, K. Poulsen, M. Kilian, J. A. Mobley, B. A. Julian, J. Mestecky, J. Novak and M. B. Renfrow, Mol. Cell. Proteomics, 2010, 9, 2545–2557 CAS.
- M. Wuhrer, J. C. Stam, F. E. van de Geijn, C. A. Koeleman, C. T. Verrips, R. J. Dolhain, C. H. Hokke and A. M. Deelder, Proteomics, 2007, 7, 4070–4081 CrossRef CAS PubMed.
- W. R. Alley Jr, Y. Mechref and M. V. Novotny, Rapid Commun. Mass Spectrom., 2009, 23, 161–170 CrossRef PubMed.
- A. Halim, U. Ruetschi, G. Larson and J. Nilsson, J. Proteome Res., 2013, 12, 573–584 CrossRef CAS PubMed.
- J. Nilsson and G. Larson, Methods Mol. Biol., 2013, 951, 79–100 CAS.
- A. Halim, J. Nilsson, U. Ruetschi, C. Hesse and G. Larson, Mol. Cell. Proteomics, 2012, 11, M111 013649 Search PubMed.
- N. E. Scott, B. L. Parker, A. M. Connolly, J. Paulech, A. V. Edwards, B. Crossett, L. Falconer, D. Kolarich, S. P. Djordjevic, P. Hojrup, N. H. Packer, M. R. Larsen and S. J. Cordwell, Mol. Cell. Proteomics, 2011, 10, M000031–MCP000201 Search PubMed.
- G. Hart-Smith and M. J. Raftery, J. Am. Soc. Mass Spectrom., 2012, 23, 124–140 CrossRef CAS PubMed.
- X. Yin, M. Bern, Q. Xing, J. Ho, R. Viner and M. Mayr, Mol. Cell. Proteomics, 2013, 12, 956–978 CAS.
- C. Sottani, M. Fiorentino and C. Minoia, Rapid Commun. Mass Spectrom., 1997, 11, 907–913 CrossRef CAS PubMed.
- J. Zhao, D. M. Simeone, D. Heidt, M. A. Anderson and D. M. Lubman, J. Proteome Res., 2006, 5, 1792–1802 CrossRef CAS PubMed.
- U. Kuzmanov, C. R. Smith, I. Batruch, A. Soosaipillai, A. Diamandis and E. P. Diamandis, Proteomics, 2012, 12, 799–809 CrossRef CAS PubMed.
- U. Kuzmanov, N. Jiang, C. R. Smith, A. Soosaipillai and E. P. Diamandis, Mol. Cell. Proteomics, 2009, 8, 791–798 CAS.
- V. Sanz-Nebot, E. Balaguer, F. Benavente, C. Neususs and J. Barbosa, Electrophoresis, 2007, 28, 1949–1957 CrossRef CAS PubMed.
- C. Neususs, U. Demelbauer and M. Pelzing, Electrophoresis, 2005, 26, 1442–1450 CrossRef PubMed.
- E. Balaguer, U. Demelbauer, M. Pelzing, V. Sanz-Nebot, J. Barbosa and C. Neususs, Electrophoresis, 2006, 27, 2638–2650 CrossRef CAS PubMed.
- K. Kakehi, M. Kinoshita, D. Kawakami, J. Tanaka, K. Sei, K. Endo, Y. Oda, M. Iwaki and T. Masuko, Anal. Chem., 2001, 73, 2640–2647 CrossRef CAS PubMed.
- E. Watson and F. Yao, Anal. Biochem., 1993, 210, 389–393 CrossRef CAS PubMed.
- M. Kinoshita, E. Murakami, Y. Oda, T. Funakubo, D. Kawakami, K. Kakehi, N. Kawasaki, K. Morimoto and T. Hayakawa, J. Chromatogr., A, 2000, 866, 261–271 CrossRef CAS PubMed.
- A. Rogowska-Wrzesinskaa, M.-C. Le Bihan, M. Thaysen-Andersen and P. Roepstorff, J. Proteomics, 2013, 88, 4–13 CrossRef PubMed.
- S. Barrabes, A. Sarrats, E. Fort, R. De Llorens, P. M. Rudd and R. Peracaula, Electrophoresis, 2010, 31, 2903–2912 CrossRef CAS PubMed.
- Y. Zhang, A. R. Sinaiko and G. L. Nelsestuen, Methods Mol. Biol., 2012, 909, 141–150 CAS.
