 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unsaturated poly(phosphoester)s via ring-opening metathesis polymerization†
Tobias
Steinbach
abc,
Evandro M.
Alexandrino
b and
Frederik R.
Wurm
*b
aInstitute of Organic Chemistry, Organic and Macromolecular Chemistry, Johannes Gutenberg-Universität Mainz (JGU), Duesbergweg 10-14, D-55128 Mainz, Germany
bMax-Planck Institute for Polymer Research (MPI-P), Ackermannweg 10, D-55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de; Fax: +49 6131 370 330; Tel: +49 6131 379 723
cGraduate School Material Science in Mainz, Staudinger Weg 9, D-55128 Mainz, Germany
First published on 24th April 2013
Abstract
For the first time, ring-opening metathesis polymerization of novel 7-membered cyclic phosphate monomers and their copolymerization with cyclooctene is presented. The monomers were investigated with respect to their metathesis behavior with different Grubbs catalysts and it was found that the Grubbs third generation catalyst gives the best results resulting in polymers with a molecular weight of up to 5000 g mol−1. Also copolymers with cyclooctene (up to a molecular weight of ca. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) were synthesized and the monomer ratios were varied. The degree of polymerization could be controlled and the polydispersity index was usually below two. Acidic hydrolysis of the copolymer showed a complete shift of the molecular weight distribution to higher elution times in SEC, indicating a random incorporation into the poly(cyclooctene) backbone of the phosphate monomers and the possible degradation of the phosphate bonds along the backbone. Further, potentially degradable nanoparticles were prepared by a solvent evaporation miniemulsion technique.
000 g mol−1) were synthesized and the monomer ratios were varied. The degree of polymerization could be controlled and the polydispersity index was usually below two. Acidic hydrolysis of the copolymer showed a complete shift of the molecular weight distribution to higher elution times in SEC, indicating a random incorporation into the poly(cyclooctene) backbone of the phosphate monomers and the possible degradation of the phosphate bonds along the backbone. Further, potentially degradable nanoparticles were prepared by a solvent evaporation miniemulsion technique.
Introduction
Degradable polymers are a growing field in modern materials science due to limitation of natural resources and due to the long half-life times of commodity plastics in nature.1 Also for the biomedical field, for example as drug carriers, in tissue engineering or also when renal clearance of (macro)molecules is necessary, degradable – or partly degradable – polymers are of high interest.1,2 Within the field of degradable polymers, polyesters are the most common materials with poly(lactide) probably being the most prominent example.2,3 In recent projects, we have been focusing on the development of novel potentially biodegradable and biocompatible polyphosphoesters (PPEs).4 PPEs can be degraded by several enzymes such as phosphatases and phosphodiestereases and/or by basic or acidic hydrolysis.5,6 In spite of this obvious benefit, PPEs are only scarcely found in recent studies, even if they are easily accessible and allow the feasible synthesis of a great variety of (functional) materials.6 In contrast polyesters based on carboxylic acids face the problem that functional cyclic lactones require multi-step syntheses or conventional polycondensation needs to be applied which also excludes many functional groups and limits the molecular weight in many cases.7,8 Almost 40 years ago, the group of Penczek developed the first strategies towards (mainly water-soluble) PPEs via polycondensation and ring-opening polymerization approaches.9,10 These materials were not investigated in detail for at least two decades but can be found in modern literature in a few elegant reports in the fields of drug delivery or DNA transfection, for example.6 We recently developed a route towards (un)saturated hydrophobic PPEs via acyclic diene metathesis polymerization (ADMET) of several phosphate monomers and we are currently investigating their performance in bioapplications.4Some benefits of PPEs over conventional polyesters will be briefly mentioned here: (1) the additional functional group that is inherently brought in by the use of a pentavalent P-center (phosphorus triesters); (2) the high tendency to generate water-soluble polymers due to the hydrophilic phosphate building block; and (3) the general low degree of crystallinity (compared with highly crystalline materials such as PLA).
The combination of phosphorus chemistry with metathesis allows tailoring of the polymer functionality due to the high functional group tolerance of modern ruthenium metathesis catalysts and is currently under investigation in our group. Herein, we present an expansion of the metathesis polymerization towards PPEs from a step-growth acyclic diene metathesis (ADMET) polymerization to the chain-growth ring opening metathesis polymerization (ROMP).11 We present different monomers, i.e. seven-membered cyclic phosphates, and their polymerization behavior is investigated. They are also copolymerized with cis-cyclooctene to yield high molecular weight polyesters with reasonable polydispersity. Further the copolymers were used in a miniemulsion solvent evaporation process12,13 to generate potentially biodegradable nanoparticles which can be used to encapsulate hydrophobic drugs or labels which are released slowly due to hydrolysis and enzymatic degradation.
Results and discussion
Monomer synthesis
For the synthesis of PPEs via ROMP, a cyclic phosphoester is mandatory. The smallest possible ring size is therefore a seven-membered ring that is readily available from cis-1,4-butenediol and phosphodichlorides. Due to the third phosphoester, a pendant group can be introduced prior to ring closure. Cyclic seven-membered phosphate monomers were synthesized by nucleophilic ring closing reaction of ethyl or phenyl dichlorophosphate with cis-1,4-butenediol. The reaction was carried out in a diluted THF solution (ca. 5 g L−1) with slow addition of the diol via a syringe pump over a period of 3–4 h to favor ring-closure over polycondensation (Scheme 1).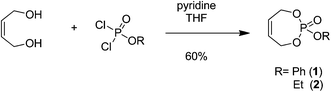 | ||
| Scheme 1 Synthetic approach to 7-membered unsaturated cyclic phosphates. | ||
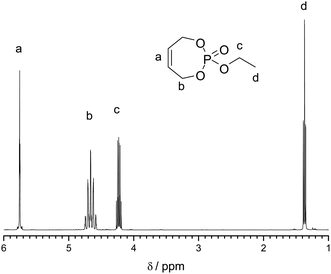
The crude reaction mixture was purified via silica gel chromatography to yield the desired unsaturated monomer in reasonable yields (higher than 60%) and high purity. Fig. 1 shows a representative 1H NMR (400 MHz) spectrum of 2 in CDCl3 (further characterization data can be found in the ESI, Fig. S1–S5†).
Polymerization
Monomers 1 and 2 were investigated with respect to their performance in ROMP. In a previous publication we have investigated the ADMET polymerization of several phosphate-based monomers to high molecular weight unsaturated PPEs.4 If diallyl-phenyl-phosphate was used as the respective monomer, no polymerization was observed. This was attributed to the negative neighboring group effect of the allylester that can complex the catalyst to form an inactive species for metathesis reactions14 and indicated by a direct color change from purple to brown and not even oligomers were observed but only the intact monomer was recovered. When only one more methylene unit was incorporated, i.e. the dibutenyl ester, the polymerization proceeded under typical ADMET conditions.4,147-Membered cyclic monomers 1 and 2 presented herein resemble very closely these allyl esters after ring-opening, so their behavior in homopolymerizations was questionable but it was envisioned that metathesis could be more effective than the acyclic derivative due to the ring strain of an unsaturated seven-membered cyclic phosphate.
We carried out homopolymerizations of monomers 1 and 2 with the Grubbs 1st generation catalyst as the respective initiator (in solution, r.t.), but almost no polymerization was observed, only the presence of ca. 10–20% oligomers (Mn < 1000 g mol−1 from SEC, also compare 1H NMR in the ESI†). When the same reaction was performed with the Grubbs 2nd generation catalyst, a slow polymerization was observed, but again without reaching 100% conversion. SEC proved a molecular weight of ca. 2000 g mol−1 with a high PDI > 2 indicating transfer reactions and quenching of the active species which can be attributed to the allyl system complexing the catalyst as mentioned above. A similar behavior was reported for different seven-membered cyclodioxepins and cyclic amides (Scheme 2).15–17
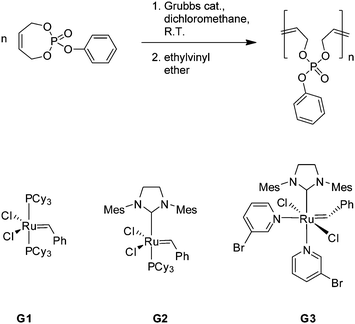 | ||
| Scheme 2 Ring-opening metathesis polymerization of 1 with different Grubbs-type catalysts. | ||
When the Grubbs 3rd generation catalyst18 was used under the same conditions, almost full monomer conversion for both monomers (1 and 2) was achieved (>90% from 1H NMR). Fig. 2 shows the zoomed-in 1H NMR spectra of polymerization mixtures of 2 with different catalysts. After ring-opening, the resonance for the methylene group of the ethyl side chain shifts to higher field (signal “c” in Fig. 1, from ca. 4.2 ppm in the monomer to ca. 4.1 ppm in the polymer) and can be used to determine the degree of ring-opening, i.e. the monomer conversion. It can be clearly seen that only the Grubbs 3rd generation catalyst results in a reasonable degree of conversion. However, SEC elugrams still showed a broad molecular weight distribution (PDI = ca. 2) and molecular weights usually lower than 5000 g mol−1, which could not be increased by changing the catalyst:monomer ratio. This molecular weight limitation could be due to transfer or back biting reactions or catalyst deactivation by coordination (negative neighboring group effect)14 which is currently under deeper investigation.
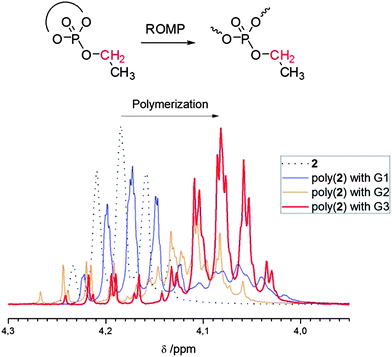 | ||
| Fig. 2 Zoom-in 1H NMR spectra of 2 and poly(2); the resonance of the methylene side chain shifts to higher field after ring-opening. | ||
These results clearly demonstrate that the seven-membered unsaturated cyclic phosphates can be polymerized via ROMP, but that the polymerization is far from a living process; this further corroborates with recent results for analogue cyclic phosphoamidates, which were used to “terminate” a living ROMP of a norbornene polymerization (i.e. oligomerization at the chain end was observed).16 The herein presented seven-membered cyclic phosphates could also be used as a second short block in other ROMPs yielding a terminal OH-group after acidic hydrolysis or enzymatic degradation; this is currently under investigation.
In the next experiments, the copolymerization of 1 and 2 with cis-cyclooctene (CO) as a comonomer was investigated.
To the best of our knowledge, there are no reports on the living polymerization of cis-cyclooctene primarily because significant chain transfer from secondary metathesis of the unhindered polymer backbone occurs during ROMP, making it difficult to polymerize in a controlled fashion. This is due to the rather low ring strain of 29 kJ mol−1 of cis-cyclooctene,19 which lowers its activity for living ROMP. However, the ROMP of CO and its derivatives represents a straightforward route towards linear polyolefins due to the availability of suitable starting materials and substantial ring strain of the eight-membered ring.20,21 A copolymerization with the cyclic unsaturated phosphate presented herein should be feasible and was investigated in the following (Scheme 3). Table 1 lists all comonomer compositions and the molecular weights as well as thermal characterization.
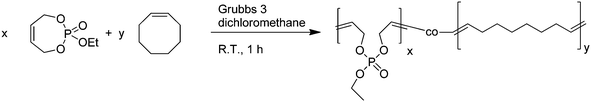 | ||
| Scheme 3 Copolymerization of 2 with cis-cyclooctene. | ||
| # | Monomer | Ratio C:Ptheoa |
Ratio C:PNMRb |
M n | PDIc | T m |
|---|---|---|---|---|---|---|
| a Monomer molar ratio between CO and 1 or 2. b Molar ratio between CO and 1 or 2 determined from 1H NMR. c Number average of the molecular weight (g mol−1) and polydispersity index determined via SEC in chloroform vs. PS standards. d Melting points determined via differential scanning calorimetry (* = no melting point observed). | ||||||
| P1 | — | 100 | 100 | 30000 |
1.75 | 62 |
| P2 | 2 | 9:1 |
10:1 |
43200 |
1.72 | 55 |
| P3 | 2 | 5:1 |
5:1 |
27200 |
1.75 | n.d. |
| P3b | 2 | 5:1 |
5:1 |
41100 |
1.75 | 45 |
| P4 | 2 | 4:1 |
4:1 |
14600 |
1.95 | 40 |
| P5 | 2 | 7:3 |
9:3 |
18700 |
1.87 | n.d. |
| P6 | 1 | 0 | 0 | 2500 | 1.95 | —* |
| P7 | 2 | 0 | 0 | 5500 | 1.90 | —* |
| P8 | 1 | 8:2 |
8:2 |
40000 |
1.90 | 54 |
Different comonomer ratios were investigated and up to 30% phosphate monomer could be incorporated into the PCO backbone with a reasonable molecular weight (up to 50000 g mol−1) and molecular weight distribution (ca. 1.7–2) and full conversion. If higher amounts of phosphate monomer were used, incomplete conversion and broad molecular weight distributions were observed. The thermal properties of the copolymers were investigated by differential scanning calorimetry. With increasing degree of incorporation of the phosphate monomer, the melting temperature of PCO is lowered from ca. 62 °C for pure PCO (with a molecular weight of 30000 g mol−1) to 40 °C when 20% of 2 are copolymerized with CO. This is reasonable as the phosphate comonomers along the polymer backbone can be regarded as defects for the crystallization of PCO, thus lowering the melting points.
Fig. 3 shows an overlay of the 1H NMR spectra of the homopolymer of 2 (P7, top), the homopolymer of cyclooctene (P1, bottom), and copolymer P4 (with a theoretical ratio 4:1 = CO:2). Clearly, the copolymer spectrum shows all resonances for PCO and P(2).
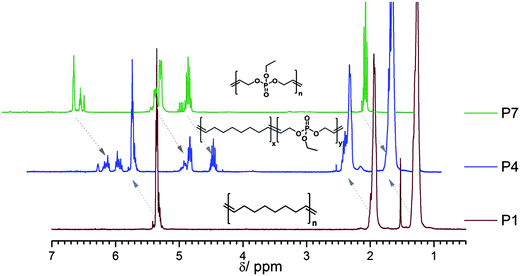 | ||
| Fig. 3
1H NMR overlay (300 MHz in CDCl3): top, homopolymer of 2; middle, copolymer of 2 and CO (ratio 1:4); bottom, homopolymer of CO. | ||
However, several other resonances can be detected. The appearance of different resonances for double bonds and methylene signals adjacent to them is expected due to the incorporation of both monomers in the polymer chain resulting in different dyad distributions. The signal pattern is similar to previously reported copolymers of CO and carborane-containing oxanorbornenes.22 From detailed 2D NMR investigations of the P(PE-co-CO) copolymers (all spectra can be found in the ESI, Fig. S9–S11†) all additional signals could be assigned. Fig. S9† shows a representative TOCSY-H-NMR of polymer P3 in CDCl3 (at 700 MHz). As expected for a ROMP, cis and trans double bonds (mainly trans) can be detected in the resulting polymers; for the PCO segments (also compare inset in Fig. S12†) these resonances are at 5.41 ppm for the trans and 5.37 ppm for the cis-oriented double bonds. Also the neighboring methylene units are affected by this orientation and two separate resonances can be detected at 1.4 and 1.3 ppm, respectively. In a CO-phosphate dyad the double bond signals shift downfield due to the proximity of the ester group to 5.8 and 5.6 ppm, respectively. For the very few double bonds between two phosphate units in the copolymer, their resonance can be detected at 5.95 ppm (lowest field due to the proximity to two ester groups). For the CP (or PC) and the PP dyad the methylene units next to the double bonds can also be distinguished at 4.6 and 4.5 ppm, respectively. The side chain ethyl group of the phosphate brings additional resonances at ca. 4.1 and 1.36 ppm. These signal assignments can be further verified by 1H-31P 2D NMR (Fig. S11†). In a high resolution 700 MHz spectrum one can also detect the signals of the initiator (aromatic peaks at ca. 7.3 ppm) and the olefinic end group at ca. 6.2 ppm. The end groups are mainly attached to CO-units as a strong coupling to the methylene units in the aliphatic region can be detected but no coupling to phosphate-resonances (Fig. S12†). Fig. 4 summarizes the chemical shifts for the three different possible dyads.
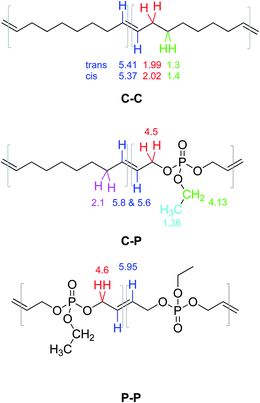 | ||
| Fig. 4 Signal assignment of different resonances in the copolymer. | ||
P2 (Mn 43200 g mol−1) was subjected to an acidic hydrolysis with hydrochloric acid in THF. The resulting crude product was dried and the molecular weight was determined via SEC. Fig. 5 shows the respective molar mass distributions before and after hydrolysis proving a complete shift of the distribution to lower molecular weights indicating a rather random incorporation of 2 in the polymer backbone with an Mn of 4000 g mol−1 after hydrolysis. These results further suggest the possibility of the hydrolytic or enzymatic degradation of the phosphoester bonds in possible (bio)applications.
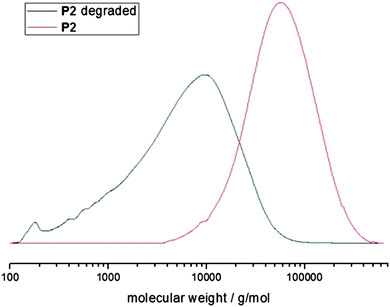 | ||
| Fig. 5 Molecular weight distributions from SEC (in CHCl3, vs. PS standards) of P2 and the hydrolyzed product. | ||
As a potential application P2 was exemplarily used in a solvent evaporation miniemulsion procedure to produce potentially biodegradable nanoparticles which could be used for the encapsulation of hydrophobic drugs.12,13,23 Heterogeneous metathesis polymerization was previously used to prepare potentially biocompatible nanoparticles.23–25 The polymer was dissolved in chloroform and dispersed in water containing SDS via ultrasound (for details compare the Experimental section) to generate a stable miniemulsion. Then the organic solvent was evaporated over a period of several hours to precipitate the polymer as a stable nanoparticle dispersion. Excess of surfactant was removed by dialysis and the particles were analyzed. By variation of the amount of surfactant and volume of the dispersed phase the particle size was varied. The hydrodynamic diameters of the particles in aqueous dispersion after dialysis were found to be 76 nm (procedure 1) and 140 nm (procedure 2) by dynamic light scattering. The stability of both systems was similar after the dialysis process with zeta-potential values of −49.2 ± 11.7 mV for the particles from procedure 1 and −65.2 ± 5.7 mV for the particles from procedure 2. Fig. 6 shows a representative SEM image of the particles (synthesized via procedure 2); the size distributions determined via dynamic light scattering can be found in the ESI (Fig. S14 and S15†). As expected, spherical particles were obtained. The size measured by SEM corresponds well to the values obtained from dynamic light scattering. As the polymer is rather soft (from DSC measurements) also the particles are soft and tend to agglomerate during the drying step.
