Straightforward synthesis of functionalized cyclic polymers in high yield via RAFT and thiolactone–disulfide chemistry†
Milan M.
Stamenović
a,
Pieter
Espeel
a,
Eisuke
Baba
b,
Takuya
Yamamoto
b,
Yasuyuki
Tezuka
b and
Filip E.
Du Prez
*a
aPolymer Chemistry Research Group, Ghent University, Department of Organic Chemistry, Krijgslaan 281 (S4-bis), 9000 Ghent, Belgium. E-mail: filip.duprez@ugent.be
bDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, O-okayama, Meguro-ku, Tokyo 152-8552, Japan
First published on 24th October 2012
Abstract
An efficient synthetic pathway toward cyclic polymers based on the combination of thiolactone and disulfide chemistry has been developed. First, heterotelechelic linear polystyrene (PS) containing an α-thiolactone (TLa) and an ω-dithiobenzoate group was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization, employing a newly designed TLa-bearing chain transfer agent (CTA). The subsequent reaction of this heterotelechelic polymer with an amine, which acts as a nucleophile for both the TLa and dithiobenzoate units, generated the α,ω-thiol-telechelic PS under ambient conditions without the need for any catalyst or other additives. The arrangement of thiols under a high dilution afforded single cyclic PS (c-PS) through an oxidative disulfide linkage. The cyclic PS (c-PS) disulfide ring formation was evidenced by SEC, MALDI-TOF MS and 1H-NMR characterization. Moreover, we demonstrated a controlled ring opening via either disulfide reduction or thiol–disulfide exchange to enable easy and clean topology transformation. Furthermore, to illustrate the broad utility of this synthetic methodology, different amines including functional ones were employed, allowing for the one-step preparation of functionalized cyclic polymers with high yields.
Introduction
Topological polymer chemistry1–3 has been expanding significantly with the growing need for fundamental research to address unusual polymer architectures having programmed chemical structures, and particularly with the development of controlled radical polymerization (CRP)4 and ‘click’ chemistry during the last decade.5–8 Nowadays, polymer chemists can use a variety of synthetic methods including robust, high yielding, simple covalent chemistries, to construct those unusual polymer topologies, yet with a precise control over the functionality, domain size, reactivity and solubility.9–11 Concomitantly, such a well-defined and discrete macromolecular design based on single-cyclic and multi-cyclic polymers results in unique physical properties and functions for the derived polymer materials.2 For example, rather than exclusively being influenced by the chemical composition, detailed investigations by Honda et al.12 have recently revealed that the thermal stability of self-assembled micelles from cyclic polymers was remarkably enhanced by a topology effect. Moreover, specific properties such as their compact hydrodynamic volume, higher glass transition temperature and distinguished dynamic behavior are some of the features that are otherwise inaccessible through linear or branched topological analogs.1Although the primitive single-cyclic form conceptually represents the simplest cyclic topology, the synthetic constraints in general prevent from the clean and easy production of uniform cyclic polymers, particularly when availability of a specific functional group13 for further topological upgrade is desired.14,15 The common syntheses of single-cyclic polymers employ end-to-end ring-closure and ring-expansion polymerization.7 The former process entails bimolecular, homo-unimolecular and hetero-unimolecular reactions. The unimolecular processes rely on telechelic structures having identical or complementary groups for the formation of a cycle. This has been realized by the use of a metathesis polymer cyclization16–18 in the case of a homo-unimolecular reaction. On the other hand, deprotection/activation treatment of the asymmetrically functional α-acetal-ω-styryl PS telechelic precursor19 and an intramolecular amidation reaction of α-amino-ω-carboxyl PS and poly(tert-butyl acrylate)20 are examples for a ring formation by the hetero-unimolecular process. However, the recent advances in CRP21 hold more promise in terms of ease of accessing various functional telechelics prior to a cyclization step. For example, Glassner et al. subjected atom transfer radical polymerization (ATRP)-derived α-(furan-protected)-maleimide-ω-cyclopentadienyl poly(methyl methacrylate) and poly(tert-butyl acrylate) to a heat treatment to afford cyclic polymers via Diels–Alder addition.22 Durmaz et al.23 also recently reported the synthesis of