 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural analysis of glycoprotein sialylation – Part I: pre-LC-MS analytical strategies†
Morten Thaysen-Andersena, Martin R. Larsenb, Nicolle H. Packera and Giuseppe Palmisano*bc
aDepartment of Chemistry and Biomolecular Sciences, Macquarie University, Sydney, Australia
bDepartment of Biochemistry and Molecular Biology, University of Southern Denmark, Campusvej 55, DK-5230, Odense, Denmark. E-mail: giuseppe@bmb.sdu.dk; Fax: +45 6550 2467; Tel: +45 6550 2342
cInstitute of Biomedical Sciences, Department of Parasitology – USP, São Paulo, Brasil
First published on 16th September 2013
Abstract
Sialic acids are carried by glycoproteins, proteoglycans and glycolipids as terminal entities of larger glycan structures and form a heterogeneous group of important monosaccharides in a wide range of biological systems in nature. Spatial and temporal structural characterisation of sialoglycoconjugates is required to understand their function. In this first of two related reviews we outline the available strategies for the analysis of mammalian N- and O-linked glycoprotein sialylation and summarise the associated sample handling methodologies that are a prerequisite for successful experimental designs including methods for enrichment, isolation, derivatisation and metabolic labelling. The downstream liquid chromatography (LC) mass spectrometry (MS) based separation and detection of N- and O-linked glycoprotein sialylation is covered in the second review. Since glycoprotein sialylation can be studied on multiple analyte levels, the analytical strategies and pre-LC-MS methodologies are covered separately for sialoglycans, sialoglycopeptides and intact sialoglycoproteins. Workflows to analyse glycoprotein sialylation at the glycomics level are particularly mature and the analytical chemist has multiple tools and technologies to acquire structural information on released glycans even at the system-wide level. The availability of analytical tools to study site-specific glycoprotein sialylation in the form of sialoglycopeptides or intact sialoglycoproteins is increasing through the development of sialic acid specific enrichment and labelling tools. However, the glycoproteomics route remains comparatively more challenging even when relatively simple protein mixtures are analysed. Evidenced by the wealth of available literature reviewed here, the glycoscience community has invested significant efforts to improve the analysis of glycoprotein sialylation.
1. Introduction
With more than 60 structurally different monosaccharides, sialic acids constitute a large subset of the even larger family of nine-carbon a-keto acids called nonulosonic acids. Although sialic acids can occur as free monomers or polymers in nature, they are more generally found as terminal entities at the non-reducing end of carbohydrates, oligosaccharides or sugars (hereafter collectively called glycans) carried by secreted or cell surface bound glycoconjugates including glycoproteins, proteoglycans and glycolipids.1,2The expression of sialic acids was previously thought to be unique to organisms of the deuterostome lineage as well as fungi and pathogenic bacteria; however, the development of more sensitive and accurate analytical techniques has found that sialic acids are distributed across a wide range of species and that they may be quite ancient in their origin.3–6 As such, it is estimated that sialic acids have populated the Earth for around 500 million years starting from the Cambrian explosion colonising the entire phylogenetic spectrum from the primitive platyhelminth Polychoerus carmelensis to eukaryotes.7
The most common mammalian sialic acids are N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc).8,9 Neu5Gc nucleotide sugars, cytidine 5-monophosphate (CMP)-Neu5Ac, are not synthesised in humans due to an irreversible mutation in the gene encoding for the CMP-Neu5Ac hydroxylase, the enzyme responsible for the conversion of CMP-Neu5Ac to CMP-Neu5Gc.10 Neu5Gc, which has been described to be antigenic for humans,11,12 has, nonetheless, been found in sialoglycoconjugates isolated from cultured human cells and tissues,12,13 in particular in fetal tissue and malignant tumours.12,14–19 However, it cannot be excluded that the Neu5Gc was introduced from exogenous sources in these studies.20 The synthesis and degradation of glycoconjugate sialylation is mediated directly by sialyltransferases and sialyl hydrolases (also known as neuraminidases and sialidases),21 but additional enzymes and transporters are responsible for the generation, regulation and availability of CMP-Neu5Ac substrates in the cell. The possibility of expression of sialoglycoconjugates in plants has been a topic of intense debate due to the importance of plants as expression systems for biotherapeutics.22,23 Recently, it was shown that plants contain trace amounts of Neu5Ac and Neu5Gc residues, which, however, may have originated from non-plant sources.23,24 Although these results indicate the absence of Neu5Ac and Neu5Gc in plants, it leaves open the possibility for the presence of other sialic acid species.
The surface of any cell in nature comprises a variety of glycoconjugates including glycoproteins, proteoglycans and glycolipids forming the cellular glycocalyx layer. Sialic acid residues of these glycoconjugates are known as the “functional ornament” of the glycocalyx.25 This functional implication is facilitated, in part, by the external cellular localisation of the sialic acid residues allowing them to directly interact with the extracellular environment and, in part, by virtue of their unique physicochemical properties including their bulkiness, hydrophilicity and negative charge. Sialylated glycoconjugates are known to play key roles in several pathophysiological processes26 including viral infection,27,28 embryogenesis,29 inflammation,30,31 cardiovascular diseases,32–34 cancer35–37 and neural development.38,39 In addition, it has been reported that alterations in glycoprotein sialylation, as a consequence of various diseases, can change the downstream intracellular signalling.40 Furthermore, the analysis of glycoprotein sialylation in body fluids is gaining attention due to the fact that most FDA approved biomarkers used in cancer staging, prognosis and treatment selection, are glycoproteins.41–44 Finally, glycoprotein sialylation has been shown to be critical for regulating blood circulation half-life,45 bioavailability, stability,46 receptor interaction47 and immunogenicity48 of biotherapeutic products such as monoclonal antibodies and other N- and O-linked glycoproteins.
Essentially, each species expresses a unique “sialome” (alternatively called “sialiome”49), a term defined as the total array of sialic acids and their related glycoconjugates expressed by a defined system e.g. cell, tissue, organ, or organism at a specified time and condition.50 As such, the sialome represents a subset of the glycoconjugate unspecific “glycome” as well as a subset of the glycoconjugate specific “glycoproteome” and “glycolipidome”. However, it should be noted that the sialome was initially coined as a term to define the mRNA transcripts and proteins expressed in the salivary glands.51 Here we use the former definition. Unlike the genome sequence, which is virtually identical in every somatic cell type of an organism and undergoes relative few changes during the lifetime of the organism, the sialome is highly cell-specific and varies markedly with regard to time, space, and environmental cues including physiological status of the cell. These fluctuations are a result of the concerted action of sialyltransferases (EC 2.4.99),37 sialidases (EC 3.2.1.18)52 and other sialic acid specific enzymes involved in the biosynthesis and post-synthesis alterations. In addition, the levels of nucleotide sugars and the cellular transit times may directly or indirectly modulate and regulate the sialome. The qualitative and quantitative analytical characterisation of sialoglycoconjugates present in a cell is an important step towards enabling us to decipher the complex glycosylation code for specific pathophysiological conditions and to obtain a better understanding of disease mechanisms.
The two major sialic acids core structures are neuraminic acid (chemical name: 5-amino-3,5-dideoxy-D-glycero-D-galacto-2-nonulopyranos-1-onic acid) and 2-keto-3-deoxynonulosonic-acid (Kdn) (chemical name: 3-deoxy-D-glycero-D-galacto-2-nonulopyranos-1-onic acid), which share a nine carbon nonulosonic acid structure from which the other sialic acids can be synthesised. Neu5Ac (chemical name: 5-acetamido-2,4-dihydroxy-6-(1,2,3-trihydroxypropyl)oxane-2-carboxylic acid) and Neu5Gc (chemical name: 2,4-dihydroxy-5-[(2-hydroxyacetyl)amino]-6-[1,2,3-trihydroxypropyl]oxane-2-carboxylic acid) are the most common sialic acid structures found in mammalian cells and, hence, are the focus of this review (see Fig. 1 for overview of common sialic acid structures and sialoconjugates). The sialic acid structures may be modified by for example acetylation, methylation and sulfation, which are covalently linked through the various hydroxyl groups on the carbon atoms of the sialic acids to generate additional layers of structural heterogeneity. Similar to the unmodified sialic acid structures, the modified counterparts are known to be involved in pathophysiological phenomena including as tumour development,53–55 immunity56,57 and hormone function.58 However, due to the lability and structural migration of these acid- and heat-sensitive sialic acid modifications during sample preparation and analysis, little is known about their function or tissue-specific expression.59,60
 | ||
| Fig. 1 Chemical structures and masses (in hydrolysed form) of the most common mammalian sialic acids (i.e. Neu5Ac, Neu5Gc and Kdn) and some examples of common mammalian sialylated N- and O-linked glycans and determinants. The core structures for the four main O-linked core types (core 1–4) have been highlighted (broken lines). The R1–9 groups in red indicate the modification position and modification moieties that form the more than 60 structurally different sialic acids. | ||
Another layer of structural diversity of sialic acids arises from the sialyl linkages to the adjacent monosaccharide by two main sialyl linkage configurations of N- and O-linked sialoglycans i.e. the α2,3- and α2,6-linkage. Both configurations are commonly used for the linkage of Neu5Ac and Neu5Gc to galactose (Gal) residues, whereas only α2,6 seems to be used for the sialyl linkage to N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) residues. Other configurations, such as the α2,8- and α2,9-sialyl linkages, which often are found in the polymeric form of sialic acids known as oligo- or polysialic acids have so far generally been limited to glycoconjugates expressed in neural tissue including the neural cell adhesion molecule.61 However, recent analytical developments have indicated their more wide-spread expression and localisation, for example in secreted epidermal growth factor receptor (EGFR) from an epidermoid carcinoma cell line.62
The structural characterisation of sialic acids and their glycoconjugates has together with functional glycobiology increased our understanding of sialylation over many decades (see timeline for significant sialic acid discoveries, ESI, Fig. S1†). Although a thorough historical review of the techniques for structural analysis of sialylated compounds is outside the scope of this review, it is well accepted that modern technology has improved the analytical depth, sensitivity and speed of such analyses. Several excellent reviews on protein glycosylation analysis in general have been published focusing on different analytical aspects.63–67 Moreover, a description of the sialome complexity from an experimental point of view has been reported68 and the recent advances in the biology and chemistry of sialic acids have been concisely reviewed69.
In the two related reviews presented here we will focus our attention on the analysis of sialic acids, in particular Neu5Ac and less on Neu5Gc, carried by N- and O-linked glycoproteins, which are the two most abundant mammalian classes of glycoproteins. In N-linked glycosylation, glycans are linked to the polypeptide chain through asparagine residues in conserved sequons (Asn-Xxx-Ser/Thr/Cys, where Xxx ≠ Pro).70–72 Lately, other non-canonical sequons have been proposed,73 but these are controversial and may turn out to be artefacts.74,75 However, recent studies have shown the presence of glutamine-linked protein glycosylation and other non-canonical asparagine-linked glycosylation motifs on recombinant human antibodies76,77 opening up for the presence of other types of protein glycosylation. Three main N-glycan types exist, all sharing a common chitobiose core; high mannose, hybrid and complex types. Of these, only the two latter types have been reported to be sialylated. In contrast, eight different core types exist for mammalian O-linked glycosylation (alternatively called mucin-type glycosylation), in which a GalNAc residue connects the O-glycan structure to the polypeptide backbone through a serine or threonine residue. All the O-glycan core types may carry sialylation as terminal modifications.
In this first of two related reviews we briefly outline the common analytical strategies and summarise in more detail the methodologies and considerations for pre-liquid chromatography (LC) and mass spectrometry (MS) sample handling for the analysis of sialylated N- and O-linked glycoproteins. Aspects covered include analyte enrichment and prefractionation, chemical derivatisation and metabolic labelling as all of these are important components of the experimental design to achieve informative structural data. The second review covers the downstream separation and detection of sialylation on N- and O-linked glycoproteins using modern LC-MS based approaches.78 Glycoprotein sialylation can be studied based on the analysis of sialoglycans, sialoglycopeptides and intact sialoglycoproteins. These three analyte levels are consequently reviewed separately in these two reviews. Evidenced by the wealth of available literature, considerable efforts have been invested to improve the analytical strategies and pre-LC-MS tools for the analysis of glycoprotein sialylation.
2. Analytical strategies for analysis of glycoprotein sialylation
Comprehensive analysis of N- and O-linked glycoprotein sialylation requires the use of well-structured experimental designs to obtain informative structural data. This section will in general terms briefly outline some analytical strategies developed to investigate the three main analytes for studying glycoprotein sialylation i.e. sialoglycans, sialoglycopeptides, and intact sialoglycoproteins.2.1 Multiple levels of structural information of glycoprotein sialylation
The choice of a specific analytical route when initiating an investigation of glycoprotein sialylation depends on the abundance of the sialylated glycoprotein of interest and most importantly on the molecular complexity and dynamic range of the mixture in which it is found. In addition, the choice of analytical strategy depends on the type of sialic acid-specific structural information that is desired since the different routes will yield different (but somewhat overlapping) subsets of the complete sialoglycoprotein structure. It is commonly accepted that no single analytical approach to date will provide the complete set of structural information on a sialoglycoprotein from a single experiment.The structural information of glycoprotein sialylation can be divided into the following categories: (i) identification and quantitation of the glycoprotein carrier of sialylation, (ii) localisation of the glycosylation site(s) and specification of the total glycan occupancy of the site(s), (iii) determination of the sialo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :asialo ratio of the glycans occupying the glycosylation site(s), (iv) characterisation of the sialoglycan structures attached to the glycosylation site(s) and finally (v) determination of the molecular relationship between sialoglycans and neutral glycans and other PTMs occupying different sites on the polypeptide backbone. The characterisation of the sialoglycan structure itself (iv) can be divided into the following sub-levels; determination of (a) monosaccharide composition e.g. determine the number of Neu5Ac/Neu5Gc residues, (b) topology e.g. establish on which N-glycan arm (3- or 6-mannose arm) the sialic acid is located, (c) sialyl linkages e.g. determine α2,3- or α2,6-sialyl linkage and (d) relative abundance of the multiple sialoglycans that often occupy a specific glycosylation site. Although the methods for initial sample preparation vary significantly for different types of biological samples (e.g. blood serum, extracts from tissues and cultured cell lysates), the subsequent analytical routes usually fall into the category of either glycomics or glycoproteomics type approaches.79–83 Several excellent reviews partly or fully devoted to glycomics67,84–88 and glycoproteomics65,89–93 analysis in general have been published, and the reader is kindly referred to these resources for an introduction to glycosylation analysis. Herein, we describe the specific analytical strategies for the study of protein N- and O-linked glycosylation with a sialic acid-centric focus.
:asialo ratio of the glycans occupying the glycosylation site(s), (iv) characterisation of the sialoglycan structures attached to the glycosylation site(s) and finally (v) determination of the molecular relationship between sialoglycans and neutral glycans and other PTMs occupying different sites on the polypeptide backbone. The characterisation of the sialoglycan structure itself (iv) can be divided into the following sub-levels; determination of (a) monosaccharide composition e.g. determine the number of Neu5Ac/Neu5Gc residues, (b) topology e.g. establish on which N-glycan arm (3- or 6-mannose arm) the sialic acid is located, (c) sialyl linkages e.g. determine α2,3- or α2,6-sialyl linkage and (d) relative abundance of the multiple sialoglycans that often occupy a specific glycosylation site. Although the methods for initial sample preparation vary significantly for different types of biological samples (e.g. blood serum, extracts from tissues and cultured cell lysates), the subsequent analytical routes usually fall into the category of either glycomics or glycoproteomics type approaches.79–83 Several excellent reviews partly or fully devoted to glycomics67,84–88 and glycoproteomics65,89–93 analysis in general have been published, and the reader is kindly referred to these resources for an introduction to glycosylation analysis. Herein, we describe the specific analytical strategies for the study of protein N- and O-linked glycosylation with a sialic acid-centric focus.
2.2 Glycomics vs. glycoproteomics oriented approaches
In the glycomics oriented approach, system-wide glycoprotein sialylation is studied in the form of sialoglycans. This typically involves the enzymatic or chemical release of the sialylated glycans from the glycoprotein carriers, followed by appropriate sample preparation steps. Single or multiple derivatisation step(s) of the sialoglycans can be included before they are separated and detected by LC-MS and tandem MS (see Fig. 2 for an overview of the general analytical approaches used to study N- and O-linked glycoprotein sialylation and the relevant sections/paragraphs covering these topics in these two reviews). The data interpretation for the glycomics approach can be assisted by the availability of a limited number of bioinformatic tools, but in most cases still fails to be fully automated. Please note that bioinformatic aspects of the glycoprotein sialylation analysis are not covered in these two reviews. Instead the reader is referred to other resources for detailed overviews and reviews.63,94–96 Although the glycomics approach suffers from being protein site-unspecific due to the release of the sialoglycans from the protein carrier components, it benefits from having more streamlined and optimised workflows to choose from compared to the current glycoproteomics approaches. In addition, a more comprehensive structural analysis of the sialoglycan structures can typically be obtained when choosing the glycomics route.97,98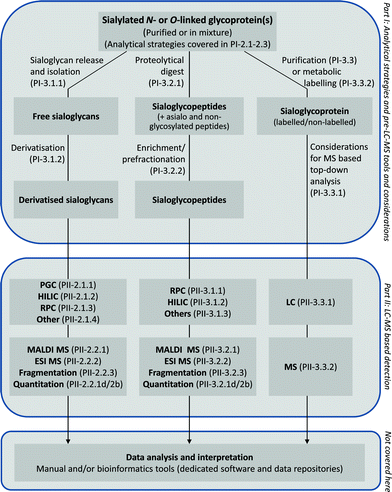 | ||
| Fig. 2 General overview of the analytical strategies for the analysis of glycoprotein sialylation and topics covered in Part I and II of these two related reviews. PI and PII refer to Part I and Part II,78 respectively, followed by paragraph numbers. Bioinformatic aspects of glycoprotein sialylation analysis are not covered by the two reviews. | ||
In the glycoproteomics oriented approach, glycoprotein sialylation is studied in the form of sialoglycopeptides or alternatively, but much less commonly, in the form of intact sialoglycoproteins. Such strategies usually consist of initial enrichment of the sialylated glycopeptides from peptide mixtures obtained by proteolysis of the glycoproteins in the sample, followed by a one or two-dimensional LC separation, detection using MS and tandem MS, and finally manual or software assisted data interpretation. Additional steps, such as metabolic and chemical labelling may be introduced to aid the enrichment and/or detection. Desialylation and even complete deglycosylation prior to LC-MS is commonly performed in experimental designs of a more proteomics type character where the aim is large-scale glycosylation site identification of previously sialylated (or previously glycosylated) peptides.99,100 In the case of intact sialoglycoprotein analysis, thorough single protein isolation/purification and appropriate sample handling including desalting and concentration are typically required prior to on- or off-line separation and detection with LC- or capillary electrophoresis (CE)-MS. Glycoprotemics approaches benefit from providing site-specific information by yielding the identity of the protein carriers of the sialoglycans and specifying the occupied glycosylation site(s), in addition to obtaining some information on the sialoglycan structure. However, this additional structural information most often comes at a cost; the sialoglycopeptides (and sialoglycoproteins) are comparatively more difficult to study than released sialoglycans, in particular, in system-wide experiments where complex protein mixtures usually are the starting material. This is, in part, due to the higher molecular complexity of the sialoglycopeptides (analysis of two conjugated biomolecules i.e. sialoglycans and peptides/proteins) and, in part, due to the fact that sialoglycopeptides are only weakly detected in MS as a result of their microheterogeneity and their lower signal intensity65,101 compared to unglycosylated peptides as discussed in the second of these two reviews.78 Employing efficient sialoglycopeptide enrichment, separation and detection techniques in the analytical workflow reduces this limitation when performing glycoproteomic type experiments.
2.3 Glycoprotein-specific vs. glycoproteome-wide approaches
As a consequence of the analytical challenges involved with the study of sialoglycopeptides, most published site-specific glycoprofiling studies have characterised glycoprotein sialylation from purified sialoglycoproteins or from simple protein mixtures46,47,102–105 (list of profiled mammalian N-glycoproteins can be found in ESI† in Thaysen-Andersen and Packer, Glycobiology, 2012, 22, 1440–1452 (ref. 106)). The sialylated glycoproteins can be isolated and purified by different biophysical and affinity methods depending on their biological origin,107 which will not be covered here. Many cell surface membrane and soluble secreted sialoglycoproteins have been targeted for characterisation since sialylation has been suggested to be involved in cell–cell and cell–matrix interactions.108Crude glycoprotein isolation by subcellular fractionation is a method for reducing the protein complexity. Organelle purification can also be used to investigate the role of sialic acids and their biosynthetic enzymes in specific organelles or sub-compartments such as the endoplasmic reticulum or the individual compartments of the Golgi apparatus, under healthy and diseased conditions.109–111 Isolation steps may, however, lead to severe analyte losses or can introduce significant analyte biases. For example, trichloroacetic acid (TCA) precipitation, which is a commonly used protein isolation technique in many workflows, is seemingly biased towards disordered/unfolded proteins112 as well as sialylated glycoproteins107 affecting, in total, more than one-third of human proteins.113 Thus, TCA precipitation is potentially disturbing the qualitative and quantitative analysis of many glycoproteins when included in the sample work-up. Hence, detailed structural analysis of N- and O-linked glycoprotein sialylation on the glycoproteome or sub-glycoproteome-wide scale without previous fractionation/purification is desirable in order to avoid any bias resulting from the isolation steps. However, this is extremely challenging when starting with complex samples of biological origin. Efficient sialic acid specific enrichment coupled with high performance modern LC-MS detection has expanded the potential for sialoglycoprotein profiling from complex samples such as cells, tissues and body fluids. However, glycoprotein sialylation analysis, in particular when performed via the glycoproteomics route, clearly still needs further development of robust techniques and integrated workflows covering all aspects from sample preparation to data acquisition. In addition, the system-wide glycoproteomic type analysis of glycoprotein sialylation is hampered by the lack of dedicated computational tools, since the characterisation of branched sialoglycans and sialoglycopeptides creates unique challenges that are not encountered in conventional proteomics and other related research areas.
3. Pre-LC-MS tools and considerations
The following describes pre-LC-MS aspects relating to the analysis of sialylated N- and O-linked glycoproteins that are important for a well-structured experimental design when downstream LC-MS analysis is employed. Pre-LC-MS aspects relating to the handling of sialoglycans, sialoglycopeptides and sialoglycoproteins are separated in this section.3.1 Sialoglycans
Sialylated N-glycans are most commonly released by N-glycosidase F (PNGase F) from Flavobacterium meningosepticum or almond N-glycosidase A (PNGase A). In contrast to PNGase F, PNGase A has the ability to cleave N-glycans containing an α1,3-linked fucose on the reducing end GlcNAc residue as found in insect and plant glycoproteins.116 The N-linked glycan release can be performed on purified sialoglycopeptides and sialoglycoproteins or on complex mixtures of such molecules on solid supports (e.g. PVDF membranes), in gels (e.g. 1D gel-electrophoresis) or in solution.82,117 The terminal sialic acid residues do not seem to alter the accessibility of the PNGase F/A to the substrates. This enables a quantitative release of sialylated N-glycans (personal unpublished observation). However, it should be noted that enzymatic release of sialoglycans from intact proteins is dependent on the sialoglycoprotein conformation since the sialoglycans need to be well exposed for complete release. Reduction and alkylation of the cysteine residues of the sialoglycoproteins and general protein denaturation will, thus, usually enhance the rate and extent of deglycosylation.
No enzymatic approach is available for the release of all O-linked sialoglycans that densely glycosylate mucins or mucin-type sialoglycoproteins. O-Glycosidase from Enterococcus faecalis removes core 1 and core 3 type O-linked glycans from glycoproteins, but only after previous desialylation. In contrast, O-linked sialoglycans can be released broadly by reductive beta-elimination,82 which promises to quantitatively release all sialoglycans while retaining the sialic acid residues on the released O-glycans. Reductive beta-elimination of sialoglycans can be performed either in-solution or from glycoproteins immobilised on solid supports. Alternatively, hydrazinolysis with anhydrous hydrazine (H2NNH2) can be used as a method for releasing sialoglycans. It was recently shown that specific hydrazinolysis reaction conditions including reaction time, temperature, chemical reactants and reaction vials are crucial to avoid significant losses of sialic acids from the sialoglycoproteins.118
The combination of high hydrophilicity and relative small size is a unique physicochemical feature that allows relative easy isolation of released N- or O-linked sialoglycans from their protein carriers and other large molecules present in samples of biological origin. For example, passing released sialoglycans over a hydrophobic reversed-phase column in a solid phase extraction (SPE) format will selectively allow the hydrophilic glycans to pass through whilst retaining the more hydrophobic protein/peptide components. Isolation methods for sialoglycans can, in addition, utilise the negative charge of the sialic acid residues for selective isolation by e.g. using TiO2 (discussed later for the enrichment of sialoglycopeptides in paragraph 3.2.2.f). Here, prior dephosphorylation is needed to avoid co-enrichment of phosphorylated peptides/proteins.49,119
In addition to permethylation, which methylates all hydroxyl and carboxyl groups of the sialoglycans, the derivatisations strategies for sialylated glycans can crudely be divided into those that specifically target the sialic acid residues at the non-reducing end and those which label the carrier glycan reducing end. The latter involves reductive amination and is a general approach in glycomics analysis.121,122 Thus, the common acid based reductive amination strategies including 2-aminobenzoic acid, 2-aminobenzamide or 2-aminopyridine labelling will only be mentioned in passing in these two reviews. Reducing end labelling under alkaline conditions e.g. with 1-(2-naphthyl)-3-methyl-5-pyrazolone is more favourable for sialoglycans than other common reagents used in reductive amination,123 due to the lower risk of loss of the acid-labile sialic acid residues. The non-reducing end derivatisation directly of the sialic acid residues aims at neutralising the carboxyl groups thus leaving the sialoglycans without any permanent charge. This can be performed using a variety of methods e.g. methyl esterification124–128 or amidation.129–131 The neutralisation stabilises the sialoglycans and limits the level of ionisation based loss of sialic acid as discussed in the second of these two reviews.78 No reactivity difference between the α2,3- and α2,6-linked NeuAc residues was reported for methyl esterification or amidation either in the derivatisation reaction itself or in the subsequent tandem MS fragmentation. In contrast, perbenzoylation132 and other types of sialic acid-specific derivatisations133,134 have different reactivities towards the two sialyl linkages, which therefore may be used to differentiate the linkage types. Permethylation is another commonly used derivatisation method in glycomics, which gives methyl esters of the acidic carboxyl group in addition to generally transforming the hydroxyl groups of the sialoglycans into methyl ethers.135,136 The sialic acid residues seem to cope with the reaction conditions without detectable losses, but permethylation reactions can suffer from significant under-methylation if reaction conditions are not optimal. In addition, permethylation is unsuitable for glycans containing O-acetylated sialic acid residues because of the chemical degradation of these groups during the harsh reaction conditions.137,138 Permethylation using heavy and light isotope labelled reagents can be introduced in the derivatisation protocol to obtain glycoform quantitation between two samples.139 Finally, acid-catalysed lactonisation i.e. internal esterification of the carboxyl to the hydroxyl group of the sialic acid residues has been employed to neutralise and discriminate between α2,8- and α2,9-linked polysialic acids for efficient MALDI MS detection.140 Irrespective of the method, sialic acid esterification has, in addition to the increased analyte stability, the advantage of allowing the analysis of sialoglycans in positive ion MS mode, where MALDI mass analyzers generally perform better in terms of sensitivity and resolution and where neutral glycans appear with higher signal strength.78
3.2 Sialoglycopeptides
| Method | Common applications | Limitations | Advantages | Examples of applications | ||
|---|---|---|---|---|---|---|
| Sialoglycans | Sialoglycopeptides | Sialoglycoproteins | ||||
| Affinity-based methods | ||||||
| Lectins (e.g. SNA, MAL, MAH, WGA) | x | x | Un-/low-specific binding of sialoglycan determinants limits enrichment of specific sialoglycoforms. | Prefractionation of crude sialoglycoprotein mixtures to lower complexity. | Enrichment of sialoglycopeptides from human serum proteins using SLAC.169 Enrichment of sialoglycoproteins from serum from pancreatic tumour patients using SNA, MAL and WGA lectins.171 | |
| Potential of sialoglycoprotein visualisation via lectin blotting and lectin histochemistry. | ||||||
| HILIC/ERLIC(e.g. ZIC, Amide-80) | (x) | x | Unspecific for sialylated species (affinity for all hydrophilic species). Multi-sialylated species difficult to elute. Relative low analyte solubility in organic mobile phase. | Possibilities for both pre-fractionation and enrichment in a variety of formats. | Separation of sialoglycopeptides from recombinant human interferon-gamma.278 Pre-fractionation of sialoglycopeptides from human platelets.279 | |
| Binding mechanism fairly well understood and can be manipulated. | ||||||
| SCX | x | x | Unspecific for sialylated species (isolate other negatively charged species). | Can separate sialylated isomers as a prefractionation step. | Prefractionation of sialoglycopeptides from human platelet membranes.188 | |
| Serotonin | x | x | Low enrichment efficiency. Binding mechanism not understood. | Can discriminate between Neu5Ac and Neu5Gc containing species by selective binding of the former. | Enrichment of sialylated N-glycans and N-glycopeptides from human serum transferrin in SPE format.186 | |
| COFRADIC | x | Low throughput due to re-chromatography. Reproducible chromatography required. | Localise sialoglycopeptide fractions by retention shift | Prefractionation of sialoglycopeptides from human serum.190 | ||
| TiO2 | x | Affinity for modified sialic acids species not well-defined (e.g. Neu5Gc and Kdn). Oligo and polysialylated species difficult to elute. | High enrichment efficiency of sialoglycopeptides (needs dephosphorylation to avoid co-enrichment of phosphopeptides) | Enrichment of sialoglycopeptides from human plasma, saliva and mouse brain.49 | ||
| Chemistry-based | ||||||
| Chemical labelling | ||||||
| Periodate oxidation/hydrazide coupling/acid release | x | Reaction needs fine tuning to avoid oxidation of asialo-species. Loss of sialic acid structure information. | High enrichment affinity due to covalent capture of oxidised cis diols. | Capture and detection of sialoglycoproteins derived from human cerebrospinal fluid.99 | ||
| Reverse glycoblotting | x | x | Reaction needs fine tuning to avoid oxidation of asialo-species. Multiple chemical reactions cause sample loss and unwanted by-products. | Covalent capture of cis diols and release of intact sialylated glycopeptides and glycans. | Capture and detections of human α-fetoprotein, bovine pancreas fibrinogen human EPO and sialoglycopeptides232 from mouse serum.280 | |
| PAL | x | Reaction needs fine tuning to avoid oxidation of asialo-species. Unknown compatibility with some cell lines. | Labelling of cell surface sialylated proteins. Fast reaction kinetics. | Capture and detection of cell surface sialoglycoproteins from B-JA-B K20 B cells.229 | ||
| Metabolic labelling | ||||||
| ManNAz labelling/Staudinger ligation with phosphine | x | Low reaction kinetic. | Selective labelling of sialylated glycoproteins applied to living organisms. | Labelling and visualisation of sialoglycoproteins of Jurkat cells.252 | ||
| ManNAz labelling/CuAAC with alkyne probes | x | Toxicity of Cu catalyst limits in vivo applications. | Efficient and selective labelling of sialylated glycoproteins. | Labelling and visualisation of sialoglycoproteins of metastatic prostate cancer cells255 and mesenchymal stem cells.264 | ||
| Alkynyl ManNAc labelling/CuAAC with azide probes | x | Toxicity of Cu catalyst limits in vivo applications. | Efficient and selective labelling of sialylated glycoproteins. | Labelling and visualisation of sialoglycoproteins of prostate cancer cell lines266 and metastatic lung cancer cells.267 | ||
3.2.2.a Lectins. Lectins form a broad class of proteins of non-immune origin that recognise glycan entities of glycoproteins. Much of our early understanding of the expression of sialic acids in cells and tissues has been enabled by chemical analysis or histochemical staining with the sialic acid-specific lectins Sambucus nigra agglutinin (SNA) and Maackia amurensis leukoagglutinin-I (MAL-I).157 Over the decades they have gained considerable popularity as histological reagents in many areas of diagnostic analysis using light and electron microscopy to visualise temporal and spatial changes in the expression of cell membrane sialoglycoconjugates. This has significantly contributed to improving our understanding of the role of sialic acids in the physiology of vertebrate tissues and human disease.157,158 The functional groups of the polypeptide backbone and metal ions of the lectins enable their interaction with glycans through hydrogen bonds and van der Waals and hydrophobic interactions.159
Lectin affinity chromatography strategies involve the binding and separation of glycoproteins or glycopeptides bearing specific glycan determinants to immobilised lectins. A detailed overview of the distribution, specificity and function of sialic acid-specific lectins focusing on those that occur in viruses, bacteria and non-vertebrate eukaryotes has been published.160 The most common plant lectins used for the isolation of sialylated glycopeptides/glycoproteins are SNA, MAL-I, Maackia amurensis hemagglutinin (MAH), and Triticum vulgaris (wheat germ) agglutinin (WGA). The specificities of these lectins have been described at different resolution: SNA binds glycoconjugates containing α2,6-linked sialic acid residues,161 MAL binds most preferably to terminal Neu5Acα2,3Galβ1,4GlcNAc entities found in N-linked glycans,162,163 MAH binds preferentially to α2,3-linked sialic acid residues of O-linked disialylated tetrasaccharides with the specific structure Neu5Acα2,3Galβ1,3(Neu5Acα2,6)GalNAc,164 MAL-I and MAH also show specificity towards nonsialylated structures such as SO4-3-Galβ1,3GalNAc165 and, finally, WGA has more broad affinity to GlcNAc and sialic acid containing glycoconjugates.166 Molecules that are unspecifically bound to the immobilised lectins after analyte loading in appropriate solvents are subsequently removed by several washing steps followed by elution of the bound sialoglycoproteins or sialoglycopeptides by low pH solvents or by using sialic acids or sialic acid analogues for competitive elution. Several different formats such as single,167 multiple168 (MLAC) and serial169 lectin affinity chromatography (SLAC) along with different immobilised supports have been used for prefractionation of sialoglycopeptides and sialoglycoproteins.170 The use of lectin combinations such as concanavalin A (ConA), WGA and jackfruit jacalin, which show different specificities for sialic acid glycoconjugates, was used to isolate the majority of glycoproteins present in human serum in an MLAC format.168 Furthermore, SLAC based on SNA and ConA was used to prefractionate sialylated N- and O-linked glycopeptides derived from proteolytic digests of human serum proteins based on their degree of branching.169 Here it was found that partly sialylated biantennary N-glycans are abundant in human serum. In order to gain deeper coverage of the sialylated glycoproteome, other combinations of sialic acid specific lectins have been used.169,171 For example, SNA, MAL-I and WGA were used to prefractionate sialylated N-linked glycoproteins in serum derived from pancreatic tumour patients (described further in paragraph 3.3.1).
One of the under-recognised disadvantages of lectins is their rather limited specificity towards glycoconjugates.172 Although the lectins selectively and reproducibly retain some glycoproteins, other glycoproteins carrying the same repertoire of glycans, may not be retained.173 Detection of sialoglycoconjugates containing α2,3- and α2,6-linked sialic acids by SNA and MAL-I are furthermore affected by sialic acid modifications. In addition, it was reported that both SNA and MAL-1 bind sialoglycoconjugates with Kdn and Kdn derivatives at the non-reducing end of the glycan. Together, the lack of glycan specificity and shared protein/glycan binding epitopes limit the capacity of lectins to perform unbiased sialoglycoprotein enrichment. Instead lectins are more valuable in the prefractionation steps and for visualisation purposes using histochemical staining techniques and lectin blotting.
3.2.2.b HILIC/ERLIC. Hydrophilic interaction liquid chromatography (HILIC) is a variant of normal-phase chromatography (NPC) useful for the separation of hydrophilic compounds.174 This is achieved on polar or charged stationary phases using water-miscible organic solvents. HILIC is as such ideal for the analysis of protein glycosylation.87,175 The retention mechanisms of HILIC in the context of glycoprotein sialylation are discussed in the second of these two reviews.78
In one of the first reports using HILIC as a tool to enrich glycopeptides from peptide mixtures, a zwitterionic (ZIC) type of HILIC resin functionalised with sulfobetaines packed in SPE format microcolumns in GeLoader tips was used as the stationary phase.176 This enrichment setup was combined with a deglycosylation step using a mixture of endo-β-N-acetylglucosaminidases, which allowed the identification of 62 N-glycosylation sites from 37 N-linked glycoproteins derived from human plasma. Moreover, ZIC-HILIC SPE showed highly efficient and unbiased enrichment of sialylated and neutral N-glycopeptides from glycoproteins purified by gel electrophoresis, enabling the N-glycoprofiling of different sources of human tissue inhibitor of metalloproteinases-1.177–180 The enrichment efficiency for both neutral and sialoglycopeptides has been further improved by the addition of an ion-pairing reagent to the mobile phase i.e. trifluoroacetic acid (TFA), which reduces the hydrophilicity of non-glycosylated peptides comparably more than the glycosylated peptides in peptide mixtures by protonating all carboxyl groups at the low pH and by ion pairing with the protonated amino groups on the peptides.181 Several HILIC solid phase materials have been shown to all yield unbiased desalting/enrichment of glycosylated peptides in SPE formats without the loss of quantitative information.182 However, the study highlighted that column capacity is a critical parameter to consider. Hydrophilic and electrostatic interactions (attraction and repulsion) between the sialic acid residues and the sulfobetaine groups on the surface of the stationary phase influence the retention behaviour and are modulated by the degree of sialylation and the type of sialic acid linkage. It should be noted that the entire complement of sialylated glycopeptides may be difficult to elute from HILIC SPE columns in common acidic (e.g. TFA/formic acid) and alkaline (e.g. ammonium bicarbonate) solvents (Fig. 3). In order to evaluate the retention behaviour of sialylated glycopeptides on a HILIC stationary phase, bovine fetuin (Uniprot entry number: P12763) was tryptic digested and the peptide map was analysed by positive ion MALDI-TOF-MS (Fig. 3A). The peptide map was dominated by signals corresponding to non-glycosylated peptides in the lower m/z region which suppressed the glycopeptide ionisation. After loading onto a ZIC-HILIC microcolumn, the glycopeptides were selectively eluted under commonly used acidic conditions using aqueous 0.1% TFA (Fig. 3B). The MS signals in the m/z 3500–5000 range were assigned to the fetuin glycopeptides bearing the L145CPDCPLLAPLNDSR159 peptide moiety. Subsequently, remaining glycopeptides were eluted from the same ZIC-HILIC micro-column with 100 mM ammonium bicarbonate (ABC) in slightly basic conditions (Fig. 3C). The same MS signals were detected indicating an electrostatic retention effect. Moreover, glycopeptides were eluted from the same ZIC-HILIC micro-column with 2,5-dihydroxybenzoic acid (DHB) matrix (Fig. 3D). Several new MS peaks appeared in the m/z 5000–7000 region; these were associated with the glycopeptides bearing the V160VHAVEVALATFNAESNGSYLQLVEISR187 and R72PTGEVYDIEIDTLETTCHVLDPTPLANCSVR103 peptide moieties. The length of the peptide moiety strongly influenced the binding affinity to ZIC-HILIC sorbent. Furthermore, fetuin tryptic peptides were treated with sialidase A and mixed with untreated fetuin tryptic peptides in a 1:1 ratio. This mixture was loaded onto a ZIC-HILIC micro-column and glycopeptides eluted with 0.1% TFA (Fig. 3E). The elution of desialylated glycopeptides was detected. Subsequently, sialylated glycopeptides were eluted with DHB matrix from the same ZIC-HILIC micro-column (Fig. 3F). Taken together, these results show that pH conditions and solvent additives, such as DHB, influence the qualitative and quantitative glycopeptide elution profile after ZIC-HILIC enrichment. Moreover sialylated glycopeptides bind ZIC-HILIC sorbent stronger than non-sialylated glycopeptides under commonly used acidic conditions.
 | ||
| Fig. 3 Positive ion MALDI-TOF-MS of tryptic peptide mixture of bovine alpha-2-HS-glycoprotein (fetuin, Uniprot entry number: P12763) in linear mode under the following conditions: (A) general peptide mass map, (B) peptide mass map of glycopeptides eluted from the ZIC-HILIC micro-column using 0.1% TFA, (C) peptide mass map of glycopeptides subsequently eluted from the same ZIC-HILIC micro-column with 100 mM ABC (pH 7.8), (D) peptide mass map of glycopeptides subsequently eluted from the same ZIC-HILIC micro-column with DHB matrix, (E) fetuin tryptic peptides treated with sialidase A and mixed with untreated fetuin tryptic peptides. This mixture was loaded onto a ZIC-HILIC micro-column and glycopeptides eluted with 0.1% TFA, (F) subsequent elution with DHB matrix from the same ZIC-HILIC micro-column. | ||
Electrostatic repulsion hydrophilic interaction chromatography (ERLIC) has been employed for the enrichment of tryptic phosphopeptides.183 For this application the secondary interaction between the negatively charged phosphate groups of the phosphopeptides and the weak anion exchange chromatography (WAX) stationary phase was used for enhanced retention in addition to the main hydrophilic interactions. ERLIC has also been used to enrich glycopeptides from human platelets using a polyWAX (PolyLC Inc.) column with a gradient of decreasing acetonitrile concentration from 70% to 60% (v/v).184 This method enabled the identification of 125 glycosylation sites from 66 glycoproteins. Possible isoform separation was proposed due to the elution of the same glycopeptide in different chromatographic fractions, however, this was not verified. The retention mechanism of ERLIC in the context of glycopeptides has not been thoroughly described; however, the combination of WAX and hydrophilic interactions in ERLIC may yield an unbiased enrichment of sialoglycopeptides and neutral glycopeptides from peptide mixtures due to their difference in size and charge. In a recent application, ERLIC was used to simultaneously enrich the mouse brain phospho- and glycoproteome.149 A total of 544 unique glycoproteins and 383 phosphoproteins were identified and this method was shown to be superior to a method based on hydrazide chemistry (see paragraph 3.2.2.g). Comparison of ERLIC and strong cation exchange chromatography (SCX) in the prefractionation of phospho- and glycopeptides from rat kidney tissue showed a higher identification rate of glycoproteins and phosphoproteins in ERLIC due to the more uniform distribution of the modified peptides in this fractionation technique.185
3.2.2.c Serotonin. Serotonin-bonded silica material has been used to enrich sialic acid containing glycopeptides in SPE formats.186 This method is based on the affinity binding between serotonin and Neu5Ac, in which the N-acetyl group, but not the N-glycolyl of Neu5Gc, seems to have a crucial role.187 This enrichment strategy was applied to sialylated N-glycans and N-glycopeptides derived from human serum transferrin. The scarce knowledge of the binding mechanism, specificity and efficiency of serotonin as an enrichment step for sialic acid containing glycoconjugates has limited its use in glycomics and glycoproteomics.
3.2.2.d SCX. Strong cation exchange chromatography (SCX) has been used to prefractionate sialic acid containing glycopeptides from human platelet membranes, which enabled the identification of 148 sialylated N-glycosylation sites from 79 N-linked glycoproteins.188 This strategy relies on the reduced net charge of sialoglycopeptides at low pH. Indeed at pH 2.7, an acidity level commonly used for SCX, the carboxyl groups of amino acid side chains are protonated while carboxyl groups of sialic acid residues are partially deprotonated (pKa of sialic acid carboxyl group is 2–2.8).189 This reduces the binding of the sialoglycopeptides to the negatively charged SCX resin, and they are consequently found in the SCX flow through fraction.
3.2.2.e COFRADIC. Selective prefractionation and identification of sialylated glycopeptides has been achieved using a chromatography method defined as combined fraction diagonal chromatography (COFRADIC).190 In this strategy tryptic peptides are first separated using reversed phase chromatography (RPC). Sialic acid containing glycopeptides are then directly identified using a broad specificity neuraminidase from Arthrobacter ureafaciens to remove sialic acids and shift the retention time of the now desialylated glycopeptides in the second round of RPC. Using this technology a total of 93 N-glycosylation sites from 53 sialylated N-linked glycoproteins derived from serum were detected. Since COFRADIC requires desialylation between highly reproducible consecutive chromatographic runs for successful application the throughput of the method is limited.
3.2.2.f TiO2. Due to its high enrichment efficiency, titanium dioxide (TiO2) purification has become the most used chemo-affinity purification technique for the enrichment of phosphorylated peptides.191–194 The loading of the sample onto TiO2 under highly acidic conditions (5% TFA (v/v)) in a solution containing non-phosphopeptide excluders is widely applied in laboratories around the world to the study the cell signalling under different pathophysiological conditions. Moreover, the high specificity towards certain negatively charged molecules has enabled the use of TiO2 to capture other negatively charged post-translational modifications including glycosylphosphatidylinositol anchors, phospholipids, nucleic acids and sialylated glycopeptides.195,196 For the latter, the ability of TiO2 resin to enrich sialic acid containing glycopeptides was first shown in studies using human plasma and saliva using SPE formats.49 Acidic mobile phases (2–5% TFA (v/v)) containing glycolic acid in high organic containing solvent content during sample loading was used to reduce the unspecific binding of acidic non-glycosylated peptides and neutral glycopeptides. High pH ammonia buffers were used to elute the enriched sialylated glycopeptides from the TiO2 stationary phase. The eluted sialoglycopeptides were treated with PNGase F prior to LC-MS detection, which identified 192 and 97 N-glycosylation sites of formerly sialylated N-glycosylated human proteins derived from plasma and saliva, respectively. It should be noted that alkaline phosphatase treatment was introduced as a crucial step in the method to remove phosphates from phosphopeptides to avoid their co-enrichment. The high enrichment capacity towards sialoglycopeptides was validated using tryptic peptides derived from bovine fetuin treated with neuraminidase and mixed with the same sample labelled with H218O without neuraminidase treatment. The interaction of phosphates (or phosphopeptides) to TiO2 has been described as a bidentate binding.197 As an extension from this study, it was speculated that sialylated species have a multidentate binding.49 Based on this hypothesis modified sialic acids would be expected to display a different binding; indeed acetylated, methylated, deoxy- and dehydro-sialic acids and sialic acids with lactone and lactam showed significantly reduced or no binding affinity to TiO2. In contrast, sulfated and phosphorylated sialic acids had increased binding strength to TiO2 sorbents.
3.2.2.g Enrichment strategies for glycosylation site identification. The highly efficient TiO2 enrichment of sialylated glycopeptides has been combined with prefractionation of deglycosylated peptides using TskGel Amide-80 HILIC (TOSOH Bioscience) prior to LC-MS and tandem MS analysis.119,198 This strategy improved the coverage of the sialoglycoproteome by identifying 1632 unique formerly sialylated N-glycopeptides from less than 107 HeLa cells. The specificity of the TiO2 enrichment in this system-wide study was evaluated at the LC-MS level by counting the ratio of higher-energy C-trap dissociation (HCD) tandem MS spectra containing the three diagnostic ions for sialoglycopeptides (m/z 274.09 (B1-H2O-ion, Neu5Ac–H2O), m/z 292.09 (B1-ion, Neu5Ac) and m/z 657.24 (B3-ion, HexNAcHexNeu5Ac)) to the tandem spectra without these diagnostic fragment ions. Here, 95.1% of the MS/MS spectra contained the three diagnostic ions for sialoglycopeptides.
N-Linked glycoproteomics of myocardial ischemia and reperfusion injury was performed with a parallel use of three available enrichment strategies: hydrazide capture (see paragraph 3.2.2.h), TiO2 and ZIC-HILIC enrichment.199 The enriched glycopeptides were all prefractionated using off-line HILIC with UV detection prior to the downstream LC-MS detection. In total, 1556 N-linked glycosylation sites were identified. In another study, the glycoproteomic analysis of the Chardonnay wine revealed five grape glycoproteins enriched by the TiO2 resin.200 This opens up for the possibility of the presence of acidic sugars in grape. However, there has not been any detailed glycan analysis performed in wine to date to document this further.
Recently, a Ti(IV)-IMAC resin201 designed with titanium ions immobilised on microspheres through a flexible linker terminated with phosphonate groups was used to enrich sialoglycopeptides from serum. The authors developed an optimised strategy based on filter digestion and enrichment identifying 217 unique N-linked sialoglycopeptides from 1 μL of human serum following deglycosylation. In another recent study, the simultaneous enrichment, identification and quantification of phosphorylated and formerly sialylated peptides during mouse brain development were reported.100 Again, a multi-dimensional separation was used combining TiO2 SPE enrichment of phosphorylated and sialylated N-linked glycopeptides and HILIC prefractionation after PNGase F treatment. Each HILIC fraction was subsequently analysed by LC-MS and a total of 7682 unique phosphopeptides and 3246 unique formerly sialylated N-linked glycopeptides were identified from 400 μg of protein starting material. The hydrophilic phosphopeptides were eluted in the early HILIC fractions whereas the formerly sialylated glycopeptides eluted later in the HILIC prefractionation. This study provide the first system-wide analysis of the dynamic changes of phosphorylated and sialylated proteins together in a biological system.
To increase the identification of sialylated glycopeptides isolated from rat liver, it was suggested to include an immobilised pH gradient-isoelectric focusing (IPG-IEF) fractionation step prior to the TiO2 enrichment.202 IPG-IEF was found to concentrate the negatively charged sialoglycopeptides in the low-pH fractions. As a result, 582 sialoglycopeptides from 322 sialylated N-linked glycoproteins were identified from rat liver tissue.
3.2.2.h Chemistry-based enrichment tools. Chemical methods to label, detect and enrich sialylated glycoconjugates are mainly based on oxidation of sialic acid cis diols before covalent addition of hydrazine, hydrazide thiosemicarbazide, or aminoxy groups and subsequent detection or enrichment of the derived hydrazones and oximes. Periodate oxidation performed under controlled conditions has been widely used to facilitate determination of the monosaccharide composition and structure of sialoglycans from different biomolecules.203 This oxidation targets specifically vicinal hydroxyl groups cleaving the C–C bond and thereby forming two aldehyde groups. Aldehydes are unnatural functional groups in almost all biomolecules in nature, and simultaneously have the benefit of having no chemical reactivity to native amino acid side chains. In addition, aldehydes can be further chemoselected using hydrazides and aminoxy groups at pH levels below physiological conditions without reaction with other biomolecules. This chemoselective ligation allows subsequent visualisation and analysis of the sialic acid containing species (see Table 1 for an overview of chemistry based enrichment methods).
The combination of hydrazide-based chemistry to enrich glycopeptides with subsequent LC-MS detection has been reported.204 The strategy involved the oxidation of cis diol containing N-glycans at room temperature before coupling to the hydrazide resin forming hydrazone bonds with the glycan portion of the glycoproteins. The non-glycosylated proteins were removed by stringent washing before performing tryptic digestion of the resin-bound glycoproteins. After removal of the non-glycosylated peptides, enzymatic N-deglycosylation was used to release the resin-bound formerly glycosylated peptides with the simultaneous conversion of the asparagine residues to aspartic acid residues. MS was used to determine this conversion as a diagnostic for the presence of a formerly occupied glycosylation site. The hydrazide strategy for enrichment of N-glycopeptides was subsequently optimised by using SPE formats in a so-called ‘SPEG setup’,205 increasing the enrichment efficiency of glycopeptides.206,207 The hydrazide chemistry strategy has been applied to identify glycoproteins isolated from several biological origins such as body fluids,208–211 the extracellular milieu,207,212 tissues108,199,213 and cell cultures.214,215 This strategy has been optimised to analyse sialylated glycoproteins as discussed below.
As compared to other monosaccharides occurring in N- and O-linked glycoproteins that are commonly oxidised by periodate at room temperature or 50 °C,216 sialic acids contain three linearly adjacent hydroxyl groups at the C7, C8 and C9 carbons, which are highly susceptible to periodate oxidation. Consequently, the periodate oxidation reaction can be carried out at much lower temperature i.e. 0–4 °C to selectively target sialic acid residues.217 This was shown in a study where 1–2 mM periodate concentration, pH 7.4, at 0 °C oxidised sialic acids within 10 min.218 The oxidation leaves an aldehyde group on the C7 carbon of the sialic acids, which has been used for subsequent radioactive218,219 and non-radioactive220,221 labelling of cell surface sialoglycoconjugates of intact cells.
A slightly different variant of the hydrazide-based chemistry approach described above is the cell surface capturing (CSC) technology,222 which enabled identification of hundreds of cell surface glycoproteins from a variety of cellular origins including Drosophila melanogaster cells, mouse myoblasts, embryonic stem cells and T and B cells.222–224 The CSC technology is based on the mild oxidation of glycans with 1.6 mM sodium meta periodate, pH 6.5, for 15 min at 4 °C to generate reactive aldehyde groups from cis diols. Subsequently, biocytin hydrazide is reacted with aldehyde groups to form hydrazone bonds. After cell lysis, the membrane glycoproteins are recovered by ultracentrifugation and proteolytically digested using trypsin. Glycopeptides are enriched with streptavidin beads and stringent washing conditions are employed to remove unspecific peptides. Finally, N-glycosylated peptides are enzymatically released from streptavidin beads by deglycosylation using PNGase F and analysed using LC-MS. It should be noted that the CSC technology does not selectively target sialylated glycoproteins; this was shown by the positive labelling and capturing of proteins from neuramidase treated cells and insect cells which has no or negligible endogenous production of sialoglycoconjugates.222,225
The development of a highly efficient and selective labelling strategy of cell surface sialylated glycoproteins was reported.226 This strategy is based on the mild periodate oxidation to generate an aldehyde on sialic acids, followed by aniline-catalysed oxime ligation (PAL) with a suitable tag. In this strategy, sialoglycoproteins residing in the cell surface were oxidised using 1 mM sodium meta periodate, pH 7.4, for 15 min at 4 °C. Subsequently, ligation was performed using aminooxy-biotin and 10 mM aniline at pH 7.4. Aniline acts as a nucleophilic catalyst for the oxime ligation227,228 allowing a highly efficient ligation of a molecular tag onto sialoglycans of the membrane proteins under mild conditions. The application of the PAL technology was recently expanded to enrich and identify a larger selection of the cell membrane tethered glycoproteins.229 Here, two complementary strategies for the enrichment of sialic acid and galactose terminated glycoproteins on living cells were reported. In addition to the PAL strategy, the introduction of aminooxy-biotin onto terminal galactose and GalNAc residues by galactose oxidase and aniline-catalysed oxime ligation (GAL) were reported to enrich different subsets of the glycoproteome. The biotin tag functioned as an affinity tag to the streptavidin resin. The enriched glycoproteins were digested and the sialic acid and galactose/GalNAc-terminating glycopeptides were analysed by LC-MS after deglycosylation. As such, PAL and GAL represent two enrichment methods that can be used to investigate different subsets of the glycoproteome. It is important to stress the small differences in reaction conditions between CSC and PAL such as the periodate concentration, temperature and reaction time. This indicates the importance of fine tuning the reaction conditions to obtain a sialic acid-selective labelling. CSC and PAL strategies can be applied to primary cells, tissues and organs but require solubilisation of viable cells for cell surface oxidation and labelling, which is not always possible. Moreover, it should be noted that CSC and PAL technologies have been used mainly to map the cell surface N-linked glycoproteins. However, it is feasible to capture O-linked glycoproteins and release the deglycosylated peptides from the hydrazide resin by β-elimination, acid hydrolysis99,230 or transoximisation.231
The reaction between glycan aldehydes and aminoxy-derivatised polymers has been used for selective capturing of both oxidised glycans and glycopeptides followed by MS detection of the acid-released species in a method named glycoblotting.232 An alternative way of releasing captured glycans via the oxyme bond is through transoximisation using excess O-substituted aminooxy derivatives under weakly acidic conditions.231 Since it is based on the similar yet opposite concept it was coined a “reverse glycoblotting” strategy. The principle is the selective oxidation of sialic acid containing glycopeptides by sodium periodate under mild conditions.230 This strategy relies on the oxidation of sialic acids using 1 mM sodium periodate at 0 °C for 15 min generating available aldehydes on the C7 of sialic acids, which are then coupled to aminooxy-functionalised polymers. After removal of unspecifically bound molecules, the sialoglycopeptides are released using 3% TFA in an aqueous solution at 100 °C for 1 h, which hydrolyses the α-glycoside bonds between the oxidised sialic acids and adjacent galactose residues and allows the identification of the desialylated glycopeptides. A similar method was later reported using hydrazide-functionalised polymers to capture sialylated glycoproteins oxidised using 2 mM sodium periodate at 0 °C for 10 min. The unspecifically bound proteins were removed with stringent washing before trypsin digestion. The captured sialoglycopeptides were released with 0.1 M formic acid at 80 °C for 1 h and the desialylated glycopeptides were analysed by LC-MSn using orthogonal fragmentation techniques.99 In total, 36 N-linked and 44 O-linked glycosylation sites formerly occupied by sialoglycans were detected from proteins derived from human cerebrospinal fluid.
Recently, the combination of glycoblotting and selected reaction monitoring (SRM) has been used to quantitate intact sialoglycopeptides derived from serum sialoglycoproteins. In the improved glycoblotting protocol, oxidised sialylated glycopeptides linked to hydrazide-resin were released in an intact form using ice-cold aqueous 1 M HCl. This treatment regenerates the aldehyde groups that initially reacted with 2-aminopyridine in the presence of 2-picoline borane and this reversible reaction caused the sialoglycopeptides to elute. Specifically, 26 sialylated glycopeptides were identified from LC-MS analysis of 50 μL mouse serum. The corresponding tandem MS spectra were then used to determine the optimal transitions for subsequent quantitative SRM experiments. The ability to elute intact sialoglycopeptides from the hydrazide resin is a highly interesting optimisation of the technique enabling the use of hydrazine chemistry in more true glycoproteomics workflows. One of the limitations of hydrazine chemistry, however, is that it requires chemical oxidation, which in addition to destroying the structure of the monosaccharide affects other vicinal amino alcohols such as N-terminal serine and threonine residues. These residues will be oxidised by the periodate and will generate glyoxylyl derivatives that may also be captured by the hydrazide resin.233 Moreover the selectivity of the hydrazide resin towards the sialylated species relies on temperature and time-dependent oxidation reactions, which will need to be described in more detail by thorough kinetic investigations in the future, before further optimisation of the workflows can be expected.
Taken together, there is an abundance of techniques available for the enrichment and prefractionation of sialoglycopeptides. This wide selection is beneficial since the presented methods are somewhat complementary in their capacity to isolate sialylated glycopeptides and are compatible with different downstream approaches. Indeed each method enriches a unique subset of the sialoglycoproteome due to their different binding mechanisms to the sialylated glycopeptides. This is supported by the limited overlap of the detected phosphoproteomes observed when different enrichment methods were compared.234 Furthermore, a comparison between affinity and chemistry-based enrichment of sialylated glycoproteins illustrated that the two methods are capable of identifying complementary subsets of the glycoproteome with some degree of overlap.235 Hence, combining these enrichment approaches is beneficial when the aim is to enhance the coverage of the sialoglycoproteome. The choice of enrichment method will, as such, largely be determined by the research question investigated, personal preference and in-house technology. Interestingly, with the exception of serotonin none of the enrichment methods developed so far has been able to discriminate between glycopeptides containing different types of sialic acids e.g. Neu5Ac and Neu5Gc.
3.3 Sialoglycoproteins
Top-down analyses of glycoproteins, and sialoglycoproteins in particular, are significantly more challenging than the analysis of their non-modified counterparts due to the extensive microheterogeneity and unfavourable MS properties (discussed in the second of these two reviews78). As a result, the top down analysis of intact sialoglycoproteins has, until now, only been performed in a few cases using purified or semi-purified glycoproteins. Hence, top-down analysis of sialoglycoproteins requires the use of efficient protein purification methods, which not only purify the protein of interest to homogeneity, but ideally also isolate or partially separate the individual sialoglycoforms. The initial isolation steps can be achieved using conventional protein purification techniques including immunoprecipitation, liquid–liquid extraction, affinity chromatography and other types of LC by considering the specific structural and physicochemical properties of the glycoprotein of interest.107 In addition, enrichment tools targeting the sialoglycans can be used to broadly purify sialoglycoproteins although one has to be aware that only a subset of the entire spectrum of glycoforms of specific glycoproteins may be represented in the enriched fraction. As an example, lectin affinity chromatography has been used to enrich broadly for sialoglycoproteins.171 In this work, lectin affinity chromatography using three different sialic acid specific lectins (SNA, MAL and WGA) followed by RPC prefractionation, enabled the identification of a total of 130 sialylated glycoproteins by downstream LC-MS of tryptic glycopeptides and released sialoglycans.
The separation of sialoglycoforms of a protein can be pursued on- or off-line and is needed, first, to allow the mass spectrometer to establish an accurate molecular mass without mass and signal suppression interference from other sialoglycoforms and, second, to allow time for the mass spectrometer to isolate and fragment the individual sialoglycoforms for additional structural validation. CE can be coupled to MS and the combination has proven powerful in the separation and detection of sialoglycoforms at high sensitivity.236,237 In comparison, various LC techniques including RPC and anion exchange chromatography yield seemingly less separation of sialoglycoforms, but may be easier to couple directly to MS for on-line data acquisition.238,239 Other non-MS detection methods can establish important structural information about the sialoglycoproteins e.g. two dimensional gel electrophoresis.83,179 Aspects dealing with the separation and detection of intact sialoglycoproteins are covered in more detail in the second of the two related reviews.78
Another important bi-orthogonal functional group for the incorporation of unnatural sialic acids is the azide, which has the advantage of being totally absent in biological systems in nature. In addition, azides do not perturbate the conformation of their substrates. The physiological precursor of all sialic acids is N-acetyl D-mannosamine (ManNAc) which undergoes a series of enzymatic transformations to generate the activated donor CMP-sialic acid substrate that transfers sialic acids to glycoproteins or glycolipids.251 The incorporation of azide containing sialic acids into sialoglycoproteins residing in the cell surface was performed in Jurkat cells by the incubation with the precursor derivative N-azidoacetylmannosamine (ManNAz).252 Using Staudinger ligation,252 the azide groups were subsequently ligated to phosphine groups bearing a wide array of probes such as biotin and various peptides e.g. FLAG, myc, and His6 for imaging and for enrichment by immunoprecipitation prior to glycoproteomics analysis.253 The phosphine-FLAG-His6 probe enabled two orthogonal purification steps before LC-MS based analysis of the glycopeptides.254 The metabolic incorporation of ManNAz was also utilised to label the cell surface sialoglycoproteins of syngeneic prostate cancer cell lines derived from non-metastatic (N2) and highly metastatic (ML2) prostate cancer cells.255 Affinity isolation of the modified sialoglycoproteins was performed using a biotinylated alkyne via click chemistry.256 This was followed by 1D gel electrophoresis and LC-MS analysis for the total identification of 324 and 372 proteins from N2 and ML2, respectively. As expected, the glycoproteins uniquely identified in the highly metastatic cancer cell line were involved in cellular migration and invasion processes. Finally, metabolic labelling of sialoglycoproteins with 9-aryl azide-substituted sialic acid (9AAzNeuAc) was used to identify the trans ligands of CD22 on opposing B cells using a protein-glycan cross-linking strategy.257 Cross-linking of CD22-Fc or CD22-expressing CHO cells to intact B cells allowed the identification of a subset of candidate trans ligands of CD22 including immunoglobulin M, CD45 and Basigin.
Staudinger ligation has been used for probing cell surface sialic acids in several biological systems ranging from cell lines258 to living animals.244,259 However, this reaction suffers from slow kinetics and phosphine oxidation in air.260 Beside their electrophile behaviour, azides are 1,3-dipoles that can react with terminal alkynes to provide stable triazoles according to [3 + 2] copper-catalysed azide–alkyne cycloaddition (CuAAC).261 However, this reaction requires high temperatures and pressures but was made biocompatible by adding catalytic amounts of Cu(I).262,263 This 1,3-dipolar CuAAC shows all the properties of a click reaction such as reaction efficiency, simplicity, and selectivity in addition to being biocompatible. Moreover this reaction is 25 times faster than the Staudinger ligation and was consequently used to label cell surface sialic acids bearing azides with alkyne probes for imaging and glycoproteomic studies. The secreted and cell surface tethered N- and O-linked sialoglycoproteins of human mesenchymal stem cells were manipulated using a membrane permeable ManNAz, which was converted to N-azidoacetylsialic acid prior to incorporation. The labelled sialoglycoproteins were separated by 1D or 2D gel electrophoresis and detected by fluorescence before the appropriate gel bands/spots were excised and analysed by MS.264
The incorporation of unnatural sialic acids was further explored using the metabolic precursor alkynyl ManNAc to image the cell surface sialoglycoproteins in several cancer cell lines.265 It was demonstrated that click-activated fluorogenic probes are useful to efficiently and selectively label alkynyl-modified glycans. This metabolic precursor was further applied to the enrichment of sialylated glycoproteins of prostate cancer cells.266 Following metabolic labelling, the cells were lysed and reacted with an azido-biotin probe through CuAAC. The tagged glycoproteins were captured by streptavidin-conjugated beads and followed by on-bead digestion with trypsin and PNGase F in order identify the formerly sialylated N-linked glycopeptides and map the previously occupied N-glycosylated sites. In total, 108 N-glycoproteins were identified from 1.5 mg of protein starting material. Another application of this strategy was to investigate the sialylation profile of isogenic lung cancer cells with low (CL1-0) and high (CL1-5) metastatic potential. MS based glycoproteomic analysis identified 157 N-linked glycoproteins and amongst the identified sialoglycoproteins, EGFR exhibited higher degree of sialylation and fucosylation in the lung cells with high metastatic potential. The sialylation turned out to be important for modulating the dimerisation and tyrosine phosphorylation, and hence the function, of EGFR.267 The sialic acid metabolic labelling efficiency with alkynyl ManNAc is superior to that of ManNAz. The successful labelling of sialic acids was shown in mice,268 however the use of copper generally limits its use in living organisms. Although new, biocompatible copper ligands, were developed to enable the use of CuAAC in living systems,269 there are still concerns regarding the toxicity of these ligands. Hence, a strain-promoted [3 + 2] cycloaddition between cyclooctynes and azides was developed that proceeds without the need for a copper catalyst.270 The sensitivity of this reaction was further improved using difluorinated cyclooctynes (DIFO) probes.271 Using this copper-free click chemistry, the spatiotemporal dynamics of the sialome in living zebrafish embryos were monitored by reactions with fluorophore-conjugated DIFO reagents.272
Finally, other unnatural sialic acids have been described as precursors for metabolic labelling of cell surface sialoglycoproteins273 including precursors using thiol containing functional groups.274 The thiol-analogue of ManNAc, Ac5ManNTGc, allowed the incorporation of N-thiolglycolyl neuraminic acid as a sialic acid substitute into sialoglycoproteins on the cell surface of Jurkat T cells and human embryoid body-derived stem cells. This allowed cell–cell clustering and attachment to biomaterials and synthetic scaffolds for tissue engineering. Based on the concept of activity based protein profiling275 that uses chemical probes to target and profile specific enzymes, new membrane-permeable alkyne-hinged 3-fluorosialyl fluoride molecules were used to covalently bind to virus, bacteria, and human sialidases.276 The alkyne group was subsequently used to covalently attach, via click chemistry, an azide containing biotin for detection and profiling. Using MS it was shown that a tyrosine residue of the sialidases was involved in the binding with the fluoride derivative of sialic acid. Moreover, it was possible to map the sialidase activity in living cells under different conditions.
In conclusion, the metabolic labelling of sialoglycoproteins has proven to be an efficient tool for their enhanced detection and isolation in several biological systems. However, several challenges remain with respect to competition from the endogenous substrates, such as natural sialic acids, which results in incomplete metabolic labelling. Moreover, the overall physiology of the system investigated may be perturbed by the addition of unnatural sialic acids. Finally, biases may be introduced in the incorporation of unnatural sialic acids into the sialoglycoproteins. Further research into the specificity of the different sialyltransferases towards the artificial sialic acid variants may generate a better understanding of this.
4. Conclusion
Well-designed analytical strategies and proper sample handling is a necessity for obtaining unbiased structural information of high quality when using MS as the downstream detection technique. This is particular true for the analysis of glycoprotein sialylation. The lability of the sialic acid residues together with its negative charge and its structural heterogeneity demands dedicated workflows. This review has outlined some available analytical strategies and summarised pre-LC-MS methodologies for the handling of sialylated glycoproteins prior to their identification. It is clear that the analytical toolbox has improved dramatically over the last decades. As such, we have now a much wider selection of more streamlined workflows to choose from when setting up an experimental design to study N- and O-linked sialoglycoproteins. Methods for sophisticated analyte manipulation where the sialoglycans, sialoglycopeptides and sialoglycoproteins are rendered more “detection friendly” by enzymatic, chemical or metabolic alteration have also played an important part in the successful analytical achievements.The use of glycoproteomics oriented approaches to study sialylation is an attractive, but challenging, analytical route to take, since it is rewarded by site-specific information of the sialoglycans. The popularity of such workflows is increasing driven, in part, by the development of efficient and robust enrichment techniques for sialoglycopeptides from complex matrices that reduce the interference from non-modified peptides and neutral glycopeptides. The negative charge of the sialic acid residues has widely been used as a physicochemical property in enrichment and prefractionation strategies, and this text has emphasised their advantages and limitations. To date, the choice of analytical route including the enrichment and derivatisation methods has mainly been based on the specific expertise in the individual laboratories and personal preferences. As a consequence, the enrichment methods still lack thorough comparison in terms of reproducibility, robustness, specificity and sensitivity. In addition, an enrichment strategy to enable the characterisation of the entire sialome is still missing since most of the methods have been directed mainly towards the Neu5Ac and Neu5Gc structures limiting our knowledge about other sialic acids and their modified counterparts. It should be noted that the majority of the enrichment methods target N-linked sialoglycopeptides and glycoproteins due to the lack of efficient methods to release and analyse O-linked glycans and O-linked glycopeptides. We expect that many of these strategies will be further improved to enable deeper investigation of the structural diversity of sialoglycoproteins using large scale glycoproteomics. Unbiased large scale glycoproteomic approaches will drive the discovery of new cellular pathways and will elucidate how different post-translational modifications regulate the protein function in a concerted way. However, to date, no large scale study has been reported targeting the entire sialoglycoproteome: System-wide sialoglycoproteomics approaches have predominantly been used to identify the sialoglycoproteins and their glycosylation sites. As such, a significant piece of structural information of the sialylated glycoproteins has not been addressed at the system-wide scale; the structure of the sialoglycans determined in a site-specific manner. We envision that the development of more dedicated bioinformatics tools and intelligent LC-MS acquisitions (discussed in the second of these two related reviews78) will allow more comprehensive investigation of the sialoglycoproteome on the site-specific level by the large-scale study of intact glycopeptides (Parker et al., submitted and Lendal S. et al., in preparation).277
In conclusion, the investigation of structural sialoglycobiology holds great promises for obtaining a better understanding of human health and diseases. Indeed sialoglycoconjugates are widely recognised targets for diagnosis, treatment, and therapy of various human diseases.
Abbreviations
| ABC | Ammonium bicarbonate |
| CE | Capillary electrophoresis |
| CSC | Cell surface capturing |
| CMP | Cytidine 5-monophosphate |
| COFRADIC | Combined fraction diagonal chromatography |
| ConA | Concanavalin A |
| CuAAC | Copper-catalysed azide-alkyne cycloaddition |
| DIFO | Difluorinated cyclooctynes |
| EGFR | Epidermal growth factor receptor |
| ERLIC | Electrostatic repulsion hydrophilic interaction chromatography |
| GAL | Galactose oxidase and aniline-catalysed oxime ligation |
| Gal | Galactose |
| GalNAc | N-Acetylgalactosamine |
| GlcNAC | N-Acetylglucosamine |
| HCD | Higher-energy C-trap dissociation |
| HILIC | Hydrophilic liquid interaction chromatography |
| IPG-IEF | Immobilised pH gradient-isoelectric focusing |
| Kdn | 2-Keto-3-deoxynonulosonic-acid |
| LC | Liquid chromatography |
| MAL | Maackia amurensis leukoagglutinin |
| MAH | Maackia amurensis hemagglutinin |
| ManNAz | N-Azidoacetylmannosamine |
| MLAC | Multiple lectin affinity chromatography |
| MS | Mass spectrometry |
| Neu5Ac | N-Acetylneuraminic acid |
| Neu5Gc | N-Glycolylneuraminic acid |
| NPC | Normal phase chromatography |
| PAL | Periodate and aniline-catalysed oxime ligation |
| PNGase A/F | N-Glycosidase A/F |
| RPC | Reversed phase chromatography |
| SCX | Strong cation exchange |
| SLAC | Serial lectin affinity chromatography |
| SNA | Sambucus nigra agglutinin |
| SPE | Solid phase extraction |
| SRM | Selected reaction monitoring |
| TCA | Trichloroacetic acid |
| TFA | Trifluoroacetic acid |
| TiO2 | Titanium dioxide |
| WAX | Weak anion exchange chromatography |
| WGA | Wheat germ agglutinin |
| ZIC | Zwitter-ionic |
Acknowledgements
M.T.-A. was funded by the Australian Research Council by a Super Science Fellowship. GP was supported by the Danish Medical Science Research Council (Grant no. 11-107551). MRL was supported by the Lundbeck foundation (Junior Group Leader Fellowship).References
- G. Blix, E. Lindberg, L. Odin and I. Werner, Nature, 1955, 175, 340–341 CrossRef CAS.
- T. Yamakawa, Trends Biochem. Sci., 1988, 13, 452–454 CrossRef CAS.
- R. Schauer and J. P. Kamerling, Chemistry, biochemistry and biology of sialic acids, Elsevier, Amsterdam, 1997 Search PubMed.
- T. Angata and A. Varki, Chem. Rev., 2002, 102, 439–469 CrossRef CAS PubMed.
- A. Varki, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(suppl. 2), 8939–8946 CrossRef CAS PubMed.
- R. Schauer, Adv. Carbohydr. Chem. Biochem., 1982, 40, 131–234 CrossRef CAS.
- L. Warren, Comp. Biochem. Physiol., 1963, 10, 153–171 CAS.
- D. G. Comb and S. Roseman, J. Biol. Chem., 1960, 235, 2529–2537 CAS.
- S. Inoue and K. Kitajima, Glycoconjugate J., 2006, 23, 277–290 CrossRef CAS PubMed.
- A. Varki, Am. J. Phys. Anthropol., 2001,(suppl. 33), 54–69 CrossRef CAS PubMed.
- A. Zhu and R. Hurst, Xenotransplantation, 2002, 9, 376–381 CrossRef.
- P. Tangvoranuntakul, P. Gagneux, S. Diaz, M. Bardor, N. Varki, A. Varki and E. Muchmore, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12045–12050 CrossRef CAS PubMed.
- A. Varki, H. H. Freeze and P. Gagneux, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor, NY, 2nd edn, 2009 Search PubMed.
- Y. N. Malykh, R. Schauer and L. Shaw, Biochimie, 2001, 83, 623–634 CrossRef CAS.
- Y. Hirabayashi, H. Kasakura, M. Matsumoto, H. Higashi, S. Kato, N. Kasai and M. Naiki, Jpn. J. Cancer Res., 1987, 78, 251–260 Search PubMed.
- H. Higashi, Y. Hirabayashi, Y. Fukui, M. Naiki, M. Matsumoto, S. Ueda and S. Kato, Cancer Res., 1985, 45, 3796–3802 CAS.
- M. Hedlund, V. Padler-Karavani, N. M. Varki and A. Varki, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18936–18941 CrossRef CAS PubMed.
- V. Padler-Karavani, N. Hurtado-Ziola, M. Pu, H. Yu, S. Huang, S. Muthana, H. A. Chokhawala, H. Cao, P. Secrest, D. Friedmann-Morvinski, O. Singer, D. Ghaderi, I. M. Verma, Y. T. Liu, K. Messer, X. Chen, A. Varki and R. Schwab, Cancer Res., 2011, 71, 3352–3363 CrossRef CAS PubMed.
- D. H. Nguyen, P. Tangvoranuntakul and A. Varki, J. Immunol., 2005, 175, 228–236 CAS.
- M. Bardor, D. H. Nguyen, S. Diaz and A. Varki, J. Biol. Chem., 2005, 280, 4228–4237 CrossRef CAS PubMed.
- R. G. Spiro, Glycobiology, 2002, 12, 43R–56R CrossRef CAS PubMed.
- M. M. Shah, K. Fujiyama, C. R. Flynn and L. Joshi, Nat. Biotechnol., 2003, 21, 1470–1471 CrossRef CAS PubMed.
- M. Seveno, M. Bardor, T. Paccalet, V. Gomord, P. Lerouge and L. Faye, Nat. Biotechnol., 2004, 22, 1351–1352 CrossRef CAS PubMed ; author reply 1352–1353.
- R. Zeleny, D. Kolarich, R. Strasser and F. Altmann, Planta, 2006, 224, 222–227 CrossRef CAS PubMed.
- A. Varki, Trends Mol. Med., 2008, 14, 351–360 CrossRef CAS PubMed.
- J. Tiralongo and I. Martinez-Duncker, in Sialic Acid Glycoconjugates in Health and Disease, ed. J. Tiralongo and I. Martinez-Duncker, Bentham Science, 2013 Search PubMed.
- P. Isa, C. F. Arias and S. Lopez, Glycoconjugate J., 2006, 23, 27–37 CrossRef CAS PubMed.
- U. Neu, J. Bauer and T. Stehle, Curr. Opin. Struct. Biol., 2011, 21, 610–618 CrossRef CAS PubMed.
- M. Schwarzkopf, K. P. Knobeloch, E. Rohde, S. Hinderlich, N. Wiechens, L. Lucka, I. Horak, W. Reutter and R. Horstkorte, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5267–5270 CrossRef CAS PubMed.
- P. Gorog and I. B. Kovacs, Agents Actions, 1978, 8, 543–545 CrossRef CAS.
- A. Hartnell, J. Steel, H. Turley, M. Jones, D. G. Jackson and P. R. Crocker, Blood, 2001, 97, 288–296 CrossRef CAS PubMed.
- S. D. Rosen, Annu. Rev. Immunol., 2004, 22, 129–156 CrossRef CAS PubMed.
- H. A. Fozzard and J. W. Kyle, Sci. STKE, 2002, 2002, 19 CrossRef PubMed.
- C. A. Ufret-Vincenty, D. J. Baro, W. J. Lederer, H. A. Rockman, L. E. Quinones and L. F. Santana, J. Biol. Chem., 2001, 276, 28197–28203 CrossRef CAS PubMed.
- J. Heimburg-Molinaro, M. Lum, G. Vijay, M. Jain, A. Almogren and K. Rittenhouse-Olson, Vaccine, 2011, 29, 8802–8826 CrossRef CAS PubMed.
- M. Audry, C. Jeanneau, A. Imberty, A. Harduin-Lepers, P. Delannoy and C. Breton, Glycobiology, 2011, 21, 716–726 CrossRef CAS PubMed.
- A. Harduin-Lepers, M. A. Krzewinski-Recchi, F. Colomb, F. Foulquier, S. Groux-Degroote and P. Delannoy, Front Biosci., 2012, 4, 499–515 Search PubMed.
- B. Wang, Adv. Nutr., 2012, 3, 465S–472S CrossRef CAS PubMed.
- G. Kochlamazashvili, O. Senkov, S. Grebenyuk, C. Robinson, M. F. Xiao, K. Stummeyer, R. Gerardy-Schahn, A. K. Engel, L. Feig, A. Semyanov, V. Suppiramaniam, M. Schachner and A. Dityatev, J. Neurosci., 2010, 30, 4171–4183 CrossRef CAS PubMed.
- R. B. Parker and J. J. Kohler, ACS Chem. Biol., 2010, 5, 35–46 CrossRef CAS PubMed.
- M. Polanski and N. L. Anderson, Biomarker Insights, 2007, 1, 1–48 Search PubMed.
- B. K. Dunn, P. D. Wagner, D. Anderson and P. Greenwald, Semin. Oncol., 2010, 37, 224–242 CrossRef CAS PubMed.
- J. A. Ludwig and J. N. Weinstein, Nat. Rev. Cancer, 2005, 5, 845–856 CrossRef CAS PubMed.
- E. P. Diamandis, J. Natl. Cancer Inst., 2010, 102, 1462–1467 CrossRef CAS PubMed.
- C. Yang, S. Mookerjea and A. Nagpurkar, Glycobiology, 1992, 2, 41–48 CrossRef CAS.
- L. Dissing-Olesen, M. Thaysen-Andersen, M. Meldgaard, P. Hojrup and B. Finsen, J. Pharmacol. Exp. Ther., 2008, 326, 338–347 CrossRef CAS PubMed.
- Z. Sumer-Bayraktar, D. Kolarich, M. P. Campbell, S. Ali, N. H. Packer and M. Thaysen-Andersen, Mol. Cell. Proteomics, 2011, 10, M111 009100 Search PubMed.
- E. Fenouillet, G. Fayet, S. Hovsepian, E. M. Bahraoui and C. Ronin, J. Biol. Chem., 1986, 261, 15153–15158 CAS.
- M. R. Larsen, S. S. Jensen, L. A. Jakobsen and N. H. Heegaard, Mol. Cell. Proteomics, 2007, 6, 1778–1787 CAS.
- A. Varki and T. Angata, Glycobiology, 2006, 16, 1R–27R CrossRef CAS PubMed.
- I. M. Francischetti, J. G. Valenzuela, V. M. Pham, M. K. Garfield and J. M. Ribeiro, J. Exp. Biol., 2002, 205, 2429–2451 CAS.
- T. Miyagi and K. Yamaguchi, Glycobiology, 2012, 22, 880–896 CrossRef CAS PubMed.
- R. Schauer, Glycoconjugate J., 2000, 17, 485–499 CrossRef CAS.
- X. Song, H. Yu, X. Chen, Y. Lasanajak, M. M. Tappert, G. M. Air, V. K. Tiwari, H. Cao, H. A. Chokhawala, H. Zheng, R. D. Cummings and D. F. Smith, J. Biol. Chem., 2011, 286, 31610–31622 CrossRef CAS PubMed.
- D. A. Cheresh, A. P. Varki, N. M. Varki, W. B. Stallcup, J. Levine and R. A. Reisfeld, J. Biol. Chem., 1984, 259, 7453–7459 CAS.
- U. Hubl, H. Ishida, M. Kiso, A. Hasegawa and R. Schauer, J. Biochem., 2000, 127, 1021–1031 CrossRef CAS.
- L. D. Bergelson, E. V. Dyatlovitskaya, T. E. Klyuchareva, E. V. Kryukova, A. F. Lemenovskaya, V. A. Matveeva and E. V. Sinitsyna, Eur. J. Immunol., 1989, 19, 1979–1983 CrossRef CAS PubMed.
- D. S. Roseman and J. U. Baenziger, Methods Enzymol., 2003, 363, 121–133 CAS.
- R. Schauer, Methods Enzymol., 1978, 50, 64–89 CrossRef CAS.
- A. Varki and S. Diaz, Anal. Biochem., 1984, 137, 236–247 CrossRef CAS.
- T. Janas, Biochimica et Biophysica Acta, 2011, 1808, 2923–2932 CrossRef CAS PubMed.
- S. L. Wu, A. D. Taylor, Q. Lu, S. M. Hanash, H. Im, M. Snyder and W. S. Hancock, Mol. Cell. Proteomics, 2013, 12, 1239–1249 CAS.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6, 713–723 CrossRef CAS PubMed.
- M. Pabst and F. Altmann, Proteomics, 2011, 11, 631–643 CrossRef CAS PubMed.
- H. Geyer and R. Geyer, Biochimica et Biophysica Acta, 2006, 1764, 1853–1869 CrossRef CAS PubMed.
- M. Wuhrer, A. M. Deelder and C. H. Hokke, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 825, 124–133 CrossRef CAS PubMed.
- M. Wuhrer, Glycoconjugate J., 2013, 30, 11–22 CrossRef CAS PubMed.
- M. Cohen and A. Varki, OMICS, 2010, 14, 455–464 CrossRef CAS PubMed.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
- R. D. Marshall, Biochem. Soc. Symp., 1974, 17–26 CAS.
- F. Schwarz and M. Aebi, Curr. Opin. Struct. Biol., 2011, 21, 576–582 CrossRef CAS PubMed.
- B. L. Schulz, in Biochemistry, Genetics and Molecular Biology, ed. S. Petrescu, 2012 Search PubMed.
- D. F. Zielinska, F. Gnad, J. R. Wisniewski and M. Mann, Cell, 2010, 141, 897–907 CrossRef CAS PubMed.
- G. Palmisano, M. N. Melo-Braga, K. Engholm-Keller, B. L. Parker and M. R. Larsen, J. Proteome Res., 2012, 11, 1949–1957 CrossRef CAS PubMed.
- P. Hao, Y. Ren, A. J. Alpert and S. K. Sze, Mol. Cell. Proteomics, 2011, 10, O111 009381 Search PubMed.
- J. F. Valliere-Douglass, P. Kodama, M. Mujacic, L. J. Brady, W. Wang, A. Wallace, B. Yan, P. Reddy, M. J. Treuheit and A. Balland, J. Biol. Chem., 2009, 284, 32493–32506 CrossRef CAS PubMed.
- J. F. Valliere-Douglass, C. M. Eakin, A. Wallace, R. R. Ketchem, W. Wang, M. J. Treuheit and A. Balland, J. Biol. Chem., 2010, 285, 16012–16022 CrossRef CAS PubMed.
- G. Palmisano, M. R. Larsen, N. H. Packer and M. Thaysen-Andersen, RSC Adv., 2013 10.1039/C3RA42969E.
- S. Pan, R. Chen, R. Aebersold and T. A. Brentnall, Mol. Cell. Proteomics, 2011, 10, R110 003251 Search PubMed.
- N. Leymarie and J. Zaia, Anal. Chem., 2012, 84, 3040–3048 CrossRef CAS PubMed.
- W. Morelle and J. C. Michalski, Nat. Protoc., 2007, 2, 1585–1602 CrossRef CAS PubMed.
- P. H. Jensen, N. G. Karlsson, D. Kolarich and N. H. Packer, Nat. Protocols, 2012, 7, 1299–1310 CAS.
- N. H. Packer and M. J. Harrison, Electrophoresis, 1998, 19, 1872–1882 CrossRef CAS PubMed.
- J. F. Rakus and L. K. Mahal, Annu. Rev. Anal. Chem., 2011, 4, 367–392 CrossRef CAS PubMed.
- Y. Mechref, Y. Hu, J. L. Desantos-Garcia, A. Hussein and H. Tang, Mol. Cell. Proteomics, 2013, 12, 874–884 CAS.
- H. Nie, Y. Li and X. L. Sun, J. Proteomics, 2012, 75, 3098–3112 CrossRef CAS PubMed.
- M. Wuhrer, A. R. de Boer and A. M. Deelder, Mass Spectrom. Rev., 2009, 28, 192–206 CrossRef CAS PubMed.
- L. R. Ruhaak, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2009, 394, 163–174 CrossRef CAS PubMed.
- S. Ito, K. Hayama and J. Hirabayashi, Methods Mol. Biol., 2009, 534, 195–203 CAS.
- S. Ongay, A. Boichenko, N. Govorukhina and R. Bischoff, J. Sep. Sci., 2012, 35, 2341–2372 CrossRef CAS PubMed.
- Z. W. Lai, E. C. Nice and O. Schilling, Proteomics, 2013, 13, 512–525 CrossRef CAS PubMed.
- H. Desaire, Mol. Cell. Proteomics, 2013, 12, 893–901 CAS.
- M. Wuhrer, M. I. Catalina, A. M. Deelder and C. H. Hokke, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 849, 115–128 CrossRef CAS PubMed.
- N. V. Artemenko, A. G. McDonald, G. P. Davey and P. M. Rudd, Methods Mol. Biol., 2012, 899, 325–350 CAS.
- F. Li, O. V. Glinskii and V. V. Glinsky, Proteomics, 2013, 13, 341–354 CrossRef CAS PubMed.
- M. P. Campbell, C. A. Hayes, W. B. Struwe, M. R. Wilkins, K. F. Aoki-Kinoshita, D. J. Harvey, P. M. Rudd, D. Kolarich, F. Lisacek, N. G. Karlsson and N. H. Packer, Proteomics, 2011, 11, 4117–4121 CrossRef CAS PubMed.
- A. V. Everest-Dass, D. Jin, M. Thaysen-Andersen, H. Nevalainen, D. Kolarich and N. H. Packer, Glycobiology, 2012, 22, 1465–1479 CrossRef CAS PubMed.
- M. Pabst, J. S. Bondili, J. Stadlmann, L. Mach and F. Altmann, Anal. Chem., 2007, 79, 5051–5057 CrossRef CAS PubMed.
- J. Nilsson, U. Ruetschi, A. Halim, C. Hesse, E. Carlsohn, G. Brinkmalm and G. Larson, Nat. Methods, 2009, 6, 809–811 CrossRef CAS PubMed.
- G. Palmisano, B. L. Parker, K. Engholm-Keller, S. E. Lendal, K. Kulej, M. Schulz, V. Schwammle, M. E. Graham, H. Saxtorph, S. J. Cordwell and M. R. Larsen, Mol. Cell. Proteomics, 2012, 11, 1191–1202 CAS.
- T. M. Annesley, Clin. Chem., 2003, 49, 1041–1044 CAS.
- Z. Sumer-Bayraktar, T. Nguyen-Khuong, R. Jayo, D. D. Chen, S. Ali, N. H. Packer and M. Thaysen-Andersen, Proteomics, 2012, 12, 3315–3327 CrossRef CAS PubMed.
- D. Kolarich, A. Weber, P. L. Turecek, H. P. Schwarz and F. Altmann, Proteomics, 2006, 6, 3369–3380 CrossRef CAS PubMed.
- A. Kondo, M. Thaysen-Andersen, K. Hjerno and O. N. Jensen, J. Sep. Sci., 2010, 33, 891–902 CrossRef CAS PubMed.
- Z. Valnickova, M. Thaysen-Andersen, P. Hojrup, T. Christensen, K. W. Sanggaard, T. Kristensen and J. J. Enghild, BMC Biochem., 2009, 10, 13 CrossRef PubMed.
- M. Thaysen-Andersen and N. H. Packer, Glycobiology, 2012, 22, 1440–1452 CrossRef CAS PubMed.
- H. H. Freeze, Current protocols in molecular biology, ed. F. M. Ausubel, et al., 2001, ch. 17, Unit17 11 Search PubMed.
- A. Lee, D. Kolarich, P. A. Haynes, P. H. Jensen, M. S. Baker and N. H. Packer, J. Proteome Res., 2009, 8, 770–781 CrossRef CAS PubMed.
- H. H. Freeze and B. G. Ng, Cold Spring Harbor Perspect. Biol., 2011, 3, a005371 CrossRef PubMed.
- H. H. Freeze, J. Biol. Chem., 2013, 288, 6936–6945 CrossRef CAS PubMed.
- B. Cox and A. Emili, Nat. Protoc., 2006, 1, 1872–1878 CrossRef CAS PubMed.
- D. Rajalingam, C. Loftis, J. J. Xu and T. K. Kumar, Protein Sci., 2009, 18, 980–993 CrossRef CAS PubMed.
- M. Sickmeier, J. A. Hamilton, T. LeGall, V. Vacic, M. S. Cortese, A. Tantos, B. Szabo, P. Tompa, J. Chen, V. N. Uversky, Z. Obradovic and A. K. Dunker, Nucleic Acids Res., 2007, 35, D786–D793 CrossRef CAS PubMed.
- Y. Wada, P. Azadi, C. E. Costello, A. Dell, R. A. Dwek, H. Geyer, R. Geyer, K. Kakehi, N. G. Karlsson, K. Kato, N. Kawasaki, K. H. Khoo, S. Kim, A. Kondo, E. Lattova, Y. Mechref, E. Miyoshi, K. Nakamura, H. Narimatsu, M. V. Novotny, N. H. Packer, H. Perreault, J. Peter-Katalinic, G. Pohlentz, V. N. Reinhold, P. M. Rudd, A. Suzuki and N. Taniguchi, Glycobiology, 2007, 17, 411–422 CrossRef CAS PubMed.
- Y. Wada, A. Dell, S. M. Haslam, B. Tissot, K. Canis, P. Azadi, M. Backstrom, C. E. Costello, G. C. Hansson, Y. Hiki, M. Ishihara, H. Ito, K. Kakehi, N. Karlsson, C. E. Hayes, K. Kato, N. Kawasaki, K. H. Khoo, K. Kobayashi, D. Kolarich, A. Kondo, C. Lebrilla, M. Nakano, H. Narimatsu, J. Novak, M. V. Novotny, E. Ohno, N. H. Packer, E. Palaima, M. B. Renfrow, M. Tajiri, K. A. Thomsson, H. Yagi, S. Y. Yu and N. Taniguchi, Mol. Cell. Proteomics, 2010, 9, 719–727 CAS.
- V. Tretter, F. Altmann and L. Marz, Eur. J. Biochem., 1991, 199, 647–652 CrossRef CAS.
- N. L. Wilson, B. L. Schulz, N. G. Karlsson and N. H. Packer, J. Proteome Res., 2002, 1, 521–529 CrossRef CAS.
- K. R. Anumula, Anal. Biochem., 2008, 373, 104–111 CrossRef CAS PubMed.
- G. Palmisano, S. E. Lendal and M. R. Larsen, Methods Mol. Biol., 2011, 753, 309–322 CAS.
- J. Zaia, Mass Spectrom. Rev., 2004, 23, 161–227 CrossRef CAS PubMed.
- L. R. Ruhaak, G. Zauner, C. Huhn, C. Bruggink, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2010, 397, 3457–3481 CrossRef CAS PubMed.
- Y. Mechref, Y. Hu, J. L. Desantos-Garcia, A. Hussein and H. Tang, Mol. Cell. Proteomics, 2013, 12, 874–884 CAS.
- J. You, X. Sheng, C. Ding, Z. Sun, Y. Suo, H. Wang and Y. Li, Anal. Chim. Acta, 2008, 609, 66–75 CrossRef CAS PubMed.
- A. K. Powell and D. J. Harvey, Rapid Commun. Mass Spectrom., 1996, 10, 1027–1032 CrossRef CAS.
- X. Liu, X. Li, K. Chan, W. Zou, P. Pribil, X. F. Li, M. B. Sawyer and J. Li, Anal. Chem., 2007, 79, 3894–3900 CrossRef CAS PubMed.
- K. S. Jang, Y. G. Kim, G. C. Gil, S. H. Park and B. G. Kim, Anal. Biochem., 2009, 386, 228–236 CrossRef CAS PubMed.
- Y. Mechref, A. G. Baker and M. V. Novotny, Carbohydr. Res., 1998, 313, 145–155 CrossRef CAS.
- Y. Miura, Y. Shinohara, J. Furukawa, N. Nagahori and S. Nishimura, Chemistry, 2007, 13, 4797–4804 CrossRef CAS PubMed.
- X. Liu, H. Qiu, R. K. Lee, W. Chen and J. Li, Anal. Chem., 2010, 82, 8300–8306 CrossRef CAS PubMed.
- J. Amano, D. Sugahara, K. Osumi and K. Tanaka, Glycobiology, 2009, 19, 592–600 CrossRef CAS PubMed.
- S. Sekiya, Y. Wada and K. Tanaka, Anal. Chem., 2005, 77, 4962–4968 CrossRef CAS PubMed.
- P. Chen, U. Werner-Zwanziger, D. Wiesler, M. Pagel and M. V. Novotny, Anal. Chem., 1999, 71, 4969–4973 CrossRef CAS.
- S. F. Wheeler, P. Domann and D. J. Harvey, Rapid Commun. Mass Spectrom., 2009, 23, 303–312 CrossRef CAS PubMed.
- W. R. Alley, Jr and M. V. Novotny, J. Proteome Res., 2010, 9, 3062–3072 CrossRef PubMed.
- I. Ciucanu, Anal. Chim. Acta, 2006, 576, 147–155 CrossRef CAS PubMed.
- P. Kang, Y. Mechref, I. Klouckova and M. V. Novotny, Rapid Commun. Mass Spectrom., 2005, 19, 3421–3428 CrossRef CAS PubMed.
- G. Kohla, E. Stockfleth and R. Schauer, Neurochem. Res., 2002, 27, 583–592 CrossRef CAS.
- X. Liu, L. Afonso, E. Altman, S. Johnson, L. Brown and J. Li, Proteomics, 2008, 8, 2849–2857 CrossRef CAS PubMed.
- G. Alvarez-Manilla, N. L. Warren, T. Abney, J. Atwood, P. Azadi, W. S. York, M. Pierce and R. Orlando, Glycobiology, 2007, 17, 677–687 CrossRef CAS PubMed.
- S. P. Galuska, R. Geyer, M. Muhlenhoff and H. Geyer, Anal. Chem., 2007, 79, 7161–7169 CrossRef CAS PubMed.
- D. Kolarich, P. L. Turecek, A. Weber, A. Mitterer, M. Graninger, P. Matthiessen, G. A. Nicolaes, F. Altmann and H. P. Schwarz, Transfusion, 2006, 46, 1959–1977 CrossRef CAS PubMed.
- C. W. Sutton, J. A. O'Neill and J. S. Cottrell, Anal. Biochem., 1994, 218, 34–46 CrossRef CAS.
- Z. Khedri, M. M. Muthana, Y. Li, S. M. Muthana, H. Yu, H. Cao and X. Chen, Chem. Commun., 2012, 48, 3357–3359 RSC.
- H. A. Chokhawala, H. Yu and X. Chen, ChemBioChem, 2007, 8, 194–201 CrossRef CAS PubMed.
- L. D. Powell and A. P. Varki, Curr. Protoc. Mol. Biol., 2001, ch. 17, Unit17 12 Search PubMed.
- L. D. Powell, A. P. Varki and H. H. Freeze, Curr. Protoc. Immunol., 2001, ch. 8, Unit 8 15 Search PubMed.
- Y. Uchida, Y. Tsukada and T. Sugimori, J. Biochem., 1977, 82, 1425–1433 CAS.
- K. Hanaoka, T. J. Pritchett, S. Takasaki, N. Kochibe, S. Sabesan, J. C. Paulson and A. Kobata, J. Biol. Chem., 1989, 264, 9842–9849 CAS.
- H. Zhang, T. Guo, X. Li, A. Datta, J. E. Park, J. Yang, S. K. Lim, J. P. Tam and S. K. Sze, Mol. Cell. Proteomics, 2010, 9, 635–647 CAS.
- R. Schauer, Methods Enzymol., 1987, 138, 611–626 CrossRef CAS.
- A. Manzi, Curr. Protoc. Mol. Biol., 2001, ch. 17, Unit17 16 Search PubMed.
- N. Leymarie, P. J. Griffin, K. Jonscher, D. Kolarich, R. Orlando, M. McComb, J. Zaia, J. Aguilan, W. R. Alley, F. Altmann, L. E. Ball, L. Basumallick, C. R. Bazemore-Walker, H. Behnken, M. A. Blank, K. J. Brown, S. C. Bunz, C. W. Cairo, J. F. Cipollo, R. Daneshfar, H. Desaire, R. R. Drake, E. P. Go, R. Goldman, C. Gruber, A. Halim, Y. Hathout, P. J. Hensbergen, D. M. Horn, D. Hurum, W. Jabs, G. Larson, M. Ly, B. F. Mann, K. Marx, Y. Mechref, B. Meyer, U. Moginger, C. Neususs, J. Nilsson, M. V. Novotny, J. O. Nyalwidhe, N. H. Packer, P. Pompach, B. Reiz, A. Resemann, J. S. Rohrer, A. Ruthenbeck, M. Sanda, J. M. Schulz, U. Schweiger-Hufnagel, C. Sihlbom, E. Song, G. O. Staples, D. Suckau, H. Tang, M. Thaysen-Andersen, R. I. Viner, Y. An, L. Valmu, Y. Wada, M. Watson, M. Windwarder, R. Whittal, M. Wuhrer, Y. Zhu and C. Zou, Mol. Cell. Proteomics, 2013 DOI:10.1074/mcp.M113.030643.
- E. D. Dodds, R. R. Seipert, B. H. Clowers, J. B. German and C. B. Lebrilla, J. Proteome Res., 2009, 8, 502–512 CrossRef CAS PubMed.
- G. Zauner, M. Hoffmann, E. Rapp, C. A. Koeleman, I. Dragan, A. M. Deelder, M. Wuhrer and P. J. Hensbergen, J. Proteome Res., 2012, 11, 5804–5814 CAS.
- M. R. Larsen, P. Hojrup and P. Roepstorff, Mol. Cell. Proteomics, 2005, 4, 107–119 CAS.
- A. Mellors and R. Y. Lo, Methods Enzymol., 1995, 248, 728–740 CAS.
- J. Roth, Histochem. Cell Biol., 2011, 136, 117–130 CrossRef CAS PubMed.
- A. Danguy, C. Decaestecker, F. Genten, I. Salmon and R. Kiss, Acta Anat., 1998, 161, 206–218 CrossRef CAS PubMed.
- T. K. Dam and C. F. Brewer, Adv. Carbohydr. Chem. Biochem., 2010, 63, 139–164 CrossRef CAS.
- F. Lehmann, E. Tiralongo and J. Tiralongo, Cell. Mol. Life Sci., 2006, 63, 1331–1354 CrossRef CAS PubMed.
- T. Sata, J. Roth, C. Zuber, B. Stamm and P. U. Heitz, Am. J. Pathol., 1991, 139, 1435–1448 CAS.
- R. N. Knibbs, I. J. Goldstein, R. M. Ratcliffe and N. Shibuya, J. Biol. Chem., 1991, 266, 83–88 CAS.
- W. C. Wang and R. D. Cummings, J. Biol. Chem., 1988, 263, 4576–4585 CAS.
- Y. Konami, K. Yamamoto, T. Osawa and T. Irimura, FEBS Lett., 1994, 342, 334–338 CrossRef CAS.
- X. Bai, J. R. Brown, A. Varki and J. D. Esko, Glycobiology, 2001, 11, 621–632 CrossRef CAS PubMed.
- J. S. Sams, H. T. Lynch, R. W. Burt, S. J. Lanspa and C. R. Boland, Cancer, 1990, 66, 502–508 CrossRef CAS.
- K. O. Lloyd, Arch. Biochem. Biophys., 1970, 137, 460–468 CrossRef CAS.
- Z. Yang and W. S. Hancock, J. Chromatogr., A, 2004, 1053, 79–88 CAS.
- R. Qiu and F. E. Regnier, Anal. Chem., 2005, 77, 7225–7231 CrossRef CAS PubMed.
- S. Fanayan, M. Hincapie and W. S. Hancock, Electrophoresis, 2012, 33, 1746–1754 CrossRef CAS PubMed.
- J. Zhao, D. M. Simeone, D. Heidt, M. A. Anderson and D. M. Lubman, J. Proteome Res., 2006, 5, 1792–1802 CrossRef CAS PubMed.
- J. C. Tercero and T. Diaz-Maurino, Anal. Biochem., 1988, 174, 128–136 CrossRef CAS.
- A. Lee, M. Nakano, M. Hincapie, D. Kolarich, M. S. Baker, W. S. Hancock and N. H. Packer, OMICS, 2010, 14, 487–499 CrossRef CAS PubMed.
- A. J. Alpert, J. Chromatogr., 1990, 499, 177–196 CrossRef CAS.
- M. Thaysen-Andersen, Merck SeQuant Technical Summary TS-001, 2010, http://www.sequant.com/files/documents/applications/zichilic/MerckSeQuant_Technical_Summary_TS-001.Glycomics_and_Glycoproteomics.pdf Search PubMed.
- P. Hagglund, J. Bunkenborg, F. Elortza, O. N. Jensen and P. Roepstorff, J. Proteome Res., 2004, 3, 556–566 CrossRef.
- M. Thaysen-Andersen, I. B. Thogersen, H. J. Nielsen, U. Lademann, N. Brunner, J. J. Enghild and P. Hojrup, Mol. Cell. Proteomics, 2007, 6, 638–647 CAS.
- M. Thaysen-Andersen, I. B. Thogersen, U. Lademann, H. Offenberg, A. M. Giessing, J. J. Enghild, H. J. Nielsen, N. Brunner and P. Hojrup, Biochimica et Biophysica Acta, 2008, 1784, 455–463 CrossRef CAS PubMed.
- A. Rogowska-Wrzesinskaa, M.-C. Le Bihan, M. Thaysen-Andersen and P. Roepstorff, J. Proteomics, 2013, 88, 4–13 CrossRef PubMed.
- L. Holten-Andersen, M. Thaysen-Andersen, S. B. Jensen, C. Buchwald, P. Hojrup, H. Offenberg, H. J. Nielsen, N. Brunner, B. Nauntofte and J. Reibel, APMIS, 2011, 119, 741–749 CrossRef CAS PubMed.
- S. Mysling, G. Palmisano, P. Hojrup and M. Thaysen-Andersen, Anal. Chem., 2010, 82, 5598–5609 CrossRef CAS PubMed.
- M. Thaysen-Andersen, S. Mysling and P. Hojrup, Anal. Chem., 2009, 81, 3933–3943 CrossRef CAS PubMed.
- A. J. Alpert, Anal. Chem., 2008, 80, 62–76 CrossRef CAS PubMed.
- K. Lohrig, U. Lewandrowski, R. P. Zahedi, D. Wolters and A. Sickmann, Clin. Proteomics, 2008, 4, 25–36 CrossRef.
- P. Hao, T. Guo and S. K. Sze, PLoS One, 2011, 6, e16884 CAS.
- M. Yodoshi, T. Ikuta, Y. Mouri and S. Suzuki, Anal. Sci., 2010, 26, 75–81 CrossRef CAS.
- R. J. Sturgeon and C. M. Sturgeon, Carbohydr. Res., 1982, 103, 213–219 CrossRef CAS.
- U. Lewandrowski, R. P. Zahedi, J. Moebius, U. Walter and A. Sickmann, Mol. Cell. Proteomics, 2007, 6, 1933–1941 CAS.