 | ||
| Fig. 6 Scanning electron micrographs of the nanoparticles obtained through the miniemulsion/solvent-evaporation approach using polymer P2. | ||
Experimental part
Chemicals
All chemicals were purchased from Sigma Aldrich and used as received if not otherwise mentioned. Dichloromethane, tetrahydrofuran and triethylamine were dried and stored under argon. Grubbs catalyst 1st generation, Grubbs catalyst 2nd generation, and Grubbs catalyst 3rd generation were purchased from Sigma Aldrich and stored under argon.Instrumentation and methods
All the NMR experiments were carried out with a 5 mm BBI 1H/X z-gradient on a 700 MHz spectrometer with a Bruker Avance III system or on a Bruker AMX400. For 1H NMR spectra, 128 transients were used with an 11 μs long 90° pulse and a 12600 Hz spectral width together with a recycling delay of 5 s. The 13C NMR (176 MHz) and 31P NMR (283 MHz) measurements were carried out with an 1H powergate decoupling method using a 30° degree flip angle, which had a 14.5 μs long 90° pulse for carbon and a 25.5 μs long 90° pulse for phosphorus. The spectral widths were 41.660 Hz (236 ppm) for 13C and 56.818 Hz (200 ppm) for 31P, both nuclei with a relaxation delay of 2 s. The spectra of proton, carbon and phosphorus were recorded in CDCl3 at 298.3 K and were referenced as follows: for the residual CHCl3 at δ (1H) = 7.26 ppm, CDCl3δ (13C triplet) = 77.0 ppm and triphenylphosphine (TPP) δ (31P) = −6 ppm. The assignment was accomplished by the 1H,1H COSY (correlated spectroscopy) 2D method. The spectroscopic widths of the homo-nuclear 2D COSY experiments were typically 14000 Hz in both dimensions (f1 and f2) and the relaxation delay was 1.2 s. The temperature was kept at 298.3 K and/or regulated by a standard 1H methanol NMR sample using the topspin 2.1 software (Bruker).
Size exclusion chromatography (SEC) measurements were carried out in CHCl3 consisting of a Waters 717 plus autosampler, a TSP Spectra Series P 100 pump, a set of three PSS SDV columns (104/500/50 Å), and RI and UV (275 nm) detectors were used. Calibration was carried out using polystyrene standards provided by Polymer Standards Service. The glass transition temperature was measured by differential scanning calorimetry (DSC) on a Mettler Toledo DSC 823 calorimeter. Three scanning cycles of heating–cooling were performed (in a N2 atmosphere, 30 mL min−1) with a heating rate of 10 °C min−1.
The average particle size and particle size distribution were obtained by dynamic light scattering (DLS) in a submicron particle sizer NICOMP® 380, equipped with a detector to measure the scattered light at 90°.
The zeta-potential of the nanoparticle dispersion was measured using a Zetasizer NanoZ using an aqueous 1 × 10−3 M KCl solution as a dispersive phase.
The particle morphology characterization was carried out on a scanning electron microscope (SEM) Zeiss LEO Gemini 1530. The sample was drop cast in a silica slice and previously covered with a thin carbon coating layer using a coating system Leica EM MED020.
Synthesis of 1 and 2
To a dried, two-necked 500 mL round bottom flask 2.4 g (11.4 mmol) of phenyl dichlorophosphate in the case of 1 (or 1.2 g (7.4 mmol) of ethyl dichlorophospahte in the case of 2) dissolved in 200 mL of dry THF, 8 eq. triethylamine was added with stirring under an argon atmosphere. The solution was cooled to 0 °C and then 1.1 equivalents of 1,4-cis-butenediol was added slowly (3–4 h) to the solution via a syringe pump in ca. 50 mL THF. The reaction was stirred overnight at room temperature. The crude mixture was concentrated, filtered and purified by silica chromatography to give a clear colorless liquid (for 1:hexanes:acetone:ethyl acetate 4:2:1 Rf = 0.5; for 2:hexanes:acetone:ethyl acetate 2:2:1 Rf = 0.55).
1: yield: 1.7 g (7.5 mmol, 66%). 1H NMR (300 MHz, CDCl3): δ (ppm) = 4.78–4.69 (m, 4H (CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2–O)), 5.77 (m, 2H (CHCH)), 7.25–7.13 (m, 5H, Ph). 13C NMR (75 MHz, CDCl3): δ (ppm) = 64.7 (CH2–CHCH–CH2–O), 120 (arom), 125.3 (arom), 126.9 (CHCH), 129.8 (arom), 150.5 (arom). 31P NMR (162 MHz, CDCl3): δ (ppm) = −2.04.
CH–CH2–O)), 5.77 (m, 2H (CHCH)), 7.25–7.13 (m, 5H, Ph). 13C NMR (75 MHz, CDCl3): δ (ppm) = 64.7 (CH2–CHCH–CH2–O), 120 (arom), 125.3 (arom), 126.9 (CHCH), 129.8 (arom), 150.5 (arom). 31P NMR (162 MHz, CDCl3): δ (ppm) = −2.04.
2: yield: 0.9 g (4.8 mmol, 68%). 1H NMR (300 MHz, CDCl3): δ (ppm) = 1.33 (t, 3H (O–CH2–CH3)), 3J = 6.0 Hz, 4.18 (m, 2H (O–CH2–CH3)), 4.63 (m, 4H (CH2–CHCH–CH2–O)), 5.70 (m, 2H (CHCH)). 13C NMR (75 MHz, CDCl3): δ (ppm) = 16.2 (O–CH2–CH3), 64.1 (CH2–CHCH–CH2–O), 64.5 (O–CH2–CH3), 127.1 (CHCH). 31P NMR (162 MHz, CDCl3): δ (ppm) = 3.78.
Representative procedure for ROMP
In a glass tube the monomers were dissolved in dry dichloromethane (100 mg of monomers in 2 mL of solvent) and the appropriate catalyst was added as a solution (ca. 5 mg (depending on the targeted molecular weight) in 100 μL dichloromethane) to the vigorously stirred mixture under an argon atmosphere. The reaction was stirred for 1 h, then 100 μL of ethyl vinyl ether was added to terminate the active chain end, concentrated in vacuo and precipitated in diethyl ether. The polymers were then dissolved in CH2Cl2, treated with activated charcoal and filtered over celite. They were isolated and then precipitated from methylene chloride into diethyl ether and finally dried. Yields are usually 80–90%.Procedure for nanoparticle preparation
30 mg of polymer P2 was dissolved in 1.25 g (procedure 1) or 0.62 g (procedure 2) of chloroform. 5 mL of Milli-Q water containing 10 mg (procedure 1) or 5 mg (procedure 2) of sodium dodecyl sulfate were added to the chloroform solution and stirred over a period of 60 min for the formation of the pre-emulsion. Then, the pre-emulsion was subjected to a pulsed ultrasonication process under an ice bath for 120 s (30 s sonication and 10 s pause) at 70% amplitude in a ¼′′ tip Brason 450 W sonifier. The obtained miniemulsion was kept at 30 °C in an oil bath over a period of 8 h to completely evaporate the organic solvent. The obtained nanoparticle dispersion was further purified by exhaustive dialysis against water for approximately 15 h before being used for further studies.Conclusion
In summary we were able to synthesize novel seven-membered cyclic phosphates which can be applied to the synthesis of degradable PPEs and copolymers with cyclooctene via ROMP. Molecular weights and comonomer ratios can be controlled by the catalyst:monomer ratio as ruthenium alkylidene is acting as the initiator. This paper is the first report for the synthesis of polyphosphoesters via ROMP. This broadens the field of biodegradable polyesters and we believe that with this approach, the previously established step growth ADMET protocol can be extended to a chain growth polymerization strategy and will allow us to synthesize telechelic materials and copolymers with other typical ROMP-monomers, for example norbornene-derivatives. We have also demonstrated with a simple approach that potentially biodegradable nanoparticles with different particle sizes and size distributions can be produced from these new polyesters, which can be an interesting option for future applications in the development of drug delivery systems, particularly for hydrophobic drugs.
