 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(ionic liquid)-derived nitrogen-doped hollow carbon spheres: synthesis and loading with Fe2O3 for high-performance lithium ion batteries†
Juan
Balach
a,
Haiping
Wu
b,
Frank
Polzer
c,
Holm
Kirmse
c,
Qiang
Zhao
a,
Zhixiang
Wei
*b and
Jiayin
Yuan
*a
aMax-Planck-Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, Am Muehlenberg 1, D-14476, Golm, Germany. E-mail: jiayin.yuan@mpikg.mpg.de; Fax: +49-3315679502; Tel: +49-3315679552
bNational Center for Nanoscience and Technology, Beiyitiao No. 11, Zhongguancun, 100190, Beijing, P. R. China. E-mail: weizx@nanoctr.cn; Fax: +86-62656765; Tel: +86-2545565
cInstitut für Physik, Humboldt-Universität zu Berlin, AG TEM, Newtonstrasse 15, D-12489, Berlin, Germany. E-mail: holm.kirmse@physik.hu-berlin.de; Fax: +49-30-20 93 77 60; Tel: +49-03020937641
First published on 19th March 2013
Abstract
Porous nitrogen-doped hollow carbon spheres (NHCSs) with tailored particle size, nitrogen content and wall thickness were prepared by grafting a thin layer of poly(ionic liquid) as carbon precursor onto silica particle surface followed by carbonization and template removal. In the next step, Fe2O3 nanoparticles were anchored onto the NHCSs via an impregnation-thermal decomposition route using Fe(NO3)3 as iron source. The as-synthesized Fe2O3–NHCS composite was utilized as the anode material for a lithium-ion battery, which exhibited a high reversible capacity of 1120 mAh g−1 at a rate of 100 mA g−1, and a coulombic efficiency of ∼98% at a rate of 500 mA g−1 after 65 cycles.
1. Introduction
In the past decades, nanostructured carbon materials have been playing an important role in the development of new electrical energy storage and conversion devices, catalyst support material for fuel cells, gas storage systems, etc.1–4 The high importance of these carbonaceous materials resides in the relatively low cost and their excellent physical and chemical properties tunable in a wide range, such as high surface area, narrow pore size, large pore volumes, good electronic conductivity, chemical inertness, and high thermal and mechanical stability.Different synthetic strategies have been devised to produce carbon materials with various structures and morphologies (nanotubes, fibers, nanosheets, spheres, etc.), including precursor-controlled carbonization,5 hydrothermal carbonization,6,7 self-assembly using soft templates,8 impregnation and surface-grafting onto hard templates,9 just to name a few. Among these strategies, polymers have been very often utilized as the carbon precursors. Poly(ionic liquid)s (PILs) are polymerized ionic liquids (ILs), which can combine some of the unique properties of ILs with the common profile of polymers.10–13 Very recently, PILs have attracted increasing attention as novel carbon precursors.14–19 The cationic and/or anionic moieties in each repeating unit of the PILs can homogeneously incorporate heteroatoms into the structure of carbon materials, in which nitrogen is the mostly used dopant. Moreover the polymeric nature of PILs allows simple processing and shaping of carbon precursor into certain geometry that is adopted in the final carbon products. From the chemical structure point of view, the presence of cyano/nitrile functionalized anions (dicyanamide or tricyanomethide) or cations tends to form highly crosslinked and thermally more stable frameworks because of the trimerization reactions of the cyano/nitrile groups at intermediate temperatures, and consequently a high carbon yield after thermal treatment is obtained.14,20,21 The doping with nitrogen atoms favorably leads to a notable increase in the electric conductivity and catalytic activity.22–24 Additionally, the introduction of nitrogen atoms into the carbon framework can stabilize metal or metal oxide nanoparticles through the generation of topological defects and the activation of nitrogen-neighboring carbon atoms due to the electron affinity of nitrogen.25–28 In this way, metal oxide-functionalized carbon can be prepared in a simple manner and employed directly in electrical energy storage devices, such as supercapacitors29 and lithium-ion batteries (LIBs).30
Among various metal oxides, Fe2O3 has attracted considerable research interest as the anode material of LIBs owing to its high theoretical specific capacity (1007 mAh g−1), low cost and high abundance.31 However, bulk Fe2O3 has low electrical conductivity and poor cyclability that is related with the large volume change during the charge–discharge process. To overcome these issues and enhance the overall electrochemical performance of Fe2O3 anodes, an effective strategy is to confine Fe2O3 nanoparticles in the cavity of hollow carbon structures, which can attune the volume change and meanwhile enhance the charge transport.32,33 For example, Yu et al.34 recently prepared Fe2O3 nanoparticles inside the hollow core (∼55 nm) of carbon nanotubes (CNTs) as the anode material of LIBs. The as-prepared Fe2O3-filled CNTs showed a high initial reversible charge capacity of 2081 mAh g−1 and a reversible capacity of 768 mAh g−1 at a rate of 35 mA g−1 after 40 cycles, with a coulombic efficiency of 95%. The improved cycling performance was attributed to the ability of the carbon to provide a physical buffering layer for the large volume change and enhance the electric conductivity. In addition, immobilization of iron oxide nanoparticles inside the matrix of single-walled CNTs or graphene aerogel has also been reported to improve the capacity and cyclability of iron oxide in LIBs or other energy-related applications.35,36
In this contribution, porous nitrogen-doped hollow carbon spheres (NHCSs) as a distinct geometry with tailored structure features were prepared via a PIL route using silica particles as template. The PIL was firstly deposited onto the silica particles through a “grafting-onto” method via polymerizing an IL monomer 3-cyanomethyl-1-vinylimidazolium bromide (CMVImBr) in the presence of silica particles in ethanol. This formed a uniform PIL coating on the silica template as carbon precursor and nitrogen source. Carbonization of the silica/PIL composite (SiO2@PCMVImBr) and subsequent removal of silica template produced nitrogen-doped porous carbon spheres with a hollow interior. The as-obtained NHCSs contained ca. 7–10 wt% of nitrogen and showed a size-dependent specific surface area up to 570 m2 g−1. To demonstrate its application in LIBs, Fe2O3 nanoparticles were then loaded in the NHCSs via simple thermal decomposition of Fe(NO3)3 at 400 °C under nitrogen atmosphere. The obtained Fe2O3–NHCS composite was tested as an anode material for LIBs. As a result of the structural synergistic effect, the cycling performance and the rate capability of the Fe2O3–NHCS composite electrode are significantly improved.
2. Experimental
2.1. Materials
1-Vinylimidazole (99%), bromoacetonitrile (97%), Fe(NO3)3·9H2O (99%), ammonia solution (28–30%, Aldrich) and tetraethoxysilane (TEOS) (97%) were obtained from Sigma-Aldrich and used without further purification. 2,2′-Azobis(2-methylpropionitrile) (AIBN) (98%, Aldrich) was recrystallized from methanol. 3-Cyanomethyl-1-vinylimidazolium bromide (CMVImBr) as IL monomer, and 1,4-butanediyl-3,3′-bis-l-vinylimidazolium dibromide (BVD) as divinylimidazolium crosslinking agent were prepared according to our previous reports.15,37 The solvents and other materials were used as received.2.2. Synthesis of SiO2 particles
Silica particles were prepared via the Stöber method.38 In a typical run, 52 g of TEOS was added to 400 mL of absolute ethanol containing 52 g of deionized water. The mixture was stirred vigorously at 800 rpm for 40 min. Subsequently, a desired amount of NH4OH solution (28–30%) was added into the above mixture dropwise, and the reaction was kept stirring for another 4 h. The diameter of the SiO2 particles was carefully adjusted by varying the amount of NH4OH solution, so that several batches of silica particles with different sizes from 70 nm to 790 nm were prepared. The obtained SiO2 suspension was washed by four centrifugation/re-dispersion cycles in deionized water. The purified silica particles were finally re-dispersed in ethanol with a solid content of about 4 wt% for further use.2.3. Synthesis of SiO2/PIL composite particles (SiO2@PCMVImBr)
The PIL layer on silica particles was synthesized via polymerization of IL monomer in the presence of silica particles in ethanol. In a general polymerization procedure, 3 g of CMVImBr monomer, 200 mg of AIBN and 5 mL of deionized water were added to 70 mL of ethanol containing 3 g of dispersed SiO2 particles. The dispersion was degassed by purging with argon for 30 min prior to polymerization. The polymerization was conducted under vigorous stirring at 80 °C overnight. After cooling down, the obtained SiO2@PCMVImBr composite particles were washed by four centrifugation/re-dispersion cycles with deionized water. A light yellow solid was obtained and dried at 60 °C under vacuum to constant weight. In order to control the NHCS size, SiO2 particles with different size were utilized. The nitrogen content of the N-doped carbon samples was adjusted by the addition of 20, 50 and 80 mol% (with regard to the CMVImBr monomer used) of acrylonitrile (AN) in the polymeric media. Furthermore, the wall thickness of hollow carbon spheres was modified by the addition of 5, 10, and 40 mol% (with respect to the CMVImBr monomer used) of the crosslinker BVD.2.4. Carbonization process
In a typical experiment, the dried SiO2@PCMVImBr samples were put into an aluminum oxide crucible in an oven and heated under a N2 atmosphere to 900 °C at a heating rate of 3.3 °C min−1. The sample was further kept at 900 °C for 1 h and cooled down slowly to room temperature. The silica templates were removed by an aqueous solution of NH4HF2 (4 M) for 5 days, followed by washing with deionized water for 6 times and finally dried at 100 °C under vacuum till constant weight. The obtained samples were denoted as NHCSx, where x represents the size of SiO2 employed to obtain the respective hollow spheres.2.5. Preparation of Fe2O3@NHCS
Fe2O3 nanoparticles were loaded onto the NHCS via simple thermal decomposition of Fe(NO3)3.31,34 Briefly, NHCS material was immersed in an ethanol solution of Fe(NO3)3 and ultrasonicated at room temperature under applied vacuum for 1 h. Then the dispersion was centrifuged and the supernatant solution of Fe(NO3)3 was discarded. The Fe(NO3)3-loaded NHCSs were heated to 80 °C and kept for 30 min. Afterwards they were rinsed with deionized water. The resulting material was put into an aluminum oxide crucible in an oven and gradually heated under a nitrogen atmosphere to 140 °C and kept for 6 h before being heated to 400 °C with a heating rate of 2 °C min−1 and held for 3 h. Following this procedure, Fe2O3 nanoparticles were obtained by the thermal decomposition of Fe(NO3)3 in the NHCSs.2.6. Characterization
The average particle size of silica template and the standard deviation was determined by dynamic light scattering (DLS) at 90° with a NICOMP particle sizer (model 370, NICOMP particle sizing systems, Santa Barbara, CA). Transmission electron microscopy (TEM) was carried out on a Zeiss EM 912 Omega microscope operating at 120 kV. For sample preparation, one drop of the sample dispersion was placed on a 200 mesh carbon coated copper grid and dried in air. Scanning electron microscopy (SEM) was performed on a GEMINI LEO 1550 microscope at 3 kV. Energy-dispersive X-ray spectra were taken on the SEM with an EDX spectrometer. Nitrogen sorption experiments were performed with a Quantachrome Autosorb-1 or Quadrasorb at liquid nitrogen temperature, and data analysis was performed by Quantachrome software. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) equation. Pore size distribution was determined by Barrett–Joyner–Halenda (BJH) method. The samples were degassed at 150 °C for 20 h before measurements. Elemental analysis (N content) was performed on a Vario Micro setup. The N content reported is the average of 4 measurements (two samples from one batch and two measurements per sample). In all cases the standard deviation is less than 10%. X-Ray diffraction (XRD) experiments were done with a Bruker D8 diffractometer using Cu-Kα radiation (λ = 1.5406 Å) and a Kevex counter. The mean crystalline size of Fe2O3 nanoparticles was estimated from the full width at the half-maximum (FWHM) of the (311) peak using the Scherrer equation with a correction for instrument line broadening. TEM studies comprising scanning transmission electron microscopy (STEM), electron energy dispersive X-ray spectroscopy (EDXS) as well as the electron energy loss spectroscopy (EELS) were carried out on a JEOL JEM2200FS microscope operating at 200 kV, and selected area electron diffraction (SAED) pattern was obtained on a JEOL JEM2100. X-Ray photoelectron spectroscopy (XPS) data were obtained on a Thermo ESCALAB250 instrument with a monochromatized Al Kα line source (200 W). Thermogravimetric analysis (TGA) was carried out using a Netzsch TG 209 with a heating rate of 10 K min−1 from room temperature to 1000 °C in an air flow.2.7. Electrochemical measurements
The electrochemical properties of the Fe2O3–NHCS composite as anode materials for LIBs were evaluated by a galvanostatic charge–discharge technique. The capacity was based on the mass of the Fe2O3–NHCS composite (Fe2O3–NHCS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :conductive carbon:polytetrafluoroethylene (PTFE) = 85:10:5). Swagelok-type cells were assembled in an argon-filled glove box with the samples as the test electrode, metallic lithium as the counter/reference electrode, a mixture of 1 M LiPF6 in ethylene carbonate, dimethyl carbonate and diethyl carbonate (1:1:1 weight) as the electrolyte and glass fiber as the separator. Charge–discharge measurements were carried out galvanostatically at various current densities over a voltage range of 0.005 to 3 V (vs. Li+/Li) using a battery testing system (Arbin-SCTS). Cyclic voltammogram (CV) measurements were carried out in the voltage range of 0.005 to 3 V (vs. Li+/Li) at a scan rate of 0.1 mV s−1.
:conductive carbon:polytetrafluoroethylene (PTFE) = 85:10:5). Swagelok-type cells were assembled in an argon-filled glove box with the samples as the test electrode, metallic lithium as the counter/reference electrode, a mixture of 1 M LiPF6 in ethylene carbonate, dimethyl carbonate and diethyl carbonate (1:1:1 weight) as the electrolyte and glass fiber as the separator. Charge–discharge measurements were carried out galvanostatically at various current densities over a voltage range of 0.005 to 3 V (vs. Li+/Li) using a battery testing system (Arbin-SCTS). Cyclic voltammogram (CV) measurements were carried out in the voltage range of 0.005 to 3 V (vs. Li+/Li) at a scan rate of 0.1 mV s−1.
3. Results and discussion
A general template-directed synthetic route to NHCSs and Fe2O3 nanoparticles loaded NHCSs is illustrated in Scheme 1. Fig. 1 shows the transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images of the as-synthesized NHCSs of different sizes by pairing several silica templates. The sizes of the silica particle templates were controlled during the synthesis by the addition of different amount of aqueous ammonia solution as the catalyst for TEOS hydrolysis and condensation. This produced uniform spherical particles of 70, 90, 130, 180, 250 and 790 nm in size, with a standard size deviation of ca 0.1, as determined by dynamic light scattering measurements and further proven by TEM (Fig. S1 and Table S1, ESI†). A thin PIL layer was grafted onto the silica particle surface via in situ polymerization of the IL monomer CMVImBr in the presence of silica particles. During this process, PCMVImBr as cationic polyelectrolyte formed and precipitated out onto the negatively charged silica particle surface due to the ionic complexation as well as its rather low solubility in ethanol. NHCSs were then successfully obtained by carbonization of the silica/PIL composite particles at 900 °C under nitrogen atmosphere and removal of silica cores.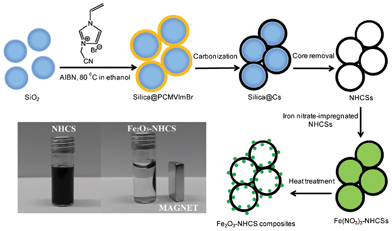 | ||
| Scheme 1 Schematic illustration of the formation of NHCSs based on the polymerization of 3-cyanomethyl-1-vinylimidazolium bromide monomer onto silica particle templates, and the further preparation of Fe2O3–NHCS composites via impregnation-thermal decomposition of iron nitrate. | ||
 | ||
| Fig. 1 TEM (a, b, d–f) and SEM images (c) of NHCSs of different sizes. (a–c) NHCS180, (d) NHCS70, (e) NHCS130 and (f) NHCS790. | ||
In general, both the shape and size of the NHCSs show good structural replication of the corresponding silica templates (Table S1, ESI†). NHCSs of 180 nm in size (NHCS180) as a representative example were visualized in both low and high magnification TEM images in Fig. 1. The overview in a large size scale (3 μm × 3 μm) in Fig. 1a, proves the mechanical stability of the NHCSs under dense packing. The NHCSs in a close view in Fig. 1b appear very uniform in both shape and contrast yet semi-transparent due to a thin carbon wall (2.1 nm). The SEM image in Fig. 1c provides a direct visual proof of the hollow interior, as it shows a characteristic open capsular morphology, frequently observed in hollow spherical objects. It should be mentioned that the smallest NHCSs prepared via this template approach is 70 nm (Fig. 1d). Below this size, the produced products either have wall structures with many defects or exist purely as irregular fragments, indicating the complete breakage of the hollow spheres. It is assumed that the amplified curvature effect at a small size scale (<70 nm) creates a high interfacial stress causing the collapse of the thin carbon wall after removing the supportive silica template. On the contrary, large sized NHCSs are free of this problem, and NHCSs as large as 790 nm in size were reached in our work. Overall, the judicious choice of the silica template can produce NHCSs in a broad diameter range of 70–790 nm.
The NHCS products were further characterized by nitrogen sorption measurements and elemental analysis. The results are summarized in Table S1, ESI.† The specific surface area derived from the BET equation varies, depending on the sphere size and can be as high as 570 m2 g−1. It can be seen that generally the larger the hollow spheres are, the lower the specific surface area is. Fig. 2a shows the nitrogen adsorption–desorption isotherms recorded from NHCS70 and NHCS180. Their specific surface areas are calculated to be 496 and 254 m2 g−1, respectively. NHCS180 is apparently macroporous due to its large shape and the intrinsic interior macropores of 180 nm. Its relatively high specific surface area of 254 m2 g−1 results from the micropores located on the carbon wall, which are very important for the NHCS applications because they allow for the mass transport between the walls and different NHCSs. When the NHCS size decreased from 180 to 70 nm, in addition to the macro- and micropores, pronounced mesopores were found as well. The mesopore size according to the BET equation is ca. 17.5 nm, far below the interior dimension of the hollow spheres (Fig. S2, ESI†). They are supposed to come from the interstitial voids generated when packing the hollow spheres densely together. It is noteworthy that the micropores in carbon hollow spheres prepared from the silica template approach seem to be very characteristic and independent of the carbon precursor type, as also found in other reports.39,40Fig. 2b shows a typical XRD pattern of NHCSs, which exhibits a sharp peak at 2θ = 26° and another at 44° attributed to the (002) and (100) reflections of a graphitic phase. The reflection peaks at (004) and (110) were somehow very weak and hardly visible, which is indicative of the lack of long range order in the crystalline structure. This is visualized in a high resolution TEM image in Fig. 2c. Bent and discontinuous graphitic textures are clearly observed. The disordered structure might stem from the heteroatom doping as atomic structure defects as well as the surface curvature in this distinctive morphology.
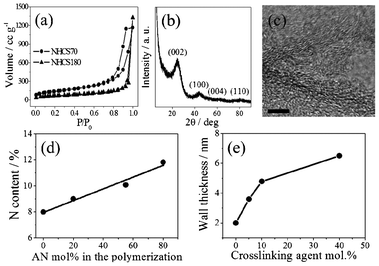 | ||
| Fig. 2 (a) Nitrogen sorption isotherms of NHCS70 and NHCS180, (b) the XRD pattern of NHCS180, (c) high resolution TEM image of NHCS180. The scale bar is 5 nm. (d) Nitrogen content of NHCS180 obtained via varying the mol% of acrylonitrile (AN) in the polymerization reaction and (e) the wall thickness variation of NHCS180 by changing the mol% of crosslinking agent BVD. | ||
NHCSs with different nitrogen contents were prepared by the addition of acrylonitrile (AN) in the CMVImBr polymerization step to form a PCMVImBr-co-PAN random copolymer shell onto the surface of silica template. PAN is a well-known and widely used carbon precursor, which has been recently used to modulate the nitrogen content during the carbonization of PIL.41 By doping PIL with PAN, an increase in the nitrogen content at 900 °C was observed. As illustrated in Fig. 2d, when more AN monomer was copolymerized with IL monomer, the nitrogen content in the NHCSs increases almost linearly up to ∼150% with respect to the NHCSs prepared from pure IL monomer. Thus, the nitrogen doping can be easily modulated via controlling the molar ratio of AN to CMVImBr. To tune the NHCS wall thickness, a dicationic vinylimidazolium crosslinker BVD, structurally more similar to the CMVImBr monomer than AN, was added in the polymerization process. The crosslinker could covalently immobilize more PIL onto the silica particle surface, which logically thickens the carbon wall in the carbon formation stage. The variation in the wall thickness of the NHCSs as a function of the increasing amount of BVD is illustrated in Fig. 2e. NHCS samples prepared without BVD usually have a wall thickness of 2.1 nm. As the amount of BVD increased from 5 to 40 mol%, the average wall thickness increased gradually from 3.8 to 6.5 nm, making the spherical shell carbon structure mechanically more stable.
The nitrogen distribution in the carbon sphere of bare NHCS180 was investigated by STEM-based electron energy loss spectroscopy (EELS) (Fig. 3). The corresponding EEL spectra of the nitrogen K-edge are shown from point A to point B (see inset in Fig. 3). In vacuum, as expected no significant nitrogen signal could be found. The EELS intensity of the nitrogen K-edge increases drastically within the area of the wall of the hollow sphere. Upon scanning towards the center of the hollow sphere, the signal of the nitrogen K-edge decreases slightly. This can be explained with a thickness effect due to the geometry of the spherical hollow particle. The result hints to a homogeneous nitrogen distribution within the hollow carbon spheres along the scanning profile.
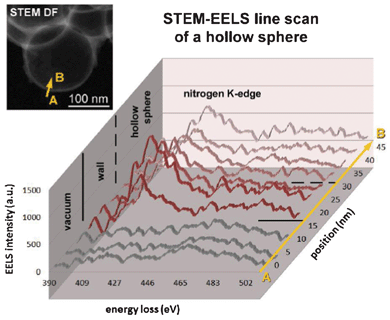 | ||
| Fig. 3 EEL spectra of nitrogen K-edge along a NHCS180. The inset shows a corresponding STEM dark field image of NHCS180 with the arrow indicating the line scan for the STEM EELS analysis. | ||
NHCS180 was applied in the next step as support material for the preparation of Fe2O3–NHCS180 composite. Fig. 4a and 4b show the TEM images of the as-obtained Fe2O3–NHCS180 composite after the impregnation-thermal decomposition process of iron nitrate in ethanol. Instead of an even contrast and a smooth contour of pure NHCSs (Fig. 1b), the Fe2O3–NHCS180 composite exhibits directly the structure heterogeneity, such as more grey dots in the sphere center area and rough surface (Fig. 4b). As observed by the TEM analysis, Fe2O3 nanoparticles on the carbon wall have an average size of 8 nm and they are uniformly distributed. Fig. 4c and 4d are two SEM images of the Fe2O3–NHCS180 composite. The Fe2O3 nanoparticles were found in fact located both on the inner- as well as outer-wall surface.
 | ||
| Fig. 4 (a, b) low and high magnification TEM images of Fe2O3–NHCS180 composite and (c, d) low and high magnification SEM images of Fe2O3–NHCS180 composite. | ||
Careful examination of more SEM images supports that the Fe2O3 nanoparticles are more concentrated in the hollow spheres than on the outer wall surface. This might be caused by the washing step, in which the rest of the iron nitrate on the outer surface was removed more completely than inside the hollow sphere. In both sites, the Fe2O3 nanoparticles were physically confined either in the hollow carbon interior or in the interstitial voids between spheres. This is important for the LIB application as the free volume in the composite buffers the volume expansion during the charge–discharge cycles.
Furthermore XRD analysis determined that Fe2O3 nanoparticles are mainly maghemite (γ-Fe2O3) with a small amount of hematite (α-Fe2O3) (Fig. 5a). High-resolution TEM (HRTEM) investigations were carried out to prove the co-existence of both phases of Fe2O3. Fig. 5b shows a representative HRTEM micrograph of the composite material. A characteristic d-spacing of about 2.68 Å of the (01![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ) plane of hematite can be found as well as a lattice spacing of about 3.00 Å of the (022) plane of maghemite. The latter finding is further supported by the presence of a perpendicular arrangement of the (022) and the (02
) plane of hematite can be found as well as a lattice spacing of about 3.00 Å of the (022) plane of maghemite. The latter finding is further supported by the presence of a perpendicular arrangement of the (022) and the (02![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) lattice plane which give evidence for the tetragonal crystal structure of maghemite. This is consistent with the SAED pattern in Fig. 5c. The corresponding SAED pattern shows (012), (01) and (024) reflections of hematite (H) as well as the characteristic (022) reflection of maghemite (M). On the basis of the XRD, TEM, and SAED results presented above support that hematite and maghemite phases are present in the composite material. The mean diameter of Fe2O3 calculated from the (311) reflection using the Scherrer equation is 7.1 nm, which agrees well with the size observed by TEM. Due to γ-Fe2O3 and Fe3O4 nanoparticles posses the inverse spinel structure and can have similar XRD patterns, further evidence for the formation of γ-Fe2O3 instead of Fe3O4 was also provided by XPS (Fig. S3, ESI†). The level of Fe2p3/2 and Fe2p1/2 are 711.1 and 724.2, respectively. In addition, XPS spectra presents a satellite peak at 719.0 eV. All these experimental results confirm that the iron oxide nanoparticles of the composite are Fe2O3 rather than Fe3O4. The content of Fe2O3 in the sample was estimated by TGA analysis (Fig. S4, ESI†). Taking into account that Fe2O3 keeps its chemical structure during the heating in air while carbon is converted into CO2 in this thermal condition, the Fe2O3 content was determined to be 57.8 wt%. On the contrary, the Fe2O3 content obtained by energy dispersive X-ray (EDX) analysis was relatively low, only 26.2 wt%. The variation of the values can be explained by the surface specificity of EDX measurements, which hints that more Fe2O3 nanoparticles are located inside the hollow sphere rather than on the outer wall surface, as discussed above.
) lattice plane which give evidence for the tetragonal crystal structure of maghemite. This is consistent with the SAED pattern in Fig. 5c. The corresponding SAED pattern shows (012), (01) and (024) reflections of hematite (H) as well as the characteristic (022) reflection of maghemite (M). On the basis of the XRD, TEM, and SAED results presented above support that hematite and maghemite phases are present in the composite material. The mean diameter of Fe2O3 calculated from the (311) reflection using the Scherrer equation is 7.1 nm, which agrees well with the size observed by TEM. Due to γ-Fe2O3 and Fe3O4 nanoparticles posses the inverse spinel structure and can have similar XRD patterns, further evidence for the formation of γ-Fe2O3 instead of Fe3O4 was also provided by XPS (Fig. S3, ESI†). The level of Fe2p3/2 and Fe2p1/2 are 711.1 and 724.2, respectively. In addition, XPS spectra presents a satellite peak at 719.0 eV. All these experimental results confirm that the iron oxide nanoparticles of the composite are Fe2O3 rather than Fe3O4. The content of Fe2O3 in the sample was estimated by TGA analysis (Fig. S4, ESI†). Taking into account that Fe2O3 keeps its chemical structure during the heating in air while carbon is converted into CO2 in this thermal condition, the Fe2O3 content was determined to be 57.8 wt%. On the contrary, the Fe2O3 content obtained by energy dispersive X-ray (EDX) analysis was relatively low, only 26.2 wt%. The variation of the values can be explained by the surface specificity of EDX measurements, which hints that more Fe2O3 nanoparticles are located inside the hollow sphere rather than on the outer wall surface, as discussed above.
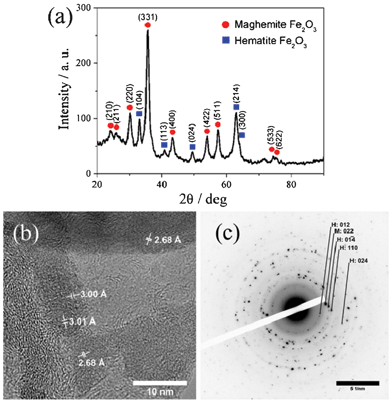 | ||
| Fig. 5 (a) XRD pattern, (b) HRTEM image, and (c) SAED pattern of Fe2O3–NHCS180 composite. Reflections of hematite and maghemite are abbreviated as H and M, respectively. | ||
Fe2O3–NHCS180 composite was employed as an anode material for LIBs. The specific capacity values reported here are calculated on the basis of the total weight of the Fe2O3–NHCS180 composite, in which Fe2O3 comprises ∼57.8% of the mass. The electrochemical behavior was characterized by cyclic voltammogram (CV) tests for the first 3 cycles at the scanning rate of 0.1 mV s−1. As shown in Fig. 6a, three peaks at voltage potentials of 1.60, 0.86 and 0.58 V are observed in the first cathodic scan, which are ascribed to the lithium ion insertion in the Fe2O3, the reduction of Fe3+ to Fe2+ and the complete reduction of iron from Fe2+ to Fe0 as well as the irreversible decomposition of the electrolyte, respectively.35,42,43 In the following cycles, the cathodic peaks shift to 0.75 V, which could be associated with reduction of Fe2+ to Fe0 and the reversible conversion reaction of lithium ion intercalation to form Li2O. The disappearance of the peaks at 1.60 V and 0.86 V from the second cathodic scan indicates a irreversible phase transformation occurred during the lithium ion insertion and extraction in the initial cycle.44 In the anodic scans, two broad overlapping peaks at 1.67 V and 1.90 V can be seen. Both peaks correspond to the reversible oxidation reactions of Fe0 to Fe2+ and Fe2+ to Fe3+, respectively. After successive cycles, the peak current and the integrated area of the anodic peak are retained, indicating the good reversible oxidation of Fe0 to Fe3+. Fig. 6b presents the discharge/charge voltage profiles of the Fe2O3–NHCS180 composite in the first 3 cycles. In accordance with the CV curve analyzed above, three voltage plateaus are observed after the first discharge/charge process, two steep plots around 1.60 and 0.90 V and widespread plateau at 0.84 V, which are attributed to the initial lithium insertion into the Fe2O3 and subsequent formation of cubic Li2Fe2O3, and the conversion of the iron from Fe2+ to Fe0.43 On the other hand, the charge curve present a sloped region from 1.40 and 2.10 V which corresponds to reversible oxidation of Fe0 to Fe3+. In the subsequent cycles, the discharge and charge capacity were kept nearly constant, suggesting excellent capacity retention of the Fe2O3–NHCS180 composite electrode.
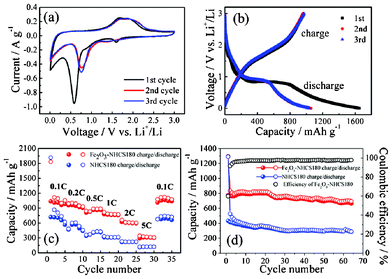 | ||
| Fig. 6 Electrochemical performance of the Fe2O3–NHCS180 composite electrode. (a) Cyclic voltammograms at a scan rate of 0.1 mV s−1 for the first three cycles. (b) Discharge and charge curves of Fe2O3–NHCS180 composite for the first 3 cycles at the current density of 100 mA g−1 in a voltage window of 0.005–3.00 V. (c) Rate performance of NHCS180 and Fe2O3–NHCS180 composite at different current densities. 1 C corresponds to 1007 mA g−1. (d) Cycling performance of NHCS180 and Fe2O3–NHCS180 composite at a current density of 500 mA g−1. | ||
Further rate capability of the Fe2O3–NHCS180 composite was investigated. Fig. 6c shows the capacity of the Fe2O3–NHCS180 composite cycled at current rate from 0.1 C to 5 C, where 1 C is defined as 1007 mA g−1 that corresponds to a maximum lithium uptake of 6 Li per Fe2O3.44 The first specific discharge capacity at a low current rate of 0.1 C is 1830 mAh g−1, which is much higher than its theoretical specific capacity of the Fe2O3 electrode (1007 mAh g−1). When the rate increases from 0.1 C to 0.2 C and 0.5 C, the discharge capacities decreased to 980 and 890 mAh g−1, respectively. Even at higher rates of 1, 2 C and 5 C, the composite can deliver a high capacity of 775, 605 and 320 mAh g−1, respectively, exhibiting a favorable high rate performance. The initial coulombic efficiency of Fe2O3–NHCS180 is 61%. This initial capacity loss can be attributed to the formation of a solid electrolyte interphase (SEI) layer on the electrode surface during the first discharge step. Nevertheless, an excellent reversible capacity of 1120 mAh g−1 with a stable coulombic efficiency of 98% was recovered when the current density was reduced back to 0.1 C. As we expected, the rate performance of NHCS180 alone is much lower compared to the Fe2O3–NHCS180 composite. At a low current rate of 0.1 C, the NHCS180 can deliver a capacity of 703 mAh g−1, roughly half of that for the composite, yet this value is surprisingly high regarding to the theoretical capacity of an ideal graphite anode (372 mAh g−1). This difference in capacity lies in that the mechanism of lithium insertion in NHCS180 is different from intercalation stage mechanism of graphite anode. CV profile of NHCS180 have not distinguishable plateaus but exhibit a hysteresis (Fig. S5, ESI†). Lithium atoms can bind on the hydrogen-terminated