- S. F. Wheeler, P. M. Rudd, S. J. Davis, R. A. Dwek and D. J. Harvey, Glycobiology, 2002, 12, 261–271 CrossRef CAS PubMed.
- E. Gimenez, F. Benavente, J. Barbosa and V. Sanz-Nebot, Rapid Commun. Mass Spectrom., 2007, 21, 2555–2563 CrossRef CAS PubMed.
- E. Gimenez, F. Benavente, J. Barbosa and V. Sanz-Nebot, Anal. Bioanal. Chem., 2010, 398, 357–365 CrossRef CAS PubMed.
- G. Stubiger, M. Marchetti, M. Nagano, C. Reichel, G. Gmeiner and G. Allmaier, Rapid Commun. Mass Spectrom., 2005, 19, 728–742 CrossRef PubMed.
- F. G. Hanisch, Anal. Chem., 2011, 83, 4829–4837 CrossRef CAS PubMed.
- M. Wilm and M. Mann, Anal. Chem., 1996, 68, 1–8 CrossRef CAS PubMed.
- M. Karas, U. Bahr and T. Dulcks, Fresenius. J. Anal. Chem., 2000, 366, 669–676 CrossRef CAS PubMed.
- K. Nagy, K. Vekey, T. Imre, K. Ludanyi, M. P. Barrow and P. J. Derrick, Anal. Chem., 2004, 76, 4998–5005 CrossRef CAS PubMed.
- D. Pousset, V. Piller, N. Bureaud, M. Monsigny and F. Piller, Cancer Res., 1997, 57, 4249–4256 CAS.
- G. A. Fragkiadakis and E. K. Stratakis, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1997, 117, 545–552 CrossRef CAS.
- M. H. Ravindranath, D. L. Morton and R. F. Irie, Cancer Res., 1989, 49, 3891–3897 CAS.
- M. Denis, P. D. Palatty, N. R. Bai and S. J. Suriya, Eur. J. Biochem., 2003, 270, 4348–4355 CrossRef CAS PubMed.
- C. Mandal, Methods Mol. Biol., 2013, 981, 57–93 CAS.
- S. L. Diaz, V. Padler-Karavani, D. Ghaderi, N. Hurtado-Ziola, H. Yu, X. Chen, E. C. Brinkman-Van der Linden, A. Varki and N. M. Varki, PLoS One, 2009, 4, e4241 Search PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- X. Ni, M. Castanares, A. Mukherjee and S. E. Lupold, Curr. Med. Chem., 2011, 18, 4206–4214 CrossRef CAS PubMed.
- S. Cho, B. R. Lee, B. K. Cho, J. H. Kim and B. G. Kim, Biotechnol. Bioeng., 2013, 110, 905–913 CrossRef CAS PubMed.
- N. Yamamoto, A. Takayanagi, T. Sakakibara, P. E. Dawson and Y. Kajihara, Tetrahedron Lett., 2006, 47, 1341–1346 CrossRef CAS.
- B. L. Wilkinson, R. S. Stone, C. J. Capicciotti, M. Thaysen-Andersen, J. M. Matthews, N. H. Packer, R. N. Ben and R. J. Payne, Angew. Chem., Int. Ed. Engl., 2012, 51, 3606–3610 CrossRef CAS PubMed.
- J. D. Tipton, J. C. Tran, A. D. Catherman, D. R. Ahlf, K. R. Durbin and N. L. Kelleher, J. Biol. Chem., 2011, 286, 25451–25458 CrossRef CAS PubMed.
- L. M. Smith, N. L. Kelleher, M. Linial, D. Goodlett, P. Langridge-Smith, Y. Ah Goo, G. Safford, L. Bonilla, G. Kruppa, R. Zubarev, J. Rontree, J. Chamot-Rooke, J. Garavelli, A. Heck, J. Loo, D. Penque, M. Hornshaw, C. Hendrickson, L. Pasa-Tolic, C. Borchers, D. Chan, N. Young, J. Agar, C. Masselon, M. Gross, F. McLafferty, Y. Tsybin, Y. Ge, I. Sanders, J. Langridge, J. Whitelegge and A. Marshall, Nat. Methods, 2013, 10, 186–187 CrossRef CAS PubMed.
- E. D. Green, G. Adelt, J. U. Baenziger, S. Wilson and H. Van Halbeek, J. Biol. Chem., 1988, 263, 18253–18268 CAS.
- M. Nakano, R. Saldanha, A. Gobel, M. Kavallaris and N. H. Packer, Mol. Cell. Proteomics, 2011, 10, M111 009001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |