 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxazol-2-ylidenes. A new class of stable carbenes?†
Zsolt
Kelemen
a,
Oldamur
Hollóczki
ab,
Julianna
Oláh
ac and
László
Nyulászi
*a
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics Szt, Gellért tér 4, Budapest, H-1111, Hungary. E-mail: nyulaszi@mail.bme.hu; Fax: +3614633408
bWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie Universität Leipzig, Linnéstr. 2, D-04103 Leipzig, Germany
cMaterials Structure and Modeling Research Group Hungarian Academy of Sciences, at Budapest University of Technology and Economics Szt, Gellért tér 4, Budapest, H-1111, Hungary
First published on 20th March 2013
Abstract
The investigation of the stability of several imidazol-2-ylidene analogue cyclic carbenes by an isodesmic reaction has revealed that the hitherto unknown oxazol-2-ylidene exhibits only slightly smaller stability than imidazol-2-ylidene, outperforming some of the already synthesized carbenes. Selenazol-2-ylidene also shows significant stability. The contribution of aromaticity to the stabilization has been analysed for the different five-membered ring carbenes, and was found to be relatively small for the oxygen containing systems. Investigation of possible reactivity/decomposition pathways reveals that properly substituted oxazol-2-ylidene is stable against dimerization. The thermodynamically feasible cycloreversion reaction yielding isocyanate and acetylene is prevented by a significant barrier, and furthermore with proper substitution (ring annellation) the ring can be stabilized thermodynamically as well. While in the presence of water a hydrolytic ring opening occurs; this reaction can be hindered if the water content of the reaction mixture is reduced to a few equivalents. This hydrolytic behaviour as well as the electrophilicity and nucleophilicity indices of several known nucleophilic carbenes were compared, revealing that oxazol-2-ylidene exhibits a reduced nucleophilicity with respect to imidazol-2-ylidene, while its electrophilicity is only slightly increased. This unique combination might result in unexpected (organo)catalytic activities, further expanding the colourful applications of NHCs.
Introduction
The chemistry of stable carbenes1 has been flourishing in the past decades. Apart from their unusual structure, the interest is generated by their catalytic applicability in various organic and organometallic reactions. As ligands they can fine-tune the activity of transition metal catalysts,2 furthermore, they can mimic3 transition metals and can be powerful organocatalysts.1b,4Perhaps the most widely investigated carbenes are imidazol-2-ylidenes (1) – see Scheme 1, which exhibit remarkable stability: these compounds can be kept for years under inert atmosphere, and the first synthesized example could even be molten at ca. 240 °C without any decomposition.5
This stability can be attributed to the effective π-electron donating character of the nitrogen atoms, and also to the 6π-aromaticity, which brings an additional stabilization of about 10 kcal mol−1,6 contributing to their reduced reactivity,7 and affecting their catalytic activity.8 The significant stabilizing effect of amino groups at the hypovalent center prompted the synthesis of further diaminocarbenes, such as imidazolidine-2-ylidene9 (2), triazol-2-ylidene10 (3) and also a stable non-cyclic bisdiisopropylaminocarbene (4).11 Despite the apparent dominance of nitrogen substitution in stabilizing the carbenes, some derivatives could also be synthesized with two significantly flattened12 phosphorus13 (5) which also benefit from aromaticity,14 or most remarkably, derivatives could be synthesized with a nitrogen and a chalcogen atom, like thiazol-2-ylidene15 (6) and non-cyclic aminothio-16 and aminooxycarbenes16 (7 and 8). Even cyclic (alkyl)(amino) carbenes (CAAC) are known.17
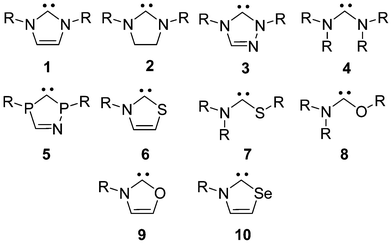 | ||
| Scheme 1 π-electron donor stabilized carbenes. | ||
To estimate the (thermodynamic) stabilization of carbenes, an isodesmic reaction I (Scheme 2) has been applied,18 exhibiting more than ΔE(carbene) = 90 kcal mol−1 stabilization for each of the known synthesized carbenes. Furthermore ΔE(carbene) has been shown to exhibit excellent correlation with the dimerization energy of carbenes.18 Very recently, we have shown19 that N-methyloxazol-2-ylidene exhibits 96.1 kcal mol−1 (B3LYP/6-311+G**) ΔE(carbene) stabilization, exceeding the 92.4 kcal mol−1 value for the analogous thiazol-2-ylidene (6) derivative. Similar conclusions were obtained in a different study, in which a singlet–triplet gap of 75.8 kcal mol−1 (B3LYP/6-311+G**) was reported20 for oxazol-2-ylidene (9) that is only slightly lower than the corresponding value for imidazol-2-ylidene (83.0 kcal mol−1 at the B3LYP/6-311+G**//B3LYP/6-31G* level).21 It was also surmised that oxazol-2-ylidene (9) benefits from 6π-aromatic stabilization, although this effect has not been quantified comprehensively.20 Considering these indications of stability, and particularly in the light of the stability of 8, it is surprising that known chalcogen-substituted N-heterocyclic carbenes (NHCs) are limited to thiazol-2-ylidene (6),15,22 and no free derivatives of oxazol-2-ylidene (9) have been prepared yet, although its complexes with transition metals23 and BPh324 are known. To assess the overall stability of a carbene, however, not only thermodynamical aspects,25 but possible chemical decomposition reactions should also be considered. The computational investigation of the cycloreversion of cyclic dioxocarbenes26 to ethyne and carbon dioxide (Scheme 3) revealed an exothermic reaction, with a low (8.8 kcal mol−1 MP2/6-31G**) barrier. This finding might indicate the tendency of cyclic chalcogen-substituted carbenes to decompose as a possible reason for the lack of success in synthesis of the derivatives.
 | ||
| Scheme 2 Isodesmic reaction to estimate the stability ΔE(carbene) of a carbene. | ||
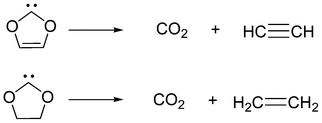 | ||
| Scheme 3 Decomposition reactions of dioxol-2-ylidene and dioxolidin-2-ylidene. | ||
On the other hand, carbon disulfide can react with strained27a or electron-deficient27b acetylenes at high temperature or under high pressure yielding 1,3-dithiolium carbenes, which can be trapped by the substrate, or dimerize to tetrathiafulvenes (Scheme 4).27 The reaction of carbon disulfide with t-BuCP was also shown to proceed via the cycloaddition intermediate.28 While for some 1,3-heterocumulenes the cycloreversion was investigated and the stability of nitrogen-containing rings was established, in that study29 neither oxazol-2-ylidene (9) nor selenazol-2-ylidene (10) was considered. Changes in the carbenes' stability might trigger changes also in their catalytic activity. We have recently demonstrated19 that the Breslow-intermediate – which is a key intermediate in many organocatalytic reactions (e.g. in the benzoin condensation) – possesses superior stabilization with thiazol-2-ylidene catalysts compared to other NHCs, affecting the catalytic activity. Also N,N′-diamidocarbenes (DAC) are engaged in reactions associated with electrophilic carbenes.30 Furthermore, even the substituents on the nitrogen of the NHC were shown to affect the catalytic effect significantly.31 In light of these results the design of new stable carbenes is of great importance in order to broaden the number of available catalysts, which might aid in choosing the most proper (and selective) one for a given reaction. As shown above, little is known about the stability of chalcogen-substituted N-heterocyclic carbenes, despite the apparent stability of the open-chain analogues. Thus, the aim of the present computational study is to explore the stability and reactivity of these species in detail, with a special emphasis on oxazol-2-ylidene.
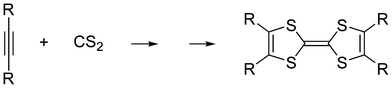 | ||
| Scheme 4 Reaction between acetylene and carbon disulfide (R: electron withdrawing group e.g. CF3). | ||
Computational methods
Unless otherwise stated, all calculations were carried out with the Gaussian 03 program package,32 using the B3LYP hybrid functional in conjunction with the 6-311+G** basis. Full geometry optimizations were performed for all molecules, and harmonic vibrational frequencies were calculated at the same level of theory to establish the nature of the stationary points obtained, as characterized by none or a single negative eigenvalue of the Hessian for minima and transition structures, respectively. Intrinsic reaction coordinate (IRC) calculations were started to locate the corresponding minima. Nucleus-independent chemical shift (NICS) values33 were calculated as the negative of the magnetic shielding at the geometric centre of the ring (NICS(0)) and also by 1 Å above the plane of the ring atoms (NICS(1)). Bird indices34 (BI) were determined from the Gordy bond orders35 of the ring using the bond distances of the simplest single- and double-bonded compounds (CH3–CH3, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, CH3–NH2, CH2NH, CH3–OH, CH2O, CH3–SH, CH2S, CH3–SeH, and CH2Se) calculated at the same level of theory as the references. Isomerisation stabilization energies (ISE, Scheme 5)36 were calculated as an energetic measure of aromaticity.
CH2, CH3–NH2, CH2NH, CH3–OH, CH2O, CH3–SH, CH2S, CH3–SeH, and CH2Se) calculated at the same level of theory as the references. Isomerisation stabilization energies (ISE, Scheme 5)36 were calculated as an energetic measure of aromaticity.
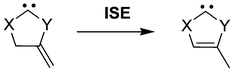 | ||
| Scheme 5 Isomerisation stabilization energy reaction. | ||
In order to verify our results, certain calculations were also performed at the B3LYP/aug-cc-PVTZ and MP2/6-311+G** levels of theory. To calculate (gas phase) Gibbs free energies the computed harmonic frequencies were used at 298 K and 1 atm. In order to get a deeper insight into the factors determining the reactivity of the investigated carbenes, we employed conceptual DFT indices,37 originating from the ground state electron density of the molecules, to estimate their hardness, electrophilic and nucleophilic characters. Pérez showed38 that a good qualitative agreement existed between the theoretical and experimental philicity of carbenes by comparing the electrophilicity index of singlet carbenes to the experimental Moss scale,39 which is based on a kinetic model of carbene addition to simple alkenes. Similarly, in a study of the divalent silicon and germanium analogues of carbenes, the electrophilicity index has been shown to be a valuable tool in assessing the reactivity of the compounds.40 The electrophilicity index (ω, eqn (1)) is calculated as
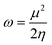 | (1) |
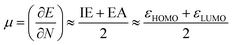 | (2) |
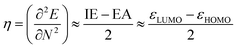 | (3) |
As direct evaluation of these indices is not straightforward we used two finite differences approaches,37 commonly applied in the literature, based on the ionization energy (IE) and electron affinity (EA) of the molecules, and on their approximations based on Koopman's theorem.42 As the trends obtained from the two formulas were identical, only results based on orbital energies are presented here, while the rest of the data are given in the SI. The nucleophilicity index (N) has been calculated as the energy difference between the HOMO energy of the compound and that of tetrachloretylene (EHOMO(TCE), eqn (4)) as was proposed by Domingo et al.43 and was successfully used earlier for carbenes as well:44
| N = εHOMO − εHOMO(TCE) | (4) |
Results and discussion
First, the thermodynamical stability has been evaluated for five-membered, chalcogen-substituted carbenes using ΔE(carbene), to find possible target molecules for synthesis. In Fig. 1. the B3LYP/aug-cc-PVTZ stability of the investigated saturated and unsaturated carbenes are presented, while the MP2/6-311+G** and B3LYP/6-311+G** data are compiled in Table S1 in the ESI.†![Stabilities of heterocyclic carbenes (ΔE(carbene) [kcal mol−1]) for different X and Y ring heteroatoms.](/image/article/2013/RA/c3ra41177j/c3ra41177j-f1.gif) | ||
| Fig. 1 Stabilities of heterocyclic carbenes (ΔE(carbene) [kcal mol−1]) for different X and Y ring heteroatoms. | ||
These results show the decreasing stabilizing effect of the heteroatoms in the order of N > O > S ∼ Se. Oxazol-2-ylidene (9) exhibits the second largest stabilization energy (98.5 and 100.7 kcal mol−1 at the B3LYP/aug-cc-pVTZ and MP2/6-311+G** levels, respectively), exceeding the stabilization of some already synthesized carbenes, such as 2 and 6 (96.2 and 95.5 kcal mol−1 at B3LYP/aug-cc-pVTZ, respectively). It should also be mentioned that the stabilization of selenazol-2-ylidene 10 is similar to that of its sulphur analogue (6). All the unsaturated compounds exhibit higher ΔE(carbene) values than their saturated counterparts, showing the stabilizing contribution of aromaticity. The difference in ΔE(carbene) values of the saturated/unsaturated pairs varies between 6 and 14 kcal mol−1 (at B3LYP/aug-cc-pVTZ) and 6 and 18 kcal mol−1 (at MP2/6-311+G**). This indicates that the extent of aromaticity is influenced by the heteroatom, therefore we decided to investigate the aromaticity of the unsaturated systems in more detail. The largest45 aromaticity is exhibited by imidazol-2-ylidene (1) according to the different measures BI (60), ISE (−14.4 kcal mol−1) and NICS(0) (−13.0 ppm) NICS(1) (−10.1 ppm) (Table 1), indicating considerable cyclic delocalisation as noted before.6 Gradual substitution of the nitrogen(s) by other heteroatoms results in a decrease of the aromaticity in the order of N > S > Se ∼ O. Accordingly, the energetic measure of aromaticity also shows the rather small (ISE −9.6 kcal mol−1) but still non-negligible26b aromatic stabilization of oxazol-2-ylidene in line with the rather small (6.8 kcal mol−1–Table S1, ESI†, Fig. 1) difference between the saturated and unsaturated ΔE(carbene) stabilization energies. It is worth noting that from the investigation of the magnetic susceptibilities of 1, 6 and 9, the smallest aromaticity was also found for 9.46
|
|
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| X | Y | NICS(0) | NICS(1) | BI | ISE | ω | N | ω | N |
| NH | NH | −13.0 | −10.1 | 60 | −14.4 | 0.0558 | 0.0509 | 0.0555 | 0.0561 |
| NH | S | −13.0 | −10.6 | 56 | −12.6 | 0.0838 | 0.0313 | 0.0703 | 0.0408 |
| NH | Se | −12.0 | −9.6 | 54 | −11.1 | 0.0926 | 0.0344 | 0.0751 | 0.0441 |
| NH | O | −12.2 | −9.8 | 41 | −9.6 | 0.0686 | 0.0244 | 0.0637 | 0.0395 |
| S | S | −16.6 | −11.9 | 53 | −13.3 | 0.1318 | 0.0158 | 0.1333 | 0.0249 |
| S | Se | −13.4 | −11.3 | 51 | −11.9 | 0.1417 | 0.0168 | 0.1419 | 0.0265 |
| Se | Se | −12.7 | −10.8 | 50 | −11.3 | 0.1507 | 0.0166 | 0.1524 | 0.0289 |
| O | O | −12.3 | −9.6 | 28 | −8.7 | 0.0963 | 0.0039 | 0.0768 | 0.0152 |
| S | O | −11.6 | −10.3 | 41 | −11.1 | 0.1170 | 0.0175 | 0.1020 | 0.0322 |
| Se | O | −10.2 | −9.5 | 37 | −10.6 | 0.1270 | 0.0235 | 0.1095 | 0.0375 |
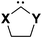
From the analysis of the effect of the carbene substituents and the aromatic stabilization it can be concluded that the exceptional stability of imidazol-2-ylidenes is mainly due to the favourable stabilizing effect of the nitrogen substituent on the carbene, supported further by aromaticity.6 Interestingly, although the oxygen substituent brings a significant stabilization in the case of oxazol-2-ylidene, this heteroatom contributes to the aromaticity to a smaller extent only. In this respect it is worth pointing out the analogy with the well-known trend in the aromaticity of five-membered heterocycles, namely that the aromaticity of furan is also significantly reduced with respect to pyrrole and thiophene.47 Clearly, the stabilization of the π-type lone pair ionization energy with respect to its saturated counterpart is the smallest for furan in the series of common five-membered heterocycles.48 A further important factor from the point of view of the carbene stability is its propensity for dimerization. While the Gibbs free energy for the dimerization of 6 is −22.1 kcal mol−1, for 9 it amounts only −12.2 kcal mol−1. The reduced dimerization energy of the (unsubstituted) 9 with respect to 6 is in accordance with the increased ΔE(carbene) value, and also with our previous results on the good correlation of these quantities.18 The N-methyl-derivative of oxazol-2-ylidene 9, however, has only −6.8 kcal mol−1 dimerization Gibbs free energy, showing that the N-substitution, even by the small methyl group, significantly reduces the propensity of dimerisation. Since N-substituted derivatives of 6 (which are less stabilized than 9) are known to exist in monomeric form,15 among N-substituted oxazol-2-ylidenes several derivatives (see below) should exist in monomeric form. A further important chemical transformation, the 1,2-hydrogen shift resulting in oxazol from 9, has been shown earlier to proceed via a high barrier, which has further increased for the synthetically most interesting alkyl substituted systems.20
The fragmentation of oxazol-2-ylidene to acetylene and an isocyanate by a cycloreversion, however, may occur and this reaction is also entropically favoured. Indeed, some of the studied reactions are exergonic, but not exothermic (Table 2). The stability against cycloreversion drops significantly for the saturated compounds. The reaction proceeds via a concerted pathway (we were not able to locate transition structures for a stepwise reaction for imidazol-2-ylidene, or for oxazol-2-ylidene). Nevertheless, the barrier in the case of the cyclic saturated diaminocarbene 2 is large enough to prevent the decomposition reaction in agreement with its synthesizability. Again, the saturated compounds have much lower decomposition barriers than their unsaturated counterparts (Table 2), and alkenes and isocyanates have been obtained from transient oxazolidin-2-ylidenes in agreement with this.49 Apparently, the effect of the heteroatoms on the cycloreversion differs from that observed for ΔE(carbene).50 In the case of the cycloreversion reaction nitrogen containing compounds still exhibit the largest stability, however, sulfur (and selenium) containing systems are less prone to cyclorevert than their oxygen analogues. For oxazol-2-ylidene 9 the cycloreversion reaction is exergonic. Nevertheless, the calculated 38.3 kcal mol−1 barrier of the concerted reaction is large, and thus we can conclude that this compound is synthesizable. It is also noteworthy that for selenazol-2-ylidene 10 the exothermicity and the significant barrier of the reaction are comparable to those of the known sulfur analogue.
| Carbene |
|
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| X | Y | ΔE | ΔE# | ΔG | ΔG# | ΔE | ΔE# | ΔG | ΔG# |
| NH | NH | −1.3 | 35.0 | −18.2 | 30.7 | 32.1 | 70.7 | 14.0 | 63.1 |
| NH | S | −12.8 | 20.7 | −28.7 | 17.6 | 16.2 | 49.6 | −0.3 | 44.7 |
| NH | Se | −10.6 | 20.6 | −26.5 | 17.6 | 16.7 | 48.2 | 0.3 | 43.4 |
| NH | O | −27.1 | 18.0 | −42.5 | 14.9 | −1.3 | 43.7 | −17.8 | 38.3 |
| S | S | −27.1 | 6.7 | −39.3 | 5.4 | −1.7 | 30.3 | −14.6 | 27.5 |
| S | Se | −24.2 | 6.6 | −34.0 | 5.4 | 0.0 | 29.4 | −10.5 | 26.7 |
| Se | Se | −21.4 | 6.6 | −32.8 | 4.7 | 1.7 | 28.7 | −10.4 | 26.1 |
| O | O | −54.7 | 7.7 | −65.4 | 6.0 | −35.2 | 26.6 | −46.6 | 23.5 |
| S | O | −47.3 | 5.7 | −57.4 | 4.1 | −24.3 | 26.2 | −34.9 | 23.3 |
| Se | O | −45.5 | 5.3 | −55.2 | 3.6 | −23.8 | 24.5 | −33.8 | 21.7 |
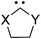
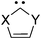
Since the substituents on the nitrogen and the carbon atoms have some apparent effect on the stability6a and reactivity31 of carbenes, we examined the effects of substituents on oxazol-2-ylidene 9 and on selenazol-2-ylidene 10. For the substitution at the nitrogen the bulky adamantyl, mesityl and supermesityl groups have been chosen, which provide extra steric shielding for the carbene centre (Table 3). These nitrogen substituents result in slightly increased stabilization energies, similar to our previous observations in case of pyridine-2-ylidene.6a It should be emphasized again that the effect of the steric shielding – which also contributes to the prevention of dimerization – is not included in the ΔE(carbene) energies, thus, the real stabilization should be larger. At the ring carbon atoms (positions 4 and 5), however, the size of the substituent is likely to be less important; thus, groups with different electronic nature (boro, methyl, trifluoromethyl, chloro, amino) have been selected. Similarly to the minor influence of substitution at the nitrogen, substituents at the carbon atoms in positions 4 and 5 exerted also a small effect; only the strong π-electron withdrawing groups (–BH2 and –CF3) resulted in a few kcal mol−1 decrease of the stability of the carbene (Table 3). Thus, the effect of the substituents on the ΔE(carbene) stabilization of oxazol-2-ylidene 9 (and its selenium analogue 10) is minor.
|
|
O | Se | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔEcarbene | ΔE | ΔE# | ΔG | ΔG# | ΔEcaebene | ΔE | ΔE# | ΔG | ΔG# | |||
| R1 | R2 | R2 | ||||||||||
| H | H | H | 96.1 | −1.3 | 43.7 | −17.8 | 38.3 | 92.3 | 16.7 | 48.2 | 0.3 | 43.4 |
| Me | H | H | 99.8 | 1.5 | 45.2 | −15.5 | 39.4 | 93.1 | 14.1 | 46.8 | −4.1 | 43.1 |
| Mes | H | H | 100.8 | −4.5 | 41.1 | −20.5 | 36.0 | 93.1 | 7.0 | 42.5 | −9.2 | 38.5 |
| Mes* | H | H | 102.6 | −10.3 | 40.5 | −29.0 | 35.6 | 93.1 | 2.6 | 40.9 | −15.2 | 36.8 |
| Ad | H | H | 101.2 | −2.2 | 44.0 | −19.4 | 39.2 | 94.8 | 9.1 | 44.1 | −5.6 | 39.8 |
| H | Cl | Cl | 95.0 | 2.7 | 47.8 | −13.2 | 42.8 | 89.9 | 22.1 | 54.6 | 6.1 | 50.2 |
| H | BH2 | BH2 | 93.4 | −14.1 | 26.3 | −31.5 | 23.0 | 87.0 | 1.6 | 26.6 | −15.9 | 23.3 |
| H | CH3 | CH3 | 98.7 | −4.6 | 44.3 | −22.5 | 39.4 | 92.1 | 9.2 | 47.0 | −8.6 | 43.0 |
| H | CF3 | CF3 | 92.6 | −1.4 | 39.1 | −18.5 | 34.8 | 88.9 | 17.5 | 42.7 | −0.9 | 37.5 |
| H | NH2 | NH2 | 95.3 | −1.1 | 46.0 | −17.9 | 41.7 | 88.9 | 13.9 | 52.4 | −3.0 | 47.4 |
| H |

|
91.7 | 43.6 | 56.9 | 25.9 | 50.7 | 87.7 | 61.2 | 67.3 | 43.4 | 60.8 | |
| H |

|
96.1 | 34.8 | 55.2 | 16.6 | 48.7 | 91.0 | 51.6 | 62.0 | 33.4 | 55.9 | |
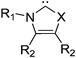
The position of the substituent has a significant effect on the cycloreversion reaction. While substitution at the nitrogen has a small impact (except for the bulky supermesityl group), having the substituent at positions 4 and 5 significantly effects both the reaction energy and the barrier (Table 3). For the boro-substituted derivates of oxazol-2-ylidene 9 and selenazol-2-ylidene 10 the cycloreversion reaction is more exothermic than for the parent carbenes, and also the reaction barrier is reduced. Ring anellation and condensation have an important effect on the reaction. First, the kinetic hindrance of cycloreversion increases significantly; furthermore, the reaction becomes endothermic due to the formation of the ring-strained cyclohexyne or benzyne, while the carbene stabilization energy exhibits only a small decrease.27a,29,51 Thus the most stable synthetic targets as free carbenes are 11 and 12 (Scheme 6).
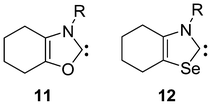 | ||
| Scheme 6 The most suitable targets for synthesis (R: bulky substituent e.g.: adamantyl or mesityl. | ||
In the assessment of the stability an important aspect is the reactivity against common reactants such as water, acids and bases. Oxazol-2-ylidenes were already considered in early works52 as alternative catalysts in the benzoin condensation to thiazol-2-ylidenes or imidazol-2-ylidenes, but the oxazolium salts did not show catalytic activity in the presence of a base.52 Haake has shown that although oxazolium salts are more acidic than their thiazolium counterparts,53 (i.e. they should produce the carbene easily) they exhibit a facile irreversible ring opening resulting in 13 (X = O, see Scheme 7) at pH values being sufficiently basic for the deprotonation.54 In contrast, thiazolium exhibits a reversible ring opening, while to open up the imidazolium ring extremely basic conditions are needed.54
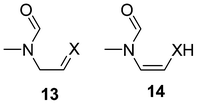 | ||
| Scheme 7 Ring opened products from the hydrolysis of azolium salts. | ||
To describe the effect of water as solvent, recently we used a microsolvatation approach by embedding the starting carbene, intermediates and product in a water cluster.7,19 For imidazol-2-ylidene, product 13 (X = NMe) was less stable than imidazolium-hydroxide by 4.7 kcal mol−1,7 while thiazol-2-ylidene 13 (X = S) was more stable than the hydroxide by 7.4 kcal mol−1, using a cluster made from 31 water molecules. Using the same approach for oxazol-2-ylidene the water microsolvated structures for the oxazol-2-ylidene, oxazolium hydroxide, 13 (X = O) and its H-shifted isomer 14 were optimized as shown in Fig. 2. For the oxazol case, 13 (X = O) is significantly more stable than oxazolium-hydroxide. All these results are in full agreement with the earlier observations54 of the irreversible ring opening in the case of the oxygen compound, and the intact nature of the imidazolium moiety in the presence of excess water.7
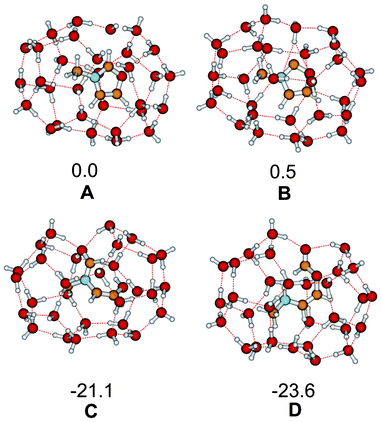 | ||
| Fig. 2 B3LYP/6-311+G**//B3LYP/6-31+G* level optimized structures and relative energies (in kcal mol−1 units) of oxazol-2-ylidene with 31 water, oxazolium-hydroxide, 13 and 14 with 30–30 water molecules. | ||
For the sake of completeness we have made similar calculations for triazol-2-ylidene and selanazole-2-ylidene. While the results (shown as Fig. S1 and S2 in the ESI†) for the former one are close to those for imidazol-2-ylidene, the selenazol-2-ylidene results are similar to those for thiazol-2-ylidene. It is worth noting that the energy of oxazol-2-ylidene in the 31 water cluster is nearly the same as for oxazolium-hydroxide, while imidazolium-hydroxide was more stable than the corresponding carbene by 6.1 kcal mol−1, in agreement with the relative basicities of these carbenes.19 This shows that the microsolvation approach applied provides a reasonably good description for the behaviour in water as solvent.
Recently, however, we have shown in a combined computational-experimental study that the mechanism of imidazol-2-ylidene hydrolysis depends on the number of participating water molecules.7 While with excess water a fast protonation takes place – as was shown by the computations using the microsolvatation approach – with a small amount of water molecules the ring-opening reaction is a kinetically-hindered slow reaction. The computational studies indicate that the open chain products (Scheme 7) are more stable than the imidazol-2-ylidene–water complex, but a significant reaction barrier hinders the reaction with one water molecule, which is only slightly reduced in the presence of two or three reacting water molecules.7
A similar conclusion was drawn for thiazol-2-ylidene,19 although the relative stability of the open chain 13 (X = S) was somewhat increased. We have carried out analogous calculations for the hydrolysis of oxazol-2-ylidene and for the sake of completeness we considered selenazol-2-ylidene and triazol-2-ylidene as well. The details of the reaction pathways are given as Fig. S3–S5 of the ESI†, while the energetically most important points of the reactions are compiled in Fig. 3 together with the previously published data for imidazol-2-ylidene7 and thiazol-2-ylidene.19 The comparison of the energetics for the different hydrolysis reactions reveals that while the activation energy for the reaction is reduced only slightly with respect to imidazol-2-ylidene and thiazol-2-ylidene, the product 13 is significantly stabilized in case of X = O. For the calculation with two and three water molecules (the reaction pathways are presented in the ESI as Fig. S4–5, S7–8, S10–11†) again similar transition structures and intermediates were obtained as in case of imidazol-2-ylidene7 and thiazol-2-ylidene.19 Since in all these calculations the barriers are similar to the imidazol-2-ylidene case, it seems reasonable to assume that the hydrolysis reaction is likewise hindered for oxazol-2-ylidene, if the water concentration is small (and the clustering is reduced).55
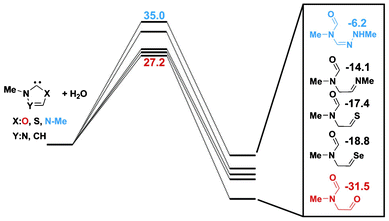 | ||
| Fig. 3 The relative energies of the products of the hydrolysis of different carbenes at B3LYP/6-311+G** level (in kcal mol−1 units). | ||
Having concluded that properly substituted oxazol-2-ylidenes (11) and selenazol-2-ylidenes (12) (Scheme 6) are viable synthetic targets, it is worth investigating their expected reactivity. It is well known that the divalent carbon centre in singlet carbenes is characterized by an empty 2p orbital and an in-plane lone pair-like orbital, which may give rise to its electrophilic and/or nucleophilic character. In general, π-electron donation from the substituents to the empty p orbital of the divalent centre is expected to reduce its electrophilic character. Furthermore, it has been shown that the nucleophilicity remains virtually unchanged upon aromatization (comparing 1 and 2),56 likewise the donating ability as measured by the Tolman electronic parameter.57 In the case of silylenes (the silicon analogues of carbenes) the relationship between the π-electron donating ability, nucleophilic and electrophilic character has been studied in detail,40,58 and a clear relationship between the π-electron donating ability of the substituent, the decrease in the electrophilicity of the silylene, and the emergence of its nucleophilic character was demonstrated. Here in this study (see data in Table 1) we did not find such a straightforward relationship. The most important aspect from the point of view of our target molecule oxazol-2-ylidine 9 is, however, that its electrophilicity is only slightly increased compared to imidazol-2-ylidene 1, while its nucleophilicity59 (and also its basicity)19,26b is much smaller, similar to that of the also widely applied organocatalysts thiazol-2-ylidene 6 which has significantly higher electrophilicity compared to both 1 and 9. Thus one may expect that oxazol-2-ylidene will show different reactivity, and organocatalytic reactivity than imidazol-2-ylidine and thiazol-2-ylidene. Selenazol-2-ylidenes (10), however, should have similar electronic properties to their sulfur analogues 6.
Conclusions
Combining the stabilizing effect of the (reasonably large) substituents (R) on the nitrogen and annellated rings on the carbon atoms, the most promising synthetic targets 11 and 12 are shown in Scheme 6. These compounds should be stable in dimerization and in cycloreversion reactions, provided that the water content of the reaction mixture is significantly reduced. The electrophilicity index of 9 indicates a comparable reactivity to the known imidazol-2-ylidene (1), while its lower nucleophilicity and basicity will result in changes in complex formation, and organocatalytic activities, opening new perspectives in the fine-tuning of catalytic reactions.Acknowledgements
Financial support from New Szechenyi Plan TAMOP-4.2.2/B-10/1-2010-0009 and OTKA K 170356 is gratefully acknowledged. J. O. acknowledges receipt of an EU Marie Curie Fellowship (Project “Oestrometab”) and O. H. the Alexander von Humboldt fellowship. Support from Sumitomo LTD for Zs. K. is also gratefully acknowledged.Notes and references
- (a) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS; (d) A. J. Arduengo III and G. Bertrand, Chem. Rev., 2009, 109, 3209 CrossRef; (e) J. Vignolle, X. Cattoen and D. Bourissou, Chem. Rev., 2009, 109, 3333 CrossRef CAS; (f) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS; (g) T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef; (h) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS.