edges of hexagonal carbon fragments in hydrogen-containing carbon.45 The capacity strongly depends on the hydrogen content of NHCS180, resulting in a higher capacity than graphite. This process causes a change from sp2 to sp3 bonding, which can lead to hysteresis formation.46 Overall, the rate performance of our Fe2O3–NHCS composite well exceeds that of structured Fe2O3 materials (note that the specific capacity values in this work are based on the total composite weight)47,48 and provides an alternative route to fabricate a high-performance anode for lithium ion batteries from Fe2O3/carbon composites.34,35,49–52 The cycling performance of the two samples was investigated as shown in Fig. 6d. After 65 cycles under a relatively high current density of 500 mA g−1, the Fe2O3–NHCS180 composite shows a specific discharge capacity of 702 mAh g−1, which is about 85% of the second cycle of discharge capacity. After 20 cycles, we noticed a slow but not yet severe decrease in the electrode performance, which might be caused by a small fraction of Fe2O3 nanoparticles located on the outside of hollow spheres that are detached during cycling and diffused out into the electrolyte. Under the same test conditions, NHCS180 only delivers 298 mAh g−1 after 65 cycles.
These results clearly show that the PIL-derived NHCS as a new type of support materials for metal oxide plays an important role in improving the electrochemical performance of anode electrodes for LIBs. The high reversible capacity and improved cycle stability and rate performance can be ascribed to the synergistic effect of both Fe2O3 and nitrogen doped carbon hollow spheres dimensionally configured at a nanoscale. Firstly, the homogeneous decoration of the carbon wall with uniform Fe2O3 nanoparticles provides a large and ideal contact interface for these two phases. The rather small nanoparticle size of Fe2O3 (∼8 nm) compared to the bulk phase facilitates additionally a rapid charge transport, greatly promoting the quick diffusion of lithium ions. Secondly, the use of PIL as novel shaped carbon precursor produced an ultrathin and highly conductive yet porous carbon layer, which additionally benefits the ion transport. The thin carbon layer was assembled in a hollow spherical morphology and densely packed into a 3D network, which builds up a fast electron transport channel and meanwhile accommodates the volume expansion/contraction of the Fe2O3 nanoparticles during the charge/discharge process.
4. Conclusions
Herein, we demonstrated the successful synthesis of nitrogen-doped hollow carbon spheres by grafting-onto method utilizing a poly(ionic liquid) as a carbon precursor and nitrogen source and silica particles as a template. The particle size of NHCSs was controlled by the choice of the silica template, while the chemistry in the poly(ionic liquid) synthesis provides a high control in the final structure properties of the carbonaceous material, such as tunable nitrogen content and wall thickness. The NHCSs are partially graphitized and exhibit hierarchical pore structures and size-dependent specific surface area up to 570 m2 g−1. Fe2O3 nanoparticles were loaded in NHCSs by thermal decomposition of iron nitrate. The as-prepared Fe2O3–NHCS180 composite exhibited a high rate performance and a good cycling stability, which can be attributed to the confinement effect of the Fe2O3 nanoparticles and the good electric property of NHCSs. The composite material and the used approach could attract great interest on the fabrication of high-performance electrode materials for lithium-ion batteries.Acknowledgements
The authors thank the Max Planck Society for the financial support. The research work was supported by an international joint laboratory program between the Max Planck institute of Colloids and Interfaces in Potsdam, Germany and the National Center for Nanoscience and Technology in Beijing, China. Holm Kirmse and Frank Polzer would like to thank the Sonderforschungsbereich 951 of the Deutsche Forschungsgemeinschaft and the Joint Lab for Structural Research of the Humboldt University Berlin and the Helmholtz-Zentrum Berlin for funding.References
- A.-H. Lu, G.-P. Hao, Q. Sun, X.-Q. Zhang and W.-C. Li, Macromol. Chem. Phys., 2012, 213, 1107–1131 CrossRef CAS.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- L. Dai, D. W. Chang, J.-B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS.
- A.-H. Lu, W.-C. Li, G.-P. Hao, B. Spliethoff, H.-J. Bongard, B. B. Schaack and F. Schüth, Angew. Chem., Int. Ed., 2010, 49, 1615–1618 CrossRef CAS.
- W.-T. Yao and S.-H. Yu, Int. J. Nanotechnol., 2007, 4, 129–162 CrossRef CAS.
- B. Hu, K. Wang, L. Wu, S.-H. Yu, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 813–828 CrossRef CAS.
- Q. Ji, S. B. Yoon, J. P. Hill, A. Vinu, J.-S. Yu and K. Ariga, J. Am. Chem. Soc., 2009, 131, 4220–4221 CrossRef CAS.
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- R. Marcilla, J. Alberto Blazquez, J. Rodriguez, J. A. Pomposo and D. Mecerreyes, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 208–212 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS.
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS.
- J. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS.
- X. Bo, J. Bai, J. Ju and L. Guo, J. Power Sources, 2011, 196, 8360–8365 CrossRef CAS.
- J. Yuan, A. G. Marquez, J. Reinacher, C. Giordano, J. Janek and M. Antonietti, Polym. Chem., 2011, 2, 1654–1657 RSC.
- J. Yuan, S. Soll, M. Drechsler, A. H. E. Mueller and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 17556–17559 CrossRef CAS.
- T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691–696 RSC.
- J. S. Lee, X. Wang, H. Luo and S. Dai, Adv. Mater., 2010, 22, 1004–1007 CrossRef CAS.
- R. Czerw, M. Terrones, J. C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab, P. M. Ajayan, W. Blau, M. Rühle and D. L. Carroll, Nano Lett., 2001, 1, 457–460 CrossRef CAS.
- K. K. R. Datta, V. V. Balasubramanian, K. Ariga, T. Mori and A. Vinu, Chem.–Eur. J., 2011, 17, 3390–3397 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- Y.-H. Li, T.-H. Hung and C.-W. Chen, Carbon, 2009, 47, 850–855 CrossRef CAS.
- A. Zamudio, A. L. Elías, J. A. Rodríguez-Manzo, F. López-Urías, G. Rodríguez-Gattorno, F. Lupo, M. Rühle, D. J. Smith, H. Terrones, D. Díaz and M. Terrones, Small, 2006, 2, 346–350 CrossRef CAS.
- Y. Ma, S. Jiang, G. Jian, H. Tao, L. Yu, X. Wang, X. Wang, J. Zhu, Z. Hu and Y. Chen, Energy Environ. Sci., 2009, 2, 224–229 CAS.
- S. Jiang, Y. Ma, G. Jian, H. Tao, X. Wang, Y. Fan, Y. Lu, Z. Hu and Y. Chen, Adv. Mater., 2009, 21, 4953–4956 CrossRef CAS.
- C. Xue, Y. Lv, F. Zhang, L. Wu and D. Zhao, J. Mater. Chem., 2012, 22, 1547–1555 RSC.
- F. Han, W.-C. Li, M.-R. Li and A.-H. Lu, J. Mater. Chem., 2012, 22, 9645–9651 RSC.
- J. Chen, L. Xu, W. Li and X. Gou, Adv. Mater., 2005, 17, 582–586 CrossRef CAS.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS.
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS.
- W.-J. Yu, P.-X. Hou, L.-L. Zhang, F. Li, C. Liu and H.-M. Cheng, Chem. Commun., 2010, 46, 8576–8578 RSC.
- G. Zhou, D.-W. Wang, P.-X. Hou, W. Li, N. Li, C. Liu, F. Li and H.-M. Cheng, J. Mater. Chem., 2012, 22, 17942–17946 RSC.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS.
- J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- Q. Zhao, T.-P. Fellinger, M. Antonietti and J. Yuan, Macromol. Rapid Commun., 2012, 33, 1149–1153 CrossRef CAS.
- A.-H. Lu, T. Sun, W.-C. Li, Q. Sun, F. Han, D.-H. Liu and Y. Guo, Angew. Chem., Int. Ed., 2011, 50, 11765–11768 CrossRef CAS.
- C. Pan, L. Qiu, Y. Peng and F. Yan, J. Mater. Chem., 2012, 22, 13578–13584 RSC.
- J. Morales, L. Sanchez, F. Martin, F. Berry and X. Ren, J. Electrochem. Soc., 2005, 152, A1748–A1754 CrossRef CAS.
- B. Sun, J. Horvat, H. S. Kim, W.-S. Kim, J. Ahn and G. Wang, J. Phys. Chem. C, 2010, 114, 18753–18761 CAS.
- S.-L. Chou, J.-Z. Wang, D. Wexler, K. Konstantinov, C. Zhong, H.-K. Liu and S.-X. Dou, J. Mater. Chem., 2010, 20, 2092–2098 RSC.
- T. Zheng, W. McKinnon and J. Dahn, J. Electrochem. Soc., 1996, 143, 2137–2145 CrossRef CAS.
- J. Dahn, T. Zheng, L. Yinghu and J. Xue, Science, 1995, 270, 590–593 CAS.
- H. S. Kim, Y. Piao, S. H. Kang, T. Hyeon and Y.-E. Sung, Electrochem. Commun., 2010, 12, 382–385 CrossRef CAS.
- B. Wang, J. S. Chen, H. B. Wu, Z. Wang and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 17146–17148 CrossRef CAS.
- Y. Zhao, J. Li, Y. Ding and L. Guan, Chem. Commun., 2011, 47, 7416–7418 RSC.
- H. Liu, G. Wang, J. Park, J. Wang, H. Liu and C. Zhang, Electrochim. Acta, 2009, 54, 1733–1736 CrossRef CAS.
- Z. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252–5256 CAS.
- X. Zhu, Y. Zhu, S. Murali, M. D. Stoller and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of silica particle templates, textural properties and nitrogen content of NHCSs, pore size distribution of NHCS70 and NHCS180, XPS spectra of Fe2O3–NHCS180 composite, TG analysis of NHCS180 and Fe2O3–NHCS180 composite, and charge-discharge profile of NHCS180 electrode. See DOI: 10.1039/c3ra41229f |
| This journal is © The Royal Society of Chemistry 2013 |