Acknowledgements
T.S. is grateful to the Max Planck Graduate Center with the Johannes Gutenberg-Universität Mainz (MPGC) for a fellowship and financial support. T.S. is a recipient of a fellowship through funding of the Excellence Initiative (DFG/GSC 266) in the context of the graduate school of excellence “MAINZ” (Materials Science in Mainz). E.M.A. is grateful to the International Max-Planck Research School (IMPRS) for a fellowship. F.W. thanks the Alexander-von-Humboldt foundation for financial support.References
- A. A. Shah, F. Hasan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- R. Mehta, V. Kumar, H. Bhunia and S. N. Upadhyay, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 45, 325–349 Search PubMed.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- H. Q. Mao and K. W. Leong, in Advances in Genetics, ed. M.-C. H. Leaf Huang and W. Ernst, Academic Press, 2005, vol. 53, pp. 275–306 Search PubMed.
- S.-W. Huang and R.-X. Zhuo, Phosphorus, Sulfur, and Silicon and the Related Elements, 2008, 183, 340–348 CrossRef CAS.
- M. Trollsås, V. Y. Lee, D. Mecerreyes, P. Löwenhielm, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619–4627 CrossRef.
- S. Ji, B. Bruchmann, F. Wurm and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 25–34 CrossRef CAS.
- G. Lapienis and S. Penczek, Macromolecules, 1974, 7, 166–174 CrossRef CAS.
- S. Penczek, J. Pretula and K. Kaluzynski, Biomacromolecules, 2005, 6, 547–551 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- A. Musyanovych, J. Schmitz-Wienke, V. Mailänder, P. Walther and K. Landfester, Macromol. Biosci., 2008, 8, 127–139 CrossRef CAS.
- M. Urban, A. Musyanovych and K. Landfester, Macromol. Chem. Phys., 2009, 210, 961–970 CrossRef CAS.
- K. B. Wagener, K. Brzezinska, J. D. Anderson, T. R. Younkin, K. Steppe and W. DeBoer, Macromolecules, 1997, 30, 7363–7369 CrossRef CAS.
- S. Hilf, R. H. Grubbs and A. F. M. Kilbinger, Macromolecules, 2008, 41, 6006–6011 CrossRef CAS.
- A. A. Nagarkar, A. Crochet, K. M. Fromm and A. F. M. Kilbinger, Macromolecules, 2012, 45, 4447–4453 CrossRef CAS.
- S. Hilf, E. Berger-Nicoletti, R. H. Grubbs and A. F. M. Kilbinger, Angew. Chem. Int. Ed., 2006, 45, 8045–8048 CrossRef CAS.
- T.-L. Choi and R. H. Grubbs, Angew. Chem., 2003, 115, 1785–1788 CrossRef.
- P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, J. Am. Chem. Soc., 1970, 92, 2377–2386 CrossRef CAS.
- J. Alonso-Villanueva, J. M. Cuevas, J. M. Laza, J. L. Vilas and L. M. León, J. Appl. Polym. Sci., 2010, 115, 2440–2447 CrossRef CAS.
- C. Liu, S. B. Chun, P. T. Mather, L. Zheng, E. H. Haley and E. B. Coughlin, Macromolecules, 2002, 35, 9868–9874 CrossRef CAS.
- Y. C. Simon and E. B. Coughlin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2557–2563 CrossRef CAS.
- L. Pichavant, C. Bourget, M.-C. Durrieu and V. Héroguez, Macromolecules, 2011, 44, 7879–7887 CrossRef CAS.
- C. Airaud, E. Ibarboure, C. Gaillard and V. Héroguez, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4014–4027 CrossRef CAS.
- D. Le, V. Montembault, S. Pascual, F. Collette, V. Héroguez and L. Fontaine, Polym. Chem., 2013, 4, 2168–2173 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py00437f |
| This journal is © The Royal Society of Chemistry 2013 |