cyclic homo- and block copolymers via Diels–Alder coupling of anthracene and maleimide end-groups. Furthermore, the advent of robust, efficient and orthogonal ‘click’ chemistries, as a toolbox complementary to CRP,24–27 enabled Laurent and Grayson28 to prepare cyclic PS, pioneering the use of a ‘click’ chemistry in the polymer cyclization field. Likewise, Sugai et al.9 employed azide–alkyne addition in conjunction with an electrostatic self-assembly and covalent fixation (ESA-CF) to effectively construct a variety of unprecedented multicyclic polymer topologies. Goldmann et al.29 described the use of RAFT polymerization, followed by end-group modification to facilitate azide–alkyne ‘click’ chemistry as a simple and effective way to generate macrocyclic PS. Recently, Touris et al.30 reported the synthesis of cyclic PS-b-polyisoprene through acetylene- and azido-‘clickable’ end-groups. Next to the azide–alkyne ‘click’ reaction, Dove and co-workers31 employed the thiol–ene chemistry, specifically the Michael addition of thiols to maleimides, as a facile methodology for the preparation of biodegradable structures. ‘Click’ chemistry also enabled a programmed polymer folding11,32 such as doubly fused tricyclic and triply fused tetracyclic polymer topologies.10
One particularly attractive feature of RAFT33–35 polymerization is the accessibility to the mercapto group as a functional handle for complementary pairing with thiol-related chemistries.36,37 Surprisingly, until now, RAFT synthetic strategies, where a direct use of thiols is exploited, have not been extensively used to produce cyclic macromolecular topologies. An example has been reported for monocyclic PS prepared via oxidation of the α,ω-thiol-containing PS to afford one disulfide linkage per cyclic polymer chain.38 The α,ω-thiol-containing PS was synthesized by polymerizing styrene in the presence of a difunctional RAFT agent and subsequent conversion of the dithioester end-groups to thiols through aminolysis. In contrast, He et al.39 prepared cyclic poly(methyl acrylate) by monomer insertion into a cyclic dithioester-based initiator using γ-ray-induced radical polymerization at low temperatures. Attention has since turned to the application of RAFT polymerization and copper-catalyzed azide–alkyne cycloaddition to form 2- and 3-arm cyclic stars.40
All methodologies described so far restrict the final cyclic structure to rather simple architectures, and the synthesis of more complex polymer topologies requires a tandem use of unlike synthetic strategies. These strategies suffer from multi-step end-group modifications of the linear polymeric precursors, often proceeding with limited yields and at high temperatures. For the formation of sophisticated single cyclic molecular nanostructures, instead, one has to choose a reaction pathway with the complementary partners at individual polymer termini. Building on this prior art, in the current work, we employed an elegant synthetic approach based on RAFT polymerization and thiolactone chemistry as a precursor to a free thiol,41 recently proposed by some of us as a facile method toward functional, linear polymers and networks. The key to this strategy is the in situ generation of thiols driven by the nucleophilic ring-opening of a thiolactone with amines, inspired by a decades-old method for the introduction of sulfhydryl groups in natural proteins.42 The initial demand to design a thiolactone-containing CTA was therefore mandatory, providing the direct access to a thiol group at both α and ω polymer termini upon the treatment with an amine (see further intermediate in Scheme 2).36,41 Then, the in situ produced thiol-telechelics can engage through a disulfide bonding in an intramolecular fashion to yield cyclic polymers, under high dilution and ambient conditions (open air, room temperature, without a need for a catalyst or any additive). The formed disulfide bridge is of particular interest because of thiol–disulfide exchange reactions and self-healing properties.43–45 Inspired by those fascinating features of disulfide bonds, this work adds on one hand an example to the growing number of cyclic polymer topologies, as recently reviewed by Monteiro and Jia.7 On the other hand, the thiolactone–disulfide approach toward c-PS presented herein widens the range of possible unusual topologies that have practical implications to be manufactured by available synthetic methods. The reason for that primarily lays in the potential use of functionalized amines allowing the c-PS to be equipped with desired functional groups for further topology upgrade, yet with the possibility to produce various topologies.
Experimental section
Materials
4,4′-Azobis(4-cyanopentanoic acid) (≥98%), phosphorus pentachloride (≥98%), n-propylamine (≥99%), ethanolamine (≥98%), and tri-n-butylphosphine (99%) were purchased from Sigma-Aldrich and used as received. Homocysteine-γ-thiolactone hydrochloride (99%) was purchased from Acros Organics and used without any further purification. Styrene (St) (99%, extra pure) (Acros Organics) was passed through a column of basic alumina to remove the radical inhibitor. 2,2′-Azobis(isobutyronitrile) (AIBN) (Sigma-Aldrich) was recrystallized twice from methanol. Dichloromethane (DCM) (≥99.9%) and triethylamine (HPLC grade) were purchased from Sigma-Aldrich and distilled from CaH2 prior to use. Ethyl acetate (EtOAc) (Aldrich, HPLC grade) was used without purification. The acid chloride of 4,4′-azobis(4-cyanopentanoic acid)46 and bis(thiobenzoyl) disulfide47 were synthesized according to literature procedures. Silica gel (ROCC, SI 1721, 60 Å, 40–63 μm) was used to perform preparative column chromatography, eluting with HPLC-grade solvents. The collected fractions were analyzed by thin layer chromatography (TLC-plates, Macherey-Nagel, SIL G-25 UV254). All other solvents employed were purchased from Sigma-Aldrich (HPLC grade) and used without further purification.Characterization
Nuclear magnetic resonance (1H- and 13C-NMR (Attached Proton Test, APT)) spectra were recorded at 300 or 500 MHz in CDCl3 (Eurisotop) solution at room temperature on a Bruker Avance 300 or Bruker AM500 spectrometer, respectively. Chemical shifts are presented in parts per million (δ) relative to CHCl3 and DMSO (7.26 ppm and 2.50 ppm in 1H- and 77.23 ppm and 39.51 ppm in 13C-NMR respectively) as an internal standard. Coupling constants (J) in 1H-NMR are given in Hz. The resonance multiplicities are described as d (doublet) or m (multiplet).Size Exclusion Chromatography (SEC) analyses were performed on a Varian PLGPC50plus instrument, using a refractive index detector, equipped with two Plgel 5 μm MIXED-D columns at 40 °C. PS standards were used for calibration and THF as an eluent at a flow rate of 1 mL min−1. Samples were injected using a PL AS RT autosampler.
An Agilent technologies 1100 series LC/MSD system equipped with a diode array detector and single quad MS detector (VL) with an electrospray source (ESI-MS) was used for classic reversed phase liquid chromatography-mass spectroscopy (LC-MS) and MS analysis. Analytic reversed phase HPLC was performed with a Phenomenex C18 (2) column (5 μ, 250 × 4.6 mm) using a solvent gradient (0 → 100% acetonitrile in H2O in 15 min) and the eluting compounds were detected via UV-detection (λ = 254 nm). High resolution mass spectra (HRMS) were collected using an Agilent 6220 Accurate-Mass time-of-flight (TOF) equipped with a multimode ionization (MMI) source.
Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) was performed on an Applied Biosystems Voyager De STR MALDI-TOF spectrometer equipped with 2 m linear and 3 m reflector flight tubes, and a 355 nm Blue Lion Biotech Marathon solid state laser (3.5 ns pulse). All mass spectra were obtained with an accelerating potential of 20 kV in positive ion mode and in linear mode. 1,8,9-Anthracenetriol (dithranol) (20 mg mL−1 in THF) was used as a matrix, CF3CO2Ag (1 mg mL−1) was used as a cationizing agent, and polymer samples were dissolved in THF (10 mg mL−1). Analyte solutions were prepared by mixing 5 μL of the matrix, 5 μL of the polymer and 10 μL of the salt solution. Subsequently, 0.5 μL of this mixture was spotted on the sample plate, and the spots were dried in air at room temperature. A poly(ethylene oxide) standard (Mn = 2000 g mol−1) was used for calibration. All data were processed using the Data Explorer 4.0.0.0 (Applied Biosystems) software package.
UV-vis measurements were performed on an Analytik Jena AG SPECORD® 200 double-beam UV-vis spectrophotometer. Absorbance was measured with the WinASPECT® software in the spectral range of 230–600 nm and a speed of 5 nm s−1. The internal wavelength was calibrated with a holmium oxide filter.
Synthesis
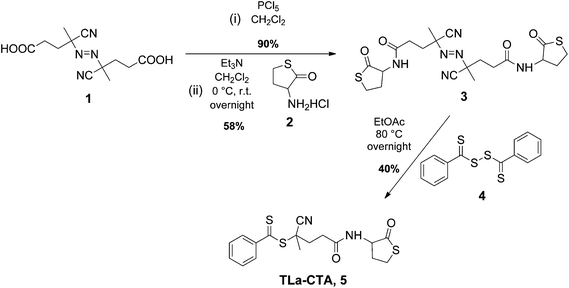 | ||
| Scheme 1 Synthesis of the thiolactone-containing CTA (TLa-CTA, 5). | ||
C20H26N6O4S2 (478.6); m/z (ESI-MS) 479.1.
1H-NMR (300 MHz, DMSO-d6, ppm) δ 8.38 (m, 2H), 4.61 (m, 2H), 3.39 (m, 4H), 3.27 (m, 4H), 2.50 → 1.97 (m, 8H), 1.70 and 1.64 (2 singlets, 6H).
13C-NMR (75 MHz, DMSO-d6, ppm) δ 205.4 (C), 170.2 (C, 2 signals), 118.2 (C, 4 signals), 71.9 (C, 4 signals), 58.2 (CH), 33.0 (CH2, 2 signals), 30.2 (CH2), 29.9 (CH2, 2 signals), 26.8 (CH2), 23.2/22.9 (CH3).
C17H18N2O2S3 (378.5); m/z (ESI-MS) 379.0. HRMS: Expected 379.0603, Found 379.0607 [M + H]+.
1H-NMR (300 MHz, CDCl3, ppm) δ 7.90 (m, 2H), 7.56 (m, 1H), 7.39 (m, 2H), 6.18 (d, 5.5 Hz, 1H), 4.52 (m, 1H), 3.40–3.22 (m, 2H), 2.88 (m, 1H), 2.71–2.36 (m, 4H), 2.04–1.89 (m, 4H).
13C-NMR (75 MHz, CDCl3, ppm) δ 226.6 (C), 205.4 (C), 171.1 (C), 144.7 (C), 133.2 (CH), 128.8 (CH), 126.9 (CH), 118.8 (C), 59.7 (CH), 46.2 (C), 34.0 (CH2), 31.9 (CH2), 31.7 (CH2), 27.7 (CH2), 24.4 (CH3).
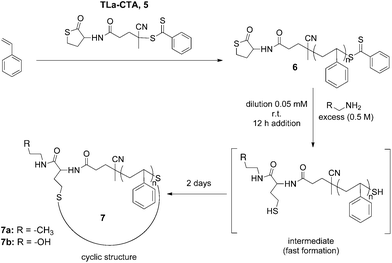 | ||
| Scheme 2 Combined RAFT and thiolactone approach toward functionalized cyclic polymers. | ||
| Entrya | Time, h | Conv.,b % | M n exp,c g mol−1 | PDIc | TLa fidelity,d % |
|---|---|---|---|---|---|
| a Reaction conditions: [St]0/[TLa-CTA]0/[AIBN]0 = 200/1/0.1; 70 °C, bulk. b Calculated from 1H-NMR. c SEC, calibrated with PS standards, THF as an eluent. d Calculated from 1H-NMR, ensuring an excellent agreement between the degree of polymerization (DP) calculated from 1H-NMR signal integration ratios and MWs observed in SEC. | |||||
| 1 | 6 | 16 | 3900 | 1.07 | 95 |
| 2 | 6 | 13 | 3300 | 1.09 | 96 |
| 3 | 8 | 22 | 4700 | 1.07 | 96 |
Reduction with tri-n-butylphosphine. To a 10 mL round-bottomed flask was added 4 mg c-PS (7) (1.54 × 10−6 mol) and dissolved in 1 mL THF. The flask was sealed and the solution was purged with nitrogen for 30 min followed by the addition of 500 μL (2.00 × 10−3 mol) reducing agent tri-n-butylphosphine. The solution was then allowed to stir overnight at room temperature before subjecting to SEC analysis.
Thiol–disulfide exchange with octanethiol. To a 10 mL round-bottomed flask was added 4 mg c-PS (7) (1.54 × 10−6 mol) and dissolved in 1 mL DMF. The flask was sealed and 1 mL octanethiol (5.76 × 10−3 mol) was added to the flask. In one example, the solution was then heated to 90 °C, and allowed to stir overnight before subjecting to SEC analysis. In another example, the solution was allowed to stir overnight at room temperature before subjecting to SEC analysis.
Results and discussion
Synthesis of the thiolactone-containing chain-transfer agent (TLa-CTA, 5, Scheme 1)
The synthesis of a thiolactone containing CTA was adapted from the procedure for the synthesis of 4-cyanopentanoic acid dithiobenzoate, a versatile RAFT controlling agent.48 Activation of the carboxylic acid group in the commercially available 4,4′-azobis(4-cyanopentanoic acid) (Scheme 1, 1)46 and subsequent treatment of the corresponding acid chlorides with homocysteine-γ-thiolactone hydrochloride (Scheme 1, 2) yielded azo-compound 3 (Scheme 1). The thermal decomposition of a 1.5 excess of 3 in the presence of bis(thiobenzoyl) disulfide 4 allowed for the preparation of the TLa-CTA (Scheme 1, 5).Synthesis and characterization of thiolactone (TLa) containing polystyrene (l-PS-TLa, 6, Scheme 2)
In the first step, to achieve a PS hetero-telechelic chain equipped with a thiolactone unit at the α-terminus, we employed the newly designed TLa-CTA (5) to mediate RAFT polymerization of St (Scheme 2). In addition, the resulting PS bears a dithiobenzoate group at the ω-terminus, having therefore two thiol groups in a latent form, each at the opposite polymer chain-end, to encode for the α,ω-thiol-telechelic upon the nucleophilic reaction with an amine (see intermediate, Scheme 2).The crucial point during the polymerization is the inertness of the thiolactone unit against the reaction temperature and carbon-centered propagating polymeric radicals.49 Previously conducted stability tests indicated that the thiolactone unit remained intact at moderate polymerization temperatures, i.e. 70 °C, and inert to a radical concentration found in a typical RAFT polymerization.49 Hence, the RAFT polymerization was conducted at 70 °C in bulk, with a monomer-to-CTA ratio of 200 that can generate PS with targeted molecular weights, yet with a low conversion (ca. 25%) to maintain high TLa end-group fidelity (Table 1, Scheme 2).
Samples, periodically taken from the reaction mixture, were analyzed by SEC and 1H-NMR to follow the progress of the polymerization (Table 1, Entry 1). A monomer conversion vs. time plot showed linear dependence and the desired conversion was achieved within 6 h with an expected molecular weight of the isolated polymer (Fig. 1a). The SEC traces indicated a linear increase of the molecular weights with conversion during the entire course of the reaction (Fig. 1b), having the apparent and theoretical MW values in a good agreement. The controlled nature of the process was further confirmed by low polydispersity indices (PDI) below 1.10. The polymerization was quenched in liquid nitrogen and the polymer was purified by precipitation from cold methanol to recover 6 (Scheme 2) as a light-pink powder (Fig. S4b†).
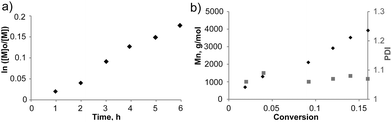 | ||
| Fig. 1 Polymerization of St using TLa-CTA, at 70 °C, in bulk and with 0.1 equiv. AIBN as the radical source, St/TLa-CTA/AIBN = 200/1/0.1 (Table 1, Entry 1): (a) first-order kinetic plot; and (b) molecular weight (◆) and PDI evolution (■) with monomer conversion. | ||
The polymer was characterized by 500 MHz 1H-NMR, UV-vis spectroscopy and SEC. Noteworthy is that MALDI-TOF MS was not suitable to study PS made by RAFT, even though a wide range of experimental conditions were examined concerning that fragmentations of the dithioester end groups under MALDI-TOF MS conditions were already reported.50,51
At this stage, it is essential to determine the TLa content, as a quantitative amount is preferred for a successful cyclization reaction. Thus, the TLa end-group fidelity of the polymer was calculated by combining the results from both high resolution 1H-NMR and SEC.52 The protons from the thiolactone group can indeed be easily distinguished. Therefore, a detailed analysis of the integration values of TLa-CTA (5) and its ring corresponding signals (see Fig. 4: a, b, c, d and e) and the PS backbone broad signal (Fig. 4: f), combined with the results obtained by SEC, revealed a TLa content at the α-terminus with 95% end-group fidelity (Table 1, Entry 1). The remaining ca. 5% counts for the polymer chains derived from the AIBN initiating adducts. Furthermore, as a member of the class of thiocarbonyls, dithiobenzoate-functional RAFT polymers show a strong UV absorption band because of the π–π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group. Consequently, the presence of the dithiobenzoate end-group was confirmed qualitatively by UV-vis spectroscopy, which showed a maximum absorbance at ca. 303 nm (Fig. S4a†).
S group. Consequently, the presence of the dithiobenzoate end-group was confirmed qualitatively by UV-vis spectroscopy, which showed a maximum absorbance at ca. 303 nm (Fig. S4a†).
Synthesis and characterization of the cyclic poly(styrene) (c-PS, 7, Scheme 2)
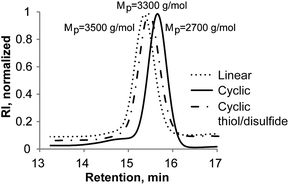
In addressing new cyclization strategies7 to yield cyclic polymers, the role of the thiolactone chemistry emerged as a powerful tool to achieve that goal in a facile and elegant manner. The covalent disulfide linking of the individual l-PS-TLa (6) chains to afford cyclic molecules is based on two nucleophilic processes: the thiolactone ring opening at the α-terminus and the aminolysis of the dithiobenzoate unit at the ω-terminus. Given that the primary amine reacts with both thiolactone and dithiobenzoate groups to yield two thiol species, our initial efforts focused on studying the cyclization involving TLa-containing PS and n-propylamine as a nucleophile. In general, to ensure complete reaction, a large excess of n-propylamine (0.5 M) was added to a flask with 600 mL DCM. Additionally, earlier observations revealed that both aminolysis and subsequent disulfide formation are promoted significantly when increasing the amine concentration in DCM.49 Bearing in mind the oxidative coupling of free thiyl radicals to afford a single c-PS, we anticipated that a quite low concentration of polymer would exclusively afford an intramolecular cyclic product, thus avoiding intermolecular coupling reactions. Therefore, the pre-solution of l-TLa-PS (6), in 10 mL DCM, was added dropwise into a reactor filled with the amine solution via a syringe pump over an extended time (10 h) with the flow rate as low as 1 mL h−1, providing the final polymer concentration of 100–150 mg L−1 (0.05 mM). Thus, stirring the excess of n-propylamine and l-TLa-PS (6) upon the polymer addition, for two days at ambient temperature and under the open air, followed by the removal of solvent and precipitation in cold methanol, afforded a cyclic polymer as a white powder product (Scheme 2, 7a and Fig. S4b†). Isolated yields of 90% or higher were obtained in a reproducible way.The influence of the amine was apparent from the color change of the isolated polymer from a distinct pink to a white colour, indicating successful aminolysis of the dithiobenzoate group (Fig. S4b†). Moreover, UV-vis measurement was performed, further confirming the quantitative removal of the RAFT end-group as evidenced by the disappearance of a characteristic absorbance at ca. 303 nm (Fig. S4a†). Other than these preliminary observations, subsequent efforts were directed toward evaluating whether and to what extent a disulfide linkage, thus individual ring formation, had occurred. In this sense, SEC, 1H-NMR and MALDI-TOF MS analysis were employed to facilitate the characterization of the synthesized cyclic polymer product. The success of the reaction was first evaluated by the SEC analysis of the starting l-PS (6) and final c-PS (7a) (Fig. 2a). Transformation from the linear into a cyclic topology via disulfide intramolecular bridging caused an increase in the retention time and, accordingly, the reduction of the measured Mp (from Mp,l = 3500 to Mp,c = 2700 g mol−1). These results are consistent with a more compact hydrodynamic volume of a cyclic polymer in comparison to a linear counterpart. The hydrodynamic volume ratio or 〈G〉 value equals 0.77, which is in good agreement with precedent literature data.53–55 Noteworthy is that a small fraction of ca. 5–7% of the intermolecular reaction product was observed at higher MWs, which corresponds to byproduct polymers lacking the TLa end groups (see above).
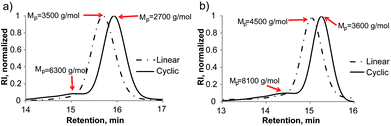 | ||
| Fig. 2 SEC profiles of TLa-terminated PS (l-PS, Scheme 2, 6) and cyclic PS (c-PS, Scheme 2, 7): (a) reaction with n-propylamine (Table 1, Entry 1 and Scheme 2, 7a); and (b) reaction with ethanolamine (Table 1, Entry 3 and Scheme 2, 7b). | ||
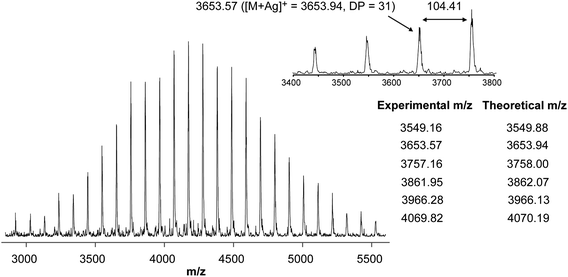
The cyclization through a disulfide bond was also clearly assessed by MALDI-TOF MS analysis, showing a well resolved spectrum with a uniform series of peaks recorded in linear mode (Fig. 3). The peaks are separated by 104 mass units corresponding to the molecular weight of a single styrene component. Moreover, each peak of the distribution matches the expected structure of a cyclic PS containing one disulfide bond and an amide group originating from a thiolactone ring (Table in Fig. 3). Importantly, only the main series attributed to the cyclic PS was observed, suggesting that events such as fragmentation or side reactions at the disulfide site occurred neither during the ring formation nor under the experimental conditions of MALDI-TOF MS measurements.56
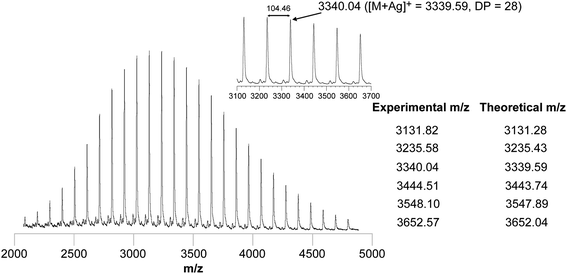 | ||
| Fig. 3 MALDI-TOF mass spectra of c-PS (Scheme 2, 7a, R = –CH3). | ||
Finally, the 1H-