- F. A. Bettelheim and B. M. Scheinthal, Carbohydr. Res., 1968, 6, 257–265 CrossRef.
- B. Ghesquiere, L. Buyl, H. Demol, J. Van Damme, A. Staes, E. Timmerman, J. Vandekerckhove and K. Gevaert, J. Proteome Res., 2007, 6, 4304–4312 CrossRef CAS PubMed.
- I. Kuroda, Y. Shintani, M. Motokawa, S. Abe and M. Furuno, Anal. Sci., 2004, 20, 1313–1319 CrossRef CAS.
- M. W. Pinkse, P. M. Uitto, M. J. Hilhorst, B. Ooms and A. J. Heck, Anal. Chem., 2004, 76, 3935–3943 CrossRef CAS PubMed.
- A. Sano and H. Nakamura, Anal. Sci., 2004, 20, 861–864 CrossRef CAS.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. Jorgensen, Mol. Cell. Proteomics, 2005, 4, 873–886 CAS.
- K. Engholm-Keller and M. R. Larsen, J. Proteomics, 2011, 75, 317–328 CrossRef CAS PubMed.
- G. Palmisano and T. E. Thingholm, Expert Rev. Proteomics, 2010, 7, 439–456 CrossRef CAS PubMed.
- A. J. McQuillan and P. A. Connor, Langmuir, 1999, 15, 2916–2921 CrossRef.
- G. Palmisano, S. E. Lendal, K. Engholm-Keller, R. Leth-Larsen, B. L. Parker and M. R. Larsen, Nat. Protoc., 2010, 5, 1974–1982 CrossRef CAS PubMed.
- B. L. Parker, G. Palmisano, A. V. Edwards, M. Y. White, K. Engholm-Keller, A. Lee, N. E. Scott, D. Kolarich, B. D. Hambly, N. H. Packer, M. R. Larsen and S. J. Cordwell, Mol. Cell. Proteomics, 2011, 10, M110 006833 Search PubMed.
- G. Palmisano, D. Antonacci and M. R. Larsen, J. Proteome Res., 2010, 9, 6148–6159 CrossRef CAS PubMed.
- J. Zhu, F. Wang, K. Cheng, J. Dong, D. Sun, R. Chen, L. Wang, M. Ye and H. Zou, Proteomics, 2013, 13, 1306–1313 CrossRef CAS PubMed.
- W. Cao, J. Cao, J. Huang, L. Zhang, J. Yao, H. Xu and P. Yang, Glycoconjugate J., 2012, 29, 433–443 CrossRef CAS PubMed.
- R. M. F. Smith, End group analysis of polysaccharides, Interscience, New York, 1956 Search PubMed.
- H. Zhang, X. J. Li, D. B. Martin and R. Aebersold, Nat. Biotechnol., 2003, 21, 660–666 CrossRef CAS PubMed.
- Y. Zhou, R. Aebersold and H. Zhang, Anal. Chem., 2007, 79, 5826–5837 CrossRef CAS PubMed.
- B. Sun, J. A. Ranish, A. G. Utleg, J. T. White, X. Yan, B. Lin and L. Hood, Mol. Cell. Proteomics, 2007, 6, 141–149 CAS.
- L. Wang, U. K. Aryal, Z. Dai, A. C. Mason, M. E. Monroe, Z. X. Tian, J. Y. Zhou, D. Su, K. K. Weitz, T. Liu, D. G. Camp, 2nd, R. D. Smith, S. E. Baker and W. J. Qian, J. Proteome Res., 2012, 11, 143–156 CrossRef CAS PubMed.
- I. Cima, R. Schiess, P. Wild, M. Kaelin, P. Schuffler, V. Lange, P. Picotti, R. Ossola, A. Templeton, O. Schubert, T. Fuchs, T. Leippold, S. Wyler, J. Zehetner, W. Jochum, J. Buhmann, T. Cerny, H. Moch, S. Gillessen, R. Aebersold and W. Krek, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3342–3347 CrossRef CAS PubMed.
- T. Ishihara, I. Fukuda, A. Morita, Y. Takinami, H. Okamoto, S. Nishimura and Y. Numata, J. Proteomics, 2011, 74, 2159–2168 CrossRef CAS PubMed.
- F. S. Berven, R. Ahmad, K. R. Clauser and S. A. Carr, J. Proteome Res., 2010, 9, 1706–1715 CrossRef CAS PubMed.
- T. Liu, W. J. Qian, M. A. Gritsenko, D. G. Camp, 2nd, M. E. Monroe, R. J. Moore and R. D. Smith, J. Proteome Res., 2005, 4, 2070–2080 CrossRef CAS PubMed.
- J. Cao, C. Shen, H. Wang, H. Shen, Y. Chen, A. Nie, G. Yan, H. Lu, Y. Liu and P. Yang, J. Proteome Res., 2009, 8, 662–672 CrossRef CAS PubMed.
- R. Chen, X. Jiang, D. Sun, G. Han, F. Wang, M. Ye, L. Wang and H. Zou, J. Proteome Res., 2009, 8, 651–661 CrossRef CAS PubMed.
- J. J. Hill, M. J. Moreno, J. C. Lam, A. S. Haqqani and J. F. Kelly, Proteomics, 2009, 9, 535–549 CrossRef CAS PubMed.
- U. Lewandrowski and A. Sickmann, Methods Mol. Biol., 2009, 534, 225–238 CAS.
- S. Honda and S. Suzuki, Anal. Biochem., 1984, 142, 167–174 CrossRef CAS.
- G. Blix, E. Lindberg, L. Odin and I. Werner, Acta Soc. Med. Ups., 1956, 61, 1–25 CAS.
- C. G. Gahmberg and L. C. Andersson, J. Biol. Chem., 1977, 252, 5888–5894 CAS.
- O. O. Blumenfeld, P. M. Gallop and T. H. Liao, Biochem. Biophys. Res. Commun., 1972, 48, 242–251 CrossRef CAS.
- E. A. Bayer, H. Ben-Hur and M. Wilchek, Anal. Biochem., 1988, 170, 271–281 CrossRef CAS.
- P. A. De Bank, B. Kellam, D. A. Kendall and K. M. Shakesheff, Biotechnol. Bioeng., 2003, 81, 800–808 CrossRef CAS PubMed.
- B. Wollscheid, D. Bausch-Fluck, C. Henderson, R. O'Brien, M. Bibel, R. Schiess, R. Aebersold and J. D. Watts, Nat. Biotechnol., 2009, 27, 378–386 CrossRef CAS PubMed.
- R. L. Gundry, K. Raginski, Y. Tarasova, I. Tchernyshyov, D. Bausch-Fluck, S. T. Elliott, K. R. Boheler, J. E. Van Eyk and B. Wollscheid, Mol. Cell. Proteomics, 2009, 8, 2555–2569 CAS.
- R. Schiess, L. N. Mueller, A. Schmidt, M. Mueller, B. Wollscheid and R. Aebersold, Mol. Cell. Proteomics, 2009, 8, 624–638 CAS.
- I. Marchal, D. L. Jarvis, R. Cacan and A. Verbert, Biol. Chem., 2001, 382, 151–159 CrossRef CAS PubMed.
- Y. Zeng, T. N. Ramya, A. Dirksen, P. E. Dawson and J. C. Paulson, Nat. Methods, 2009, 6, 207–209 CrossRef CAS PubMed.
- E. H. Cordes and W. P. Jencks, Biochemistry, 1962, 1, 773–778 CrossRef CAS.
- A. Dirksen and P. E. Dawson, Bioconjugate Chem., 2008, 19, 2543–2548 CrossRef CAS PubMed.
- T. N. Ramya, E. Weerapana, B. F. Cravatt and J. C. Paulson, Glycobiology, 2013, 23, 211–221 CrossRef CAS PubMed.
- M. Kurogochi, M. Amano, M. Fumoto, A. Takimoto, H. Kondo and S. Nishimura, Angew. Chem., Int. Ed., 2007, 46, 8808–8813 CrossRef CAS PubMed.
- H. Shimaoka, H. Kuramoto, J. Furukawa, Y. Miura, M. Kurogochi, Y. Kita, H. Hinou, Y. Shinohara and S. Nishimura, Chemistry, 2007, 13, 1664–1673 CrossRef CAS PubMed.
- S. Nishimura, K. Niikura, M. Kurogochi, T. Matsushita, M. Fumoto, H. Hinou, R. Kamitani, H. Nakagawa, K. Deguchi, N. Miura, K. Monde and H. Kondo, Angew. Chem., Int. Ed., 2004, 44, 91–96 CrossRef PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS.
- B. Bodenmiller, L. N. Mueller, M. Mueller, B. Domon and R. Aebersold, Nat. Methods, 2007, 4, 231–237 CrossRef CAS PubMed.
- C. A. McDonald, J. Y. Yang, V. Marathe, T. Y. Yen and B. A. Macher, Mol. Cell. Proteomics, 2009, 8, 287–301 CAS.
- K. P. Bateman, J. F. Kelly, P. Thibault, L. Ramaley and R. L. White, Methods Mol. Biol., 2003, 213, 219–239 CAS.
- S. Amon, A. D. Zamfir and A. Rizzi, Electrophoresis, 2008, 29, 2485–2507 CrossRef CAS PubMed.
- C. Sottani, M. Fiorentino and C. Minoia, Rapid Commun. Mass Spectrom., 1997, 11, 907–913 CrossRef CAS.
- U. Kuzmanov, C. R. Smith, I. Batruch, A. Soosaipillai, A. Diamandis and E. P. Diamandis, Proteomics, 2012, 12, 799–809 CrossRef CAS PubMed.
- P. Datta, Biochemistry, 1974, 13, 3987–3991 CrossRef CAS.
- S. Diaz and A. Varki, Anal. Biochem., 1985, 150, 32–46 CrossRef CAS.
- H. Kayser, R. Zeitler, C. Kannicht, D. Grunow, R. Nuck and W. Reutter, J. Biol. Chem., 1992, 267, 16934–16938 CAS.
- O. T. Keppler, P. Stehling, M. Herrmann, H. Kayser, D. Grunow, W. Reutter and M. Pawlita, J. Biol. Chem., 1995, 270, 1308–1314 CrossRef CAS PubMed.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- N. J. Agard and C. R. Bertozzi, Acc. Chem. Res., 2009, 42, 788–797 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS.
- K. J. Yarema, L. K. Mahal, R. E. Bruehl, E. C. Rodriguez and C. R. Bertozzi, J. Biol. Chem., 1998, 273, 31168–31179 CrossRef CAS PubMed.
- C. L. Jacobs, K. J. Yarema, L. K. Mahal, D. A. Nauman, N. W. Charters and C. R. Bertozzi, Methods Enzymol., 2000, 327, 260–275 CAS.
- N. W. Charter, L. K. Mahal, D. E. Koshland, Jr and C. R. Bertozzi, Glycobiology, 2000, 10, 1049–1056 CrossRef CAS PubMed.
- A. Varki, FASEB J., 1991, 5, 226–235 CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- S. T. Laughlin, N. J. Agard, J. M. Baskin, I. S. Carrico, P. V. Chang, A. S. Ganguli, M. J. Hangauer, A. Lo, J. A. Prescher and C. R. Bertozzi, Methods Enzymol., 2006, 415, 230–250 CAS.
- S. T. Laughlin and C. R. Bertozzi, Nat. Protoc., 2007, 2, 2930–2944 CrossRef CAS PubMed.
- L. Yang, J. O. Nyalwidhe, S. Guo, R. R. Drake and O. J. Semmes, Mol. Cell. Proteomics, 2011, 10, M110 007294 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- T. N. Ramya, E. Weerapana, L. Liao, Y. Zeng, H. Tateno, J. R. Yates, 3rd, B. F. Cravatt and J. C. Paulson, Mol. Cell. Proteomics, 2010, 9, 1339–1351 CAS.
- E. Saxon, S. J. Luchansky, H. C. Hang, C. Yu, S. C. Lee and C. R. Bertozzi, J. Am. Chem. Soc., 2002, 124, 14893–14902 CrossRef CAS PubMed.
- A. A. Neves, H. Stockmann, R. R. Harmston, H. J. Pryor, I. S. Alam, H. Ireland-Zecchini, D. Y. Lewis, S. K. Lyons, F. J. Leeper and K. M. Brindle, FASEB J., 2011, 25, 2528–2537 CrossRef CAS PubMed.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Brase, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- C. Hart, L. G. Chase, M. Hajivandi and B. Agnew, Methods Mol. Biol., 2011, 698, 459–484 CAS.
- T. L. Hsu, S. R. Hanson, K. Kishikawa, S. K. Wang, M. Sawa and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2614–2619 CrossRef CAS PubMed.
- S. R. Hanson, T. L. Hsu, E. Weerapana, K. Kishikawa, G. M. Simon, B. F. Cravatt and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 7266–7267 CrossRef CAS PubMed.
- Y. C. Liu, H. Y. Yen, C. Y. Chen, C. H. Chen, P. F. Cheng, Y. H. Juan, K. H. Khoo, C. J. Yu, P. C. Yang, T. L. Hsu and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11332–11337 CrossRef CAS PubMed.
- P. V. Chang, X. Chen, C. Smyrniotis, A. Xenakis, T. Hu, C. R. Bertozzi and P. Wu, Angew. Chem., Int. Ed., 2009, 48, 4030–4033 CrossRef CAS PubMed.
- D. Soriano Del Amo, W. Wang, H. Jiang, C. Besanceney, A. C. Yan, M. Levy, Y. Liu, F. L. Marlow and P. Wu, J. Am. Chem. Soc., 2010, 132, 16893–16899 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- K. W. Dehnert, J. M. Baskin, S. T. Laughlin, B. J. Beahm, N. N. Naidu, S. L. Amacher and C. R. Bertozzi, ChemBioChem, 2012, 13, 353–357 CrossRef CAS PubMed.
- J. Du, M. A. Meledeo, Z. Wang, H. S. Khanna, V. D. Paruchuri and K. J. Yarema, Glycobiology, 2009, 19, 1382–1401 CrossRef CAS PubMed.
- S. G. Sampathkumar, A. V. Li, M. B. Jones, Z. Sun and K. J. Yarema, Nat. Chem. Biol., 2006, 2, 149–152 CrossRef CAS PubMed.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS.
- C. S. Tsai, H. Y. Yen, M. I. Lin, T. I. Tsai, S. Y. Wang, W. I. Huang, T. L. Hsu, Y. S. Cheng, J. M. Fang and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2466–2471 CrossRef CAS PubMed.
- X. Yin, M. Bern, Q. Xing, J. Ho, R. Viner and M. Mayr, Mol. Cell. Proteomics, 2013, 12, 956–978 CAS.
- J. Zhang and D. I. Wang, J. Chromatogr., B: Biomed. Sci. Appl., 1998, 712, 73–82 CrossRef CAS.
- U. Lewandrowski, S. Wortelkamp, K. Lohrig, R. P. Zahedi, D. A. Wolters, U. Walter and A. Sickmann, Blood, 2009, 114, e10–19 CrossRef CAS PubMed.
- M. Kurogochi, T. Matsushista, M. Amano, J. Furukawa, Y. Shinohara, M. Aoshima and S. Nishimura, Mol. Cell. Proteomics, 2010, 9, 2354–2368 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra42960a |
| This journal is © The Royal Society of Chemistry 2013 |