 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of poly(vinyl pivalate)-based stabiliser architecture on CO2-solubility and stabilising ability in dispersion polymerisation of N-vinyl pyrrolidone†
Natasha A.
Birkin
,
Oliver J.
Wildig
and
Steven M.
Howdle
*
School of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: steve.howdle@nottingham.ac.uk; Tel: +44 (0)1159513486
First published on 16th May 2013
Abstract
We report the comparison of block and statistical copolymers of poly(vinyl pivalate) (PVPi), synthesised via RAFT polymerisation, as hydrocarbon stabilisers for use in dispersion polymerisations in CO2. Block copolymer stabilisers are synthesised, and their relative effectiveness in terms of solubility and stabilising ability is compared to statistical copolymer equivalents. The effect of stabiliser molecular weight on dispersion polymerisation of poly(N-vinyl pyrrolidone) PNVP in scCO2 is also explored. Finally, the RAFT end-group incorporated into the stabiliser is completely removed using radical-induced reduction to produce a hydrogen-capped stabiliser in order to investigate the significance of the xanthate in the final stabiliser structure, and whether this component plays a key role in anchoring to the growing polymer particles in scCO2.
Introduction
Supercritical carbon dioxide (scCO2) has attracted considerable attention in recent years as a potentially viable ‘green’ alternative to traditional organic solvents currently used in polymer synthesis.1–3 Most importantly, it is relatively non-toxic, non-flammable and is inert towards reactive compounds including radicals. scCO2 is an ideal solvent for dispersion polymerisation because of the miscibility of the vast majority of monomers in the continuous phase at modest pressures, and the low solubility of most high molecular weight materials.4 With the aid of a stabiliser, dispersion polymerisation enables the production of high yields of free-flowing powder polymers composed of uniform, spherical particles. However, employing a suitable stabiliser for dispersion polymerisation in scCO2 is crucial. The stabiliser must possess a CO2-soluble component, and this is an issue with most polymeric surfactants, which are insoluble in CO2. Fluorinated and silicone-based polymers are the two classes of polymer that have been shown to have appreciable solubility. For this reason, a wide range of fluorine and silicone containing polymers have been adopted successfully as surfactants in dispersion polymerisation of a range of monomers in scCO2.5–9One of the continuing challenges in the application of scCO2 as a solvent for polymerisation is the design of CO2-soluble stabilisers which are cost-effective, non-toxic and efficient. As a result of this, considerable academic research has focused on the development of inexpensive hydrocarbon stabilisers. A number of polymers have already been identified as possessing promising CO2-philicity, with the potential to act as effective stabilisers.10–14
Recently we reported the development of a series of poly(vinyl alkylate) hydrocarbon stabilisers which show favourable CO2-solubility, and are able to successfully support dispersion polymerisation of N-vinyl pyrrolidone (NVP).15 Further to this, Destarac et al. recently compared the relative solubilities of vinyl acetate (VAc), vinyl pivalate (VPi) and vinyl trifluoroacetate (VTFAc) polymers in CO2, looking at the strength of the polymer–polymer interactions and their impact on CO2-solubility.16 Taken together, these solubility studies highlight the real potential of hydrocarbon stabilisers such as poly(vinyl alkylates) for future applications using scCO2.
One of the most appealing characteristics of RAFT polymerisation for stabiliser synthesis is the ability to access a wide range of architectures and to control well the chain lengths and hence solubilities. In this paper, the impact of modifying the architecture of PVPi-based stabilisers is explored; from statistical to block copolymer stabilisers of varying molecular weight. In addition, post-polymerisation strategies are employed to manipulate the thiocarbonylthio end-group and to assess the impact upon stabiliser performance.
Experimental section
Materials
Deuterated chloroform, potassium ethyl xanthate (96%), ethyl 2-bromopropionate (99%), magnesium sulfate (>99.5%), aluminium oxide, and HPLC grade tetrahydrofuran were supplied by Sigma Aldrich and used as received. N-Ethylpiperidine hypophosphite (EPHP) (95%) and sodium hydroxide (99%), and monomers vinyl acetate (VAc) (99%) and vinyl pivalate (VPi) (99%) were also obtained from Sigma Aldrich. N-Vinyl pyrrolidone (NVP) (97%) was obtained from Fluka. All monomers were stored at 3–4 °C and purified prior to use by passing through a column of activated aluminium oxide, and subsequently degassing via three freeze–pump–thaw cycles. The initiator 2,2′-azobis(isobutyronitrile) (AIBN) was obtained from Acros and purified by recrystallisation twice from cold methanol. Initiator 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) (WAKO, 95%) was used as received. Dry CO2 (99.99%) and nitrogen (99.99%) were purchased from BOC.Typical stabiliser synthesis
The RAFT agent S (1-ethoxycarbonylethyl) O-ethyl xanthate (X1) was synthesised as previously reported.17 Statistical copolymers were prepared in a controlled manner via RAFT polymerisation, using the xanthate X1. Block copolymer synthesis was carried out by first producing one homopolymer chain (e.g. PVAc-X) via the xanthate X1. This was then employed as a macromonomer in a second polymerisation reaction in which the homopolymer acted as the RAFT/MADIX agent, as the xanthate end-groups were incorporated in the final polymer produced in the first step and therefore able to reinitiate RAFT polymerisation. A schematic of a typical block copolymer synthesis is shown (Fig. 1).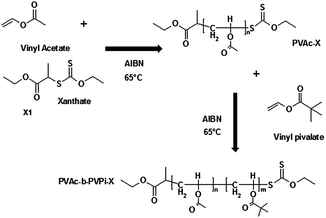 | ||
| Fig. 1 Typical block copolymer synthesis employing xanthate X1, and carrying out successive polymerisation of VAc, followed by VPi. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), filtered and dried in the oven. Mn: 4.2 kg mol−1, Đ: 1.28, conversion: 59%.
:1), filtered and dried in the oven. Mn: 4.2 kg mol−1, Đ: 1.28, conversion: 59%.
In the second stage, VPi (5.00 g, 3.9 × 10−2 mol), PVAc macro-xanthate polymer (3.09 g, 7.4 × 10−4 mol), AIBN (0.01 g, 7.4 × 10−5 mol) and dry toluene (5 mL) were added to a 50 mL round bottomed flask with stirrer bar and three-way stop cock. The flask was degassed using three freeze–pump–thaw cycles and immersed in an oil bath at 65 °C for 24 h. The product was precipitated into a mixture of ice cold methanol–water and vacuum dried overnight. Mn: 11.9 kg mol−1, Đ: 1.45, ratio: 46:54, conversion: 75% (Table 2, entry 5). Evidence of block copolymer formation was confirmed via comparison of GPC trace before and after block extension, and analysis of the glass transition temperature (Tg).†
Radical-induced reduction
For removal of the RAFT end-group from the copolymer stabilisers, and introduction of a C–H end group, EPHP was employed in a 20-fold excess w.r.t polymer, and 0.5 equivalents of AIBN, resulting in [polymer] :[EPHP]:[I] of 1:20:0.5, following the method described in the literature.18
:1) and vacuum dried overnight.
Phase behaviour measurements
Phase behaviour measurements for both the homopolymers and statistical copolymers were determined using the high pressure variable volume view cell procedure as outlined previously.19 All cloud point measurements were determined in a CO2–NVP mixture (15 wt% NVP w.r.t CO2, 5 wt% stabiliser w.r.t monomer). Once the desired quantities of CO2, monomer and stabiliser were added, the volume of the cell was decreased to a point where a single phase mixture was obtained. The volume was then gradually increased, decreasing the pressure and density of the CO2 phase, until the cloud point pressure was reached and the stabiliser precipitated out. All cloud points were repeated three times and an average of the values taken (±0.5–1.0 bar). Cloud point measurements were recorded from 35 to 75 °C (±0.3 °C).High pressure polymerisation of NVP
The stabilising ability of the polymeric surfactants was determined by employing the materials in dispersion polymerisations in scCO2 to produce poly(vinyl pyrrolidone) (PNVP). A low temperature initiator 2,2′-azobis-(2,4-dimethyl-4-methoxyvaleronitrile) (V-70) was employed. In a typical high pressure polymerisation, the initiator was added to a custom built 60 mL Mk III clamp sealed autoclave.20 Hydrocarbon stabiliser (5 wt%) was dissolved in NVP monomer (8 mL) and injected through the safety valve into the autoclave. The vessel was filled with CO2 (∼55 bar) and heated to 35 °C, which resulted in an observed increase in pressure to ∼83 bar. At this point, the autoclave pressure was increased by addition of scCO2 to ∼276 bar and the reaction proceeded for 48 h with stirring, using a mechanically driven overhead stirrer. The autoclave was then cooled to room temperature and the CO2 vented from the vessel to yield the polymeric product.Characterisation techniques
Molecular weight and polydispersity of the hydrocarbon stabiliser samples were determined using Gel Permeation Chromatography (PL-GPC 120, Polymer Labs) with differential refractometer detection. THF was employed as eluent, with two columns (30 cm, PolarGel-M) in series calibrated against polystyrene standards. Molecular weight of PNVP dispersion products was analysed using CHCl3 with 5 wt% triethylamine as eluent, and PS standards.Differential Scanning Calorimetry (DSC) analysis was obtained using a DSC Q2000 for thermal analysis. Measurements were run at 10 °C min−1 with a nitrogen flow rate of 50 mL min−1, and a temperature range between 0 °C and 100 °C.
Determination of the composition ratios for the statistical copolymers, and the monomer conversion for all polymers, was calculated from the relevant peaks of the 1H NMR spectra, recorded using a Bruker DPX-300 Spectrometer in CDCl3.
SEM analysis of PNVP products was carried out using a JEOL 6060LV variable pressure scanning electron microscope. Samples were prepared using a Balzers SCD 030 gold sputter coater. Mean particle diameter (Dn, μm) of the samples was determined by measuring ∼100 particles from SEM data using ImageJ analysis software, and calculating a mean value from the results. The coefficient of variance (Cv) is a statistical measure of the dispersion of the data points in a series around the mean, and was determined using the equation Cv = (σ/Dn) × 100, where σ corresponds to the standard deviation of the particle diameter (μm).
Results and discussion
Designing a suitable stabiliser for use in scCO2 requires careful consideration of a range of different parameters, all of which are likely to impact on the overall ability of a material to both anchor to the growing polymer particles and provide sufficient solubility/steric stabilisation to afford a successful dispersion polymerisation. Previous studies have considered this, looking at the impact of adjustments to the molecular architecture and anchor group on phase behaviour and dispersion ability in scCO2.6,21–26 Herein, a series of modifications to the stabiliser architecture are considered to assess the stabiliser requirements for dispersion polymerisation in scCO2 using the new PVPi-based materials. It is important to point out that the addition of NVP clearly increases the solubility of the stabiliser, but is also representative of the true conditions in which the stabiliser will be active, i.e. during a dispersion polymerisation where initially monomer is present. The effect of the NVP monomer on the cloud point of such a stabiliser is shown clearly (Fig. ESI-2 and ESI-3).†Variation of statistical copolymer stabiliser molecular weight
When considering polymeric stabilisers, it is important to evaluate the effect of molecular weight, which will have an impact on both the solubility and stabilising ability of the material. Lower molecular weight stabilisers should act to improve solubility, but will also provide decreased steric stabilisation because of the reduced chain length. Previously, we reported the synthesis of a series of PVAc-s-PVPi-X copolymer stabilisers of similar molecular weight and varying composition ratio. We showed that the optimum ratio of VAc:VPi afforded high CO2-solubility. The impact of molecular weight on solubility and stabilising ability is considered in depth, for a range of copolymer stabilisers. Both ∼50:50 and 10:90 PVAc:PVPi ratios were studied (Table 1).
| Stabiliser | PNVP product | ||||||
|---|---|---|---|---|---|---|---|
| M n (kg mol−1) | Đ | Ratio VAc:VPi |
Cloud pointc (bar) | D n (μm) | C v (%) | Yieldf (%) | App. |
| a Polymerisation conditions: scCO2 polymerisation at 35 °C for 48 hours with V-70 initiator and 5 wt% of stabiliser. b Experimental Mn and Đ of stabiliser determined via GPC-RI with THF eluent and PS standards, ratio determined from 1H NMR. c Stabiliser cloud point determined using variable volume view cell (15 wt% NVP w.r.t CO2, 5 wt% stabiliser w.r.t monomer). d Mean particle diameter as determined from sampling of ∼100 particles of a typical SEM image. e Coefficient of variance as determined by equation Cv = (σ/Dn) × 100. f Yield determined gravimetrically. g Agglomerated and non-spherical particles were unsuitable for particle size measurement. | |||||||
| 4.7 | 1.29 | 48:52 |
106.3 | 7.0 | 36.3 | 58 | Solid |
| 5.7 | 1.35 | 46:54 |
131.9 | 3.1 | 20.8 | 74 | Powder |
| 9.4 | 1.50 | 44:56 |
154.2 | 1.4 | 30.2 | 86 | Powder |
| 13.8 | 1.50 | 48:52 |
179.6 | 2.5 | 23.2 | 91 | Powder |
| 15.6 | 1.43 | 47:53 |
193.2 | 2.5 | 31.0 | 91 | Powder |
| 20.6 | 1.38 | 48:52 |
206.1 | 2.6 | 23.0 | 93 | Powder |
| 21.8 | 1.47 | 48:52 |
213.7 | 3.1 | 24.6 | 83 | Powder |
| 29.4 | 1.42 | 50:50 |
233.4 | 3.2 | 24.1 | 57 | Solid |
| 4.5 | 1.22 | 10:90 |
101.2 | n/ag | n/ag | 82 | Powder |
| 7.4 | 1.42 | 10:90 |
117.4 | 3.0 | 48.7 | 87 | Powder |
| 8.9 | 1.53 | 10:90 |
133.9 | 2.0 | 28.6 | 84 | Powder |
| 16.5 | 1.49 | 8:92 |
169.1 | n/ag | n/ag | 78 | Powder |
As anticipated, following an increase in the molecular weight of the stabiliser, the solubility in scCO2 decreases, reflected by an increase in the cloud point pressure for each of the stabilisers. As the chain length is shortened, dissolution of the stabilisers in scCO2 occurs more easily (Table 1, column 3). This highlights the significant effect molecular weight of the stabiliser will exert on phase behaviour in scCO2. The same cloud point trend for molecular weight was observed for both ratios of PVAc:PVPi.
Following determination of the relative solubilities of the materials, dispersion polymerisations were conducted in scCO2 using the monomer NVP. The majority of stabilisers yielded a polymeric powder, and the products were analysed using SEM. Considering first the series of stabilisers with a 50:50 ratio, it is evident that there is a clear dependence on molecular weight in relation to stabilising ability (Table 1, entries 1–8).
At low molecular weight, the stabiliser is highly CO2-soluble, but chain length is insufficient to provide steric stabilisation, leading to a highly agglomerated structure and low yield, indicative of unsuccessful dispersion polymerisation (Fig. 2a).
 | ||
| Fig. 2 Variation of particle morphology with different molecular weight stabilisers of 50:50 ratio (Table 1; entries 1–6): (a) Mn = 4.7k; (b) Mn = 9.4k; (c) Mn = 15.6k; (d) Mn = 20.6k; (e) Mn = 21.8k; and (f) Mn = 29.4k. | ||
As the molecular weight of the stabiliser is increased, the stabiliser chain length is also increased, and subsequently steric stabilisation is improved. This results in the production of PNVP particles of increasing particle diameter, and indeed, using a stabiliser of 21.8 kg mol−1 results in significantly larger PNVP particles (Fig. 2b–e).
However, solubility in scCO2 is adversely affected at higher molecular weights, and eventually this will have a negative impact on the stabilising ability. This is observed when employing a higher molecular weight stabiliser of 29.4 kg mol−1. In this case, a powder product is no longer obtained, and control over particle morphology is lost (Fig. 2f and Table 1, entry 8).
Studies on stabiliser molecular weight by DeSimone et al. also led to the conclusion that increasing the molecular weight of the stabiliser resulted in larger particles.27 The PVPi-based stabilisers show the same trend; the average particle size diameter increases with increasing molecular weight and backbone length of the stabiliser (Table 1, entries 1–8, column 4). This is attributed to the presence of fewer stabiliser molecules at higher molecular weight, leading to decreased surface coverage and subsequently larger particles. In addition, it is important to emphasise that increased backbone length will reduce the solubility of the stabiliser in scCO2.
Polymerisation using a series of 10:90 ratio stabilisers was also considered, and the range of molecular weights at which these stabilisers could function successfully was found to be significantly more limited (Fig. 3 and Table 1, entries 9–13). For both 10:90 and 50:50 PVAc:PVPi stabilisers, a molecular weight of ∼10.0 kg mol−1 is required for well-defined, spherical particle formation (Fig. 2b and 3c). However, the upper molecular weight limit is very restricted in the case of a 10:90 stabiliser, with control of spherical particle morphology being lost when a stabiliser reaches only 12.8 kg mol−1 (Fig. 3d).
 | ||
| Fig. 3 Variation of particle morphology with different molecular weight copolymer stabilisers of 10:90 ratio (Table 1; entries 9–13): (a) Mn = 4.5k; (b) Mn = 7.4k; (c) Mn = 8.9k; (d) Mn = 12.8k; and (e) Mn = 15.6k. | ||
These results suggest that as the proportion of PVAc in the structure is increased (e.g. 50:50 ratio rather than 10:90), in addition to producing smaller particles, the stabilisers also possess a wider molecular weight range at which they can operate successfully (∼6 to 22 kg mol−1, Fig. 2). Conversely, the 10:90 ratio stabilisers possess a higher proportion of PVPi and are more soluble in CO2, yet they are less successful at stabilising dispersion polymerisations of NVP, operating within a very limited molecular weight range of ∼10 kg mol−1 (Fig. 3).
Clearly solubility is not the only important parameter. Increasing the level of VPi within the structure minimises self-interaction of the stabiliser and increases free volume and hence solubility in scCO2. However, VAc confers additional flexibility to the polymer chain, allowing for improved interaction with scCO2, optimising the solubility of the stabilisers,28 but also enhancing steric stabilisation around the growing particles of PNVP. To try to better understand these two different effects of PVAc and PVPi we turned to investigation of block copolymers as stabilisers.
Comparison of block and statistical stabilisers
Block copolymers are known to act successfully as stabilisers in scCO2. Such stabilisers are typically composed of a CO2-philic block and a polymer-philic/anchoring block. A range of block copolymer structures such as PS-b-PFOA and PS-b-PDMS have been extensively employed in dispersion polymerisations in CO2, utilising one of the blocks within the structure as the anchor group.8,29–31Architecture can have a significant influence on the ability of a polymer to act as a stabiliser material.6,26,30,32 In order to understand this relationship further a series of statistical and block copolymers of equivalent molecular weight (∼12 kg mol−1) and ratio were synthesised and employed in phase behaviour studies and dispersion polymerisations of NVP in scCO2 to compare their relative effectiveness (Table 2).
| Stabiliser | PNVP product | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Polymer | M n (kg mol−1) | Đ | Ratioc (VAc:VPi) |
Cloud pointd (bar) | M n (kg mol−1) | Đ | D n (μm) | C v (%) | Yieldh (%) | Appearance |
| a Polymerisation conditions: scCO2 polymerisation at 35 °C for 48 hours with V-70 initiator and 5 wt% of stabiliser. b Experimental Mn and Đ of stabiliser determined via GPC-RI with THF eluent and PS standards. c Ratio determined from 1H NMR. d Stabiliser cloud point determined using variable volume view cell (15 wt% NVP w.r.t CO2, 5 wt% stabiliser w.r.t monomer). e PNVP Experimental Mw and Đ obtained from GPC-RI detector in chloroform with 5% triethylamine using PS standards. f Mean particle diameter as determined from sampling of ∼100 particles of a typical SEM image. g Coefficient of variance as determined by equation Cv = (σ/Dn) × 100. h Yield determined gravimetrically. | ||||||||||
| PVAc-s-PVPi-X | 12.1 | 1.46 | 24:76 |
153.9 | 223 | 4.3 | 2.4 | 25.2 | 89 | Powder |
| 11.6 | 1.40 | 51:49 |
170.1 | 231 | 5.3 | 2.1 | 23.0 | 83 | Powder | |
| 11.7 | 1.38 | 74:26 |
194.4 | 259 | 5.9 | 1.9 | 20.7 | 84 | Powder | |
| PVAc-b-PVPi-X | 12.3 | 1.54 | 27:73 |
171.2 | 260 | 5.3 | 3.5 | 40.7 | 74 | Powder |
| 11.9 | 1.45 | 46:54 |
182.8 | 261 | 5.3 | 3.2 | 41.2 | 82 | Powder | |
| 11.8 | 1.52 | 70:30 |
214.9 | 223 | 4.8 | 3.5 | 41.0 | 89 | Powder | |
| PVPi-b-PVAc-X | 11.7 | 1.53 | 48:52 |
209.0 | n/a | n/a | 2.9 | 40.7 | 85 | Powder |
Initially, a series of block copolymers were produced by synthesising a PVAc-X macroRAFT agent, followed by growth of the PVPi block. This order of synthesis was chosen because it was clear that there was an observed improvement in control of molecular weight and overall dispersity of the resulting polymer when synthesising the PVAc block first, followed by extension of this block using VPi monomer. For comparison, we have also investigated the reversed order of block synthesis and those data are discussed in a later section. In all cases block copolymer formation was confirmed via GPC, which showed a distinct shift of the polymer peak to higher molecular weight, evidence of chain extension and block copolymer formation.† Following the successful synthesis of the block copolymer stabiliser materials, their relative solubility in a CO2 + NVP mixture was determined. The phase behaviour measurements at 35 °C show quite clearly the impact of stabiliser architecture on the CO2-solubility of the materials (Table 2, column 3). The phase behaviour of the blocks was determined, and an improvement in solubility was observed as the proportion of PVPi is increased within the sample, following the same trend exhibited by the statistical copolymers in terms of compositional ratio. Comparison of the block copolymer with the equivalent statistical copolymer of a similar ratio shows that statistical copolymers are more soluble (Fig. 4)
 | ||
| Fig. 4 Phase behaviour curve of PVPi-based copolymers of comparable molecular weight and composition, with different stabiliser architectures (Table 2, entries 2, 5 and 7). Stabiliser cloud points determined using variable volume view cell (15 wt% NVP w.r.t CO2, 5 wt% stabiliser w.r.t monomer). | ||
The PVAc-b-PVPi-X block copolymer shows slightly reduced affinity for CO2, and indeed for all block copolymers of varying composition at ∼12 kg mol−1, there is ∼10 to 20 bar difference in the cloud point values. This is expected, as the more soluble PVPi component is concentrated at one end of the chain in the block copolymers, rather than statistically arranged throughout the structure. This segregation of the two monomer types will likely result in decreased solubility overall.
For block arrangements, only the PVPi portion of the block will have improved CO2-solubility compared to the PVAc unit, whilst in the case of the statistical copolymer, the VPi units are randomly interspersed throughout the structure and will improve dissolution of the whole polymer chain.
The statistical and block copolymer stabilisers were also compared and contrasted in the dispersion polymerisation of NVP in scCO2. All PNVP polymers produced using the block copolymer stabilisers were obtained in the form of a free-flowing, polymer powder, with high conversion. These results are indicative of successful stabilisation and dispersing ability, and comparable to PNVP products obtained using a statistical copolymer stabiliser (Table 2, column 4–7).
However, whilst stabilisation using the block copolymers was successful, it is clear that the block copolymer architecture was somewhat less effective in the dispersion polymerisation of NVP when compared to a statistical copolymer. It was observed that the resulting particle size diameter and distribution was larger when using a block copolymer stabiliser compared to the statistical copolymer equivalent of similar ratio and molecular weight. The extent of the differences in particle morphology and size distribution using the two stabiliser types can be observed in the corresponding SEM micrographs (Fig. 5).
 | ||
| Fig. 5 Comparison of PNVP products using block and statistical stabilisers of ∼12 kg mol−1 and varying composition. (a) PVAc-s-PVPi-X 24:76; (b) PVAc-s-PVPi-X 51:49; (c) PVAc-s-PVPi-X 74:26; (d) PVAc-b-PVPi-X 46:54; (e) PVAc-b-PVPi-X 27:73; (f) PVAc-b-PVPi-X 70:30. Polymerisation conditions were kept constant for all reactions, varying only the type of stabiliser employed. | ||
Whilst the difference in size between block and statistical copolymer equivalents is evident, there is also a clear difference in the PNVP particle morphology. All of the statistical stabilisers allow for spherical particle formation, but when employing block copolymer stabilisers, the surface of the resulting particles also appears less well-defined, with an apparent increase in the proportion of distorted spherical particles.
Employing statistical copolymers as stabilisers was also found to result in PNVP microspheres with a relatively tight distribution of particle size. By contrast, the block copolymer stabilisers produce a much broader, relatively bimodal PNVP particle size distribution. This effect is also seen in polymerisations using PVAc-b-PVPi-X stabilisers of varying molecular weight.† Evidence of a second crop of polymer particles, and consequently a bimodal size distribution, has also been observed in previous studies of dispersion polymerisations in scCO2.5,30,33 A number of reasons have been suggested for such findings, including secondary nucleation processes towards the end of the reaction and excess/insufficient stabiliser concentration. However, variation of stabiliser concentration has previously been studied and no such second crop was observed for the PVPi-based statistical copolymer stabilisers, suggesting this is not the reason for such observations in the case of these stabilisers.15
It has been established in our previous work that the increase of free volume and the concomitant decrease of backbone self-interactions is obtained through the statistical incorporation of a bulky vinyl ester such as VPi.15 Thus, the statistical copolymer arrangement should provide better overall solubility compared to the block copolymer stabilisers. Clearly, it might be anticipated that separation of the VAc and VPi units would result in lower overall solubility, but might this also improve partitioning of the stabiliser to the growing PNVP surface?
It is reasonable to assume that the least CO2-philic portion of the stabiliser might start to collapse first, perhaps as the NVP monomer is consumed in the polymerisation process, and there might be some degree of phase separation because of the solubility differences between the VAc and VPi blocks. This is one possibility that could be investigated by in situ small angle X-ray scattering experiments in the future.
In addition, we have assumed also that the RAFT agent makes a strong contribution to anchoring of the stabiliser. Since each block does have a different scCO2 solubility, it may well be important to ascertain whether the order of synthesis of the PVAc and PVPi units, and hence their proximity to the RAFT functionality, could have an effect upon CO2-solubility and stabilisation. We have thus synthesised the block copolymers starting with the PVAc and also by an alternative route with the PVPi block. Both of these block copolymer stabilisers can also be used to stabilise the growth of PNVP particles in a dispersion (Table 2, entry 7).
The phase behaviour data for the PVPi-b-PVAc-X stabiliser were compared to those of the PVAc-b-PVPi-X block copolymers of comparable molecular weight and composition. The results showed a slight decrease in solubility in comparison for PVPi-b-PVAc-X compared to the PVAc-b-PVPi-X block copolymer stabiliser (Fig. 4).
The PVPi-b-PVAc-X stabiliser was also employed in dispersion polymerisation of PNVP and the resulting microparticles were carefully analysed (Fig. 6).
 | ||
| Fig. 6 SEM micrographs comparing PNVP particles synthesised using block copolymer stabilisers with different order of synthesis: (a) PVAc-b-PVPi-X (11.9k, 1.45, 46:54); and (b) PVPi-b-PVAc-X (11.7k, 1.53, 48:52). | ||
The PNVP powder obtained using the PVPi-b-PVAc-X block copolymer stabiliser exhibited no distinct differences in the overall microparticle morphology when compared to those obtained from the PVAc-b-PVPi-X stabiliser. It was also observed that both the mean particle size diameter and Cv for the PNVP microparticles were also comparable for both reactions using each of the block copolymer stabiliser, again showing no clear differences in the dispersion polymerisation reaction product, regardless of the order of synthesis of the block copolymer stabiliser.
If these polymers do indeed perform as reactive stabilisers, anchoring through the RAFT end group, then it should follow that employing PVPi-b-PVAc-X as a stabiliser would position the most soluble PVPi portion in the outer region of the particle, improving CO2-philicity and overall dispersion ability. However, the dispersion polymerisation studies using these two contrasting stabilisers do not appear to show significant differences in terms of stabilising ability, and CO2-solubility. Therefore, whilst order of synthesis could be important when employing block copolymers as stabilisers, there appears to be no distinct impact in this case.
Overall, the comparison of block and statistical stabiliser architecture clearly confirms that the PVAc/PVPi statistical copolymers are able to stabilise dispersion polymerisations of NVP in scCO2 more effectively than the corresponding block copolymers, leading to better control of the particle size distribution and increasingly uniform, monodisperse particles.
Effect of RAFT end-group
The role of the stabiliser in anchoring to a polymer particle is crucial in facilitating a successful dispersion polymerisation.5,21,25 A key question was to determine the mode of action of the stabiliser. We initially reported the RAFT end-group had the potential to act as an anchor group in dispersion polymerisation, adhering to the growing polymer particle in scCO2. In this section, radical-induced reduction has been used to cleave the RAFT end-group functionality from the hydrocarbon polymer structure, in order to determine the impact of the RAFT end-group, and consequently the anchor group of the stabiliser, on the dispersing ability of the scCO2 stabiliser. The removal of this group from the stabiliser both a block and statistical copolymer stabiliser were reduced using this method, and their solubility and stabilising ability considered (Table 3).| Stabiliser | PVP product | ||||
|---|---|---|---|---|---|
| (Mn, Đ, ratio)b | Cloud pointc (bar) | D n (μm) | C v (%) | Yieldf (%) | Appearance |
| a Polymerisation conditions: scCO2 polymerisation at 35 °C for 48 hours with V-70 initiator and 5 wt% of stabiliser. b Stabiliser Mn and Đ determined via GPC-RI with THF eluent and PS standards, Ratio determined from 1H NMR. c Stabiliser cloud point determined using variable volume view cell (15 wt% NVP w.r.t CO2, 5 wt% stabiliser w.r.t monomer). d Mean particle diameter as determined from sampling of ∼100 particles of a typical SEM image. e Coefficient of variance as determined by equation Cv = (σ/Dn) × 100. f Yield determined gravimetrically. | |||||
| PVAc-s-PVPi-X (9.4k, 1.45, 54:46) |
163 | 2.4 | 25.2 | 89 | Powder |
| PVAc-s-PVPi-H (8.8k, 1.40, 54:46) |
147 | 3.7 | 42.9 | 84 | Powder |
| PVAc-b-PVPi-X (11.9k, 1.45, 46:54) |
180 | 3.2 | 41.2 | 82 | Powder |
| PVAc-b-PVPi-H (11.9k, 1.44, 46:54) |
171 | 9.3 | 23.2 | 87 | Powder |
1H NMR and GPC-UV analyses were employed to demonstrate the removal of the xanthate group from the polymer. After reduction, the characteristic thiocarbonylthio absorbance is diminished, evidence that the reduction was effective leading to a terminal C–H end group.†
Phase behaviour measurements demonstrated the reduced forms of the stabilisers to be more soluble in scCO2, by >10 bar (Table 3, column 3). This contradicts the work of Destarac et al. to some extent, where solubility of PVAc3.8k–Xa and a xanthate-free equivalent, PVAc4.2k–H, obtained by a radical-induced reduction of the xanthate group, showed no substantial differences in the solubility in scCO2 and the polymers were found to be respectively soluble in proportions of 0.89 wt% and 0.87 wt% at 35 MPa.34
The polymerisation of NVP in scCO2 was conducted using the reduced stabilisers and compared with the polymerisation results using the xanthate-terminated polymers (Table 3). Somewhat surprisingly, all polymerisations resulted in a free-flowing powder product and comparable conversions. However, SEM analysis showed that discrete spherical microparticle formation was not observed using the reduced stabilisers (Fig. 7b and d) in complete contrast to the RAFT-terminated stabiliser (Fig. 7a and c).