- (a) D. R. Anderson, V. Lavallo, D. J. O’Leary, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2007, 46, 7262–7265 CrossRef CAS; (b) R. H. Crabtree, J. Organomet. Chem., 2005, 690, 5451–5457 CrossRef CAS; (c) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951–961 CrossRef CAS; (d) S. Leuthaöusser, D. Schwarz and H. Plenio, Chem.–Eur. J., 2007, 13, 7195–7203 CrossRef; (e) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS; (b) N. Marion, S. Díez- González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (c) P.-C. Chiang and J. W. Bode, N-Heterocyclic Carbene Catalyzed Reactions of alpha-Functionalized Aldehydes, Science of Synthesis: Asymmetric Organocatalysis, vol. 1, 2012, Georg Thieme Verlag, pp. 639–672 Search PubMed.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- (a) O. Hollóczki and L. Nyulászi, J. Org. Chem., 2008, 73, 4794–4799 CrossRef; (b) C. Boehme and G. Frenking, J. Am. Chem. Soc., 1996, 118, 2039–2046 CrossRef CAS; (c) C. Heinemann, T. Müller, Y. Apeloig and H. Schwartz, J. Am. Chem. Soc., 1996, 118, 2023–2038 CrossRef CAS; (d) M.-J. Cheng and C.-H. Hu, Chem. Phys. Lett., 2001, 349, 477–482 CrossRef CAS; (e) S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef; (f) A. K. Guha, S. Sarmah and A. K. Phukan, Dalton Trans., 2010, 39, 7374–7383 RSC; (g) S. Gronert, J. R. Keeffe and R. A. More O’Ferrall, J. Am. Chem. Soc., 2011, 133, 3381–3389 CrossRef CAS; (h) S. Gronert, J. R. Keeffe and R. A. More O’Ferrall, J. Am. Chem. Soc., 2011, 133, 11817–11818 CrossRef CAS.
- The hydrolysis of imidazol-2-ylidene was studied by O. Hollóczki, P. Terleczky, D. Szieberth, G. Mourgas, D. Gudat and L. Nyulászi, J. Am. Chem. Soc., 2011, 133, 780–789 CrossRef . Much higher activation barriers were found than for the saturated counterpart imidazolidine-2-ylidene.
- B. Maji, M. Horn and H. Mayr, Angew. Chem., Int. Ed., 2012, 51, 6231–6235 CrossRef CAS.
- A. J. Arduengo, J. R. Goerlich and W. Marshall, J. Am. Chem. Soc., 1995, 117, 11027 CrossRef CAS.
- D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J. P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed. Engl., 1995, 34, 1021–1023 CrossRef CAS.
- P. R. Allen, M. Murray and G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123 CrossRef.
- (a) A. Fekete and L. Nyulászi, J. Organomet. Chem., 2002, 643–644, 278–284 CrossRef CAS; (b) L. Nyulászi, Tetrahedron, 2000, 56, 79–84 CrossRef.
- D. Martin, A. Baceiredo, H. Gornitzka, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 1700–1703 CrossRef CAS.
- L. Nyulászi, Chem. Rev., 2001, 101, 1229–1246 CrossRef.
- A. J. Arduengo, J. R. Goerlich and W. Marshall, Liebigs Ann./Recl., 1997, 365–369 CrossRef CAS.
- (a) R. W. Alder, C. P. Butts and A. G. Orpen, J. Am. Chem. Soc., 1998, 120, 11526–11527 CrossRef CAS; (b) N. Merceron-Saffon, A. Baceiredo, H. Gornitzka and G. Bertrand, Science, 2003, 301, 1223 CrossRef CAS.
- V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS.
- L. Nyulászi, T. Veszprémi and A. Forró, Phys. Chem. Chem. Phys., 2000, 2, 3127–3129 RSC.
- O. Hollóczki, Zs. Kelemen and L. Nyulászi, J. Org. Chem., 2012, 77, 6014–6022 CrossRef.
- F. Freeman, D. J. Lau, A. R. Patel, P. R. Pavia and J. D. Willey, J. Phys. Chem. A, 2008, 12, 8775–8784 CrossRef.
- M.-J. Cheng and C.-H. Hu, Chem. Phys. Lett., 2000, 322, 83–90 CrossRef CAS.
- The synthesis of a stable isothiazol carbene has been claimed ( J. Wolf, W. Bohlmann, M. Findeisen, T. Gelbrich, H.-J. Hofmann and B. Schulze, Angew. Chem., Int. Ed. Engl., 2007, 46, 3118–21 CrossRef CAS ), but was subsequently shown to be a rearranged product ( A. DeHope, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed. Engl., 2007, 46, 6922–6925 CrossRef ), formed from the intermediate isothiazol carbene ( J. Wolf, W. Bohlmann, M. Findeisen, T. Gelbrich, H.-J. Hofmann and B. Schulze, Angew. Chem., Int. Ed. Engl., 2007, 46, 6926 CrossRef.
- (a) K. R. Rundy and W. R. Roper, J. Organomet. Chem., 1975, 91, C61–C64 CrossRef; (b) A. C. Tubaro, A. Biffs, M. Basato, F. Benetollo, K. J. Cavell and L. Ooi, Organometallics, 2005, 24, 4153–4158 CrossRef; (c) J. A. Cabeza, I. del Río, D. Miguel, E. Pérez-Carreño and M. G. Sánchez-Vega, Organometallics, 2008, 27, 211–217 CrossRef CAS; (d) J. Zhang, J. Fu, X. Su, X. Wang, S. Song and M. Shi, Chem.–Asian J., 2013, 3, 552–555 CrossRef.
- (a) C. Lambert, I. Lopez-Solera and P. R. Raithby, Organometallics, 1996, 15, 452–455 CrossRef CAS; (b) F. E. Hahn, T. Lugger and M. Z. Beinhoff, Z. Naturforsch., 2004, 59b, 196–201 Search PubMed.
- R. Hoffmann, P. V. R. Schleyer and H. F. Schaefer III, Angew. Chem., Int. Ed., 2008, 47, 7164–7167 CrossRef.