 | ||
| Fig. 7 SEM micrographs of PNVP microparticles synthesised using xanthate capped and reduced stabilisers: (a) PVAc-s-PVPi-X; (b) PVAc-s-PVPi-H; (c) PVAc-b-PVPi-X; and (d) PVAc-b-PVPi-H. | ||
These results indicate that both block and statistical stabilisers in which the xanthate anchor group has been cleaved and replaced with a C–H end group, are still able to support dispersion polymerisation to the extent that a free flowing powder product with high conversion was obtained. However, the defined microparticle structure obtained when employing the RAFT-synthesised stabilisers was not present when using a reduced stabiliser. This suggests that the xanthate end-group of the stabiliser does play a significant role in successfully anchoring to the growing polymer particle. Some form of stabilisation is clearly present with the reduced polymers, as observed by the production of high yielding powder products, but there are likely to be differences in the mechanism by which the stabiliser anchors to the PNVP particles.
We assume that the PVAc-s-PVPi-X stabilisers are anchoring by chemically interacting with the PNVP particles, as a result of the compatible xanthate RAFT agent attached to the stabiliser. Although they are not of course exerting any RAFT control of the growth of the PNVP. For the reduced stabilisers, some degree of stabilisation must still be provided through a weak physical adsorption via the backbone of the stabiliser, similar to the anchoring mechanism of stabilisers such as PFOA.3,33 This could account for the differences observed in the dispersion polymerisations when employing the two types of stabiliser end-groups.† By contrast, when stabilisers were synthesised by a conventional free radical polymerisation method, in the absence of RAFT agent, it was found that they were unable to support dispersion polymerisation of NVP in scCO2 to any extent, and result in low yields of highly agglomerated solids with no defined PNVP particle morphology.† Therefore, the RAFT polymerisation process is essential for synthesising successful PVPi-based hydrocarbon stabilisers which are able to aid dispersion polymerisations in scCO2, presumably because the RAFT method yields much tighter dispersity, in which all of the chains are soluble, and the method also ensures the correct anchoring group is located at the end of the stabiliser chain.
Conclusions
A series of statistical copolymers of PVAc and PVPi of varying molecular weight were synthesised using RAFT polymerisation and their solubility and stabilising ability in CO2 was determined. Stabilisers of ∼10 kg mol−1 were typically observed to be of sufficient length to impart steric stabilisation, whilst being of suitably low molecular weight to maintain good CO2-solubility. Moreover, it has been shown that adjustment of the molecular weight of the stabiliser results in variation of particle morphology, with larger particles at higher molecular weight, attributed to decreased surface coverage. In addition, the stabilisers with a greater proportion of PVPi were observed to have a wider molecular weight range at which they can operate successfully (6–22 kg mol−1).Secondly, a range of block copolymers of varying composition and molecular weight were produced, and their solubility determined. Block copolymer architectures were observed to be less soluble than comparable statistical copolymer stabilisers, which was attributed to the segregated arrangement of the VAc and VPi components. These block copolymers were found to be successful in dispersion polymerisation of NVP in scCO2, with SEM analysis revealing the production of discrete, spherical microparticles, albeit with larger particle size diameter and a broader particle size distribution than those obtained from the statistical copolymers of the same chain length.
Finally, end-group transformation of the polymeric stabiliser was utilised to produce polymers without a xanthate end-group. All of the subsequent C–H capped polymers were found to be slightly more soluble than the stabilisers with a xanthate end-group. These C–H capped stabilisers could be employed in polymerisations of NVP in scCO2 and were found to stabilise to some degree. Following SEM analysis, it was observed that the resulting particle morphology was ill-defined and irregular, demonstrating that the xanthate group is essential. In addition, stabilisers synthesised in the absence of RAFT agent were found to be unsuccessful at supporting dispersion polymerisations of NVP.
Whilst we have sought to identify the means by which the architecture can be varied, there is much additional work to be completed in this area. Variation of the end-group functionality, and understanding of the mechanism of anchoring of the PVPi-based stabilisers to polymers in scCO2, could be key to extending the range of monomers suitable for use with such stabilisers. Application of these PVPi-based materials to the polymerisation of other monomers in scCO2, such as styrene and MMA, is the next challenge which must be addressed if PVAc-PVPi-X stabilisers are to be considered as viable and versatile hydrocarbon stabilisers.
Acknowledgements
We gratefully acknowledge the financial support of the EPSRC and The University of Nottingham for studentship support (NAB), and thank Mr M. Guyler, Mr P. Fields and Mr R. Wilson for their continued technical support.Notes and references
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- M. F. Kemmere and T. Meyer, Supercritical Carbon Dioxide In Polymer Reaction Engineering, Wiley-VCH, 2006 Search PubMed.
- A. I. Cooper, J. Mater. Chem., 2000, 10, 207–234 RSC.
- K. E. J. Barrett, Dispersion Polymerization in Organic Media, Wiley-VCH, 1975 Search PubMed.
- K. A. Shaffer, T. A. Jones, D. A. Canelas and J. A. DeSimone, Macromolecules, 1996, 29, 2704–2706 CrossRef CAS.
- M. R. Giles, R. M. T. Griffiths, A. Aguiar-Ricardo, M. Silva and S. M. Howdle, Macromolecules, 2001, 34, 20–25 CrossRef CAS.
- H. Shiho and J. M. DeSimone, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2429–2437 CrossRef CAS.
- D. A. Canelas and J. M. DeSimone, Macromolecules, 1997, 30, 5673–5682 CrossRef CAS.
- Z. Ma and P. Lacroix-Desmazes, Polymer, 2004, 45, 6789–6797 CrossRef CAS.
- H. Lee, J. W. Pack, W. X. Wang, K. J. Thurecht and S. M. Howdle, Macromolecules, 2010, 43, 2276–2282 CrossRef CAS.
- T. Sarbu, T. Styranec and E. J. Beckman, Nature, 2000, 405, 165–168 CrossRef CAS.
- Z. Shen, M. A. McHugh, J. Xu, J. Belardi, S. Kilic, A. Mesiano, S. Bane, C. Karnikas, E. Beckman and R. Enick, Polymer, 2003, 44, 1491–1498 CrossRef CAS.
- C. Drohmann and E. J. Beckman, J. Supercrit. Fluids, 2002, 22, 103–110 CrossRef CAS.
- E. J. Park, A. P. Richez, N. A. Birkin, H. Lee, N. Arrowsmith, K. J. Thurecht and S. M. Howdle, Polymer, 2011, 52, 5403–5409 CrossRef CAS.
- N. A. Birkin, N. J. Arrowsmith, E. J. Park, A. P. Richez and S. M. Howdle, Polym. Chem., 2011, 2, 1293–1299 RSC.
- E. Girard, T. Tassaing, S. Camy, J.-S. Condoret, J.-D. Marty and M. Destarac, J. Am. Chem. Soc., 2012, 134, 11920–11923 CrossRef CAS.
- M. Destarac, C. Brochon, J. M. Catala, A. Wilczewska and S. Z. Zard, Macromol. Chem. Phys., 2002, 203, 2281–2289 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- P. Licence, M. P. Dellar, R. G. M. Wilson, P. A. Fields, D. Litchfield, H. M. Woods, M. Poliakoff and S. M. Howdle, Rev. Sci. Instrum., 2004, 75, 3233–3236 CrossRef CAS.
- H. M. Woods, Hydrocarbon Stabilisers for Use in Supercritical Carbon Dioxide, Ph.D thesis, University of Nottingham, 2005.
- H. M. Woods, C. Nouvel, P. Licence, D. J. Irvine and S. M. Howdle, Macromolecules, 2005, 38, 3271–3282 CrossRef CAS.
- C. Lepilleur, E. J. Beckman, H. Schonemann and V. J. Krukonis, Fluid Phase Equilib., 1997, 134, 285–305 CrossRef CAS.
- T. Ribaut, J. Oberdisse, B. Annighofer, B. Fournel, S. Sarrade, H. Haller and P. Lacroix-Desmazes, J. Phys. Chem. B, 2011, 115, 836–843 CrossRef CAS.
- I. Stoychev, F. Peters, M. Kleiner, S. Clerc, F. Ganachaud, M. Chirat, B. Fournel, G. Sadowski and P. Lacroix-Desmazes, J. Supercrit. Fluids, 2012, 62, 211–218 CrossRef CAS.
- W. X. Wang, A. Naylor and S. M. Howdle, Macromolecules, 2003, 36, 5424–5427 CrossRef CAS.
- M. R. Giles, S. J. O'Connor, J. N. Hay, R. J. Winder and S. M. Howdle, Macromolecules, 2000, 33, 1996–1999 CrossRef CAS.
- J. M. Desimone, E. E. Maury, Y. Z. Menceloglu, J. B. McClain, T. J. Romack and J. R. Combes, Science, 1994, 265, 356–359 CAS.
- T. Sarbu, T. J. Styranec and E. J. Beckman, Ind. Eng. Chem. Res., 2000, 39, 4678–4683 CrossRef CAS.
- D. A. Canelas, D. E. Betts, J. M. DeSimone, M. Z. Yates and K. P. Johnston, Macromolecules, 1998, 31, 6794–6805 CrossRef CAS.
- D. A. Canelas, D. E. Betts and J. M. DeSimone, Macromolecules, 1996, 29, 2818–2821 CrossRef CAS.
- W. P. Hems, T. M. Yong, J. L. M. van Nunen, A. I. Cooper, A. B. Holmes and D. A. Griffin, J. Mater. Chem., 1999, 9, 1403–1407 RSC.
- H. S. Ganapathy, H. S. Hwang, M. Y. Lee, Y. T. Jeong, Y. S. Gal and K. T. Lim, J. Mater. Sci., 2008, 43, 2300–2306 CrossRef CAS.
- Y. L. Hsiao, E. E. Maury, J. M. Desimone, S. Mawson and K. P. Johnston, Macromolecules, 1995, 28, 8159–8166 CrossRef CAS.
- E. Girard, T. Tassaing, J.-D. Marty and M. Destarac, Polym. Chem., 2011, 2, 2222–2230 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthesis data. Phase behaviour curves. Proton NMR and GPC-UV data for reduced stabilisers. Molecular weight distributions of typical block copolymer. Histograms for particle size analysis. Additional SEM data. See DOI: 10.1039/c3py00275f |
| This journal is © The Royal Society of Chemistry 2013 |