- (a) R. R. Sauers, Tetrahedron Lett., 1994, 35, 7213–7216 CrossRef CAS; (b) R. R. Sauers, Tetrahedron Lett., 1996, 37, 149–152 CrossRef CAS.
- (a) A. Krebs and H. Kimling, Angew. Chem., Int. Ed. Engl., 1971, 10, 509–510 CrossRef CAS; (b) H. D. Hartzler, J. Am. Chem. Soc., 1973, 95, 4379–4387 CrossRef CAS; (c) C. U. Pittman and M. Narita, Bull. Chem. Soc. Jpn., 1976, 49, 1996–1999 CrossRef CAS (the adduct of trialkylphosphine and carbon disulfide was reacted with electon deficient acetylene).
- S. E. d’Arbeloff-Wilson, P. B. Hitchcock, S. Krill, J. F. Nixon, L. Nyulászi and M. Regitz, J. Am. Chem. Soc., 2000, 122, 4557–4562 CrossRef.
- J. Fabian, A. Krebs, D. Schoönemann and W. Schaefer, J. Org. Chem., 2000, 65, 8940–8947 CrossRef CAS.
- (a) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS; (b) G. A. Blake, J. P. Moerdyk and C. W. Bielawski, Organometallics, 2012, 31, 3373–3378 CrossRef CAS.
- J. Mahatthananchai and J. W. Bode, Chem. Sci., 2012, 3, 192–197 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) NICS(0) is the magnetic shielding in the geometrical center of the ring. P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van. Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS; (b) NICS(1) is the magnetic shielding 1 Å above the geometrical center of the ring. P. V. R. Schleyer, H. Jiao, N. J. R. van Eikema Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- C. W. Bird, Tetrahedron, 1985, 41, 1409 CrossRef CAS.
- W. Gordy, J. Chem. Phys., 1947, 15, 305–310 CrossRef CAS.
- (a) P. V. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4, 2873–2876 CrossRef CAS introduced ISE for six membered rings; (b) For five membered rings: L. Nyulászi, O. Hollóczki, C. Lescop, M. Hissler and R. Réau, Org. Biomol. Chem., 2006, 4, 996–998 RSC; (c) O. Hollóczki and L. Nyulászi, Struct. Chem., 2011, 22, 1385–1392 CrossRef . ISE was calculated for some NCH-s in ref. 6a.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS.
- P. Pérez, J. Phys. Chem. A, 2003, 107, 522–525 CrossRef.
- (a) R. A. Moss, Acc. Chem. Res., 1989, 22, 15–21 CrossRef CAS; (b) R. A. Moss, Acc. Chem. Res., 1980, 13, 58–64 CrossRef CAS.
- J. Oláh, F. D. Proft, T. Veszprémi and P. Geerlings, J. Phys. Chem. A, 2005, 109, 1608–1615 CrossRef.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr, W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989.
- T. Koopmans, Physica, 1934, 1, 104 CrossRef.
- L. R. Domingo, E. Chamorro and P. J. Perez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS.
- M. Z. Kassaee, F. A. Shakib, M. R. Momeni, M. Ghambarian and S. M. Musavi, J. Org. Chem., 2010, 75, 2539–2545 CrossRef CAS.
- With the exception of the NICS values of some sulfur containing carbenes.
- Yu. B. Vysotsky, V. S. Bryantsev and O. A. Gorban, Chem. Heterocycl. Compd., 2002, 38, 1451–1468 CrossRef CAS.
- The aromaticity of the five membered heterocycles has been compared many times. For a work using several aromaticity measures see e.g. L. Nyulászi and P. V. R. Schleyer, J. Am. Chem. Soc., 1999, 121, 6872–75 CrossRef . For evaluation of the ISE see ref. 36c.
- L. Nyulászi, G. Keglevich and L. D. Quin, J. Org. Chem., 1996, 61, 7808–7812 CrossRef.
- P. Couture, J. K. Terlouw and J. Warkentin, J. Am. Chem. Soc., 1996, 118, 4214–4215 CrossRef CAS.
- This difference is likely to be attributable to the different stability of the reaction product isocyanate analogues.
- A. Krebs and J. Wilke, Top. Curr. Chem., 1983, 190–233 Search PubMed.
- R. Breslow, J. Am. Chem. Soc., 1958, 40, 3719–3726 CrossRef.
- P. Haake and W. B. Miller, J. Am. Chem. Soc., 1963, 85, 4044–4045 CrossRef CAS.
- J. M. Duclos and P. Haake, Biochemistry, 1974, 13, 5358–5362 CrossRef CAS.
- It is known that clustering of water is significantly reduced in ionic liquids. See e.g. C. G. Hanke and R. M. Lynden-Bell, J. Phys. Chem. B, 2003, 107, 10873–10878 CrossRef CAS.
- B. Maji, M. Breugst and H. Mayr, Angew. Chem., Int. Ed., 2011, 50, 6915–6919 CrossRef CAS.
- R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef.
- (a) J. Oláh, F. De Proft, T. Veszpremi and P. Geerlings, J. Phys. Chem. A, 2007, 111, 10815–10823 CrossRef; (b) J. V. Correa, P. Jaque, J. Oláh, A. Toro-Labbe and P. Geerlings, Chem. Phys. Lett., 2009, 470, 180–186 CrossRef CAS.
- In a study of the related thiazol-2-ylidene the nucleophilic character of oxazol-2-ylidene was also considered. (a) D. B. DuPre and J. L. Wong, J. Phys. Chem. A, 2005, 109, 7606–7612 CrossRef CAS; (b) D. B. DuPre and J. L. Wong, J. Phys. Chem. A, 2007, 111, 2172–2181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra41177j |
| This journal is © The Royal Society of Chemistry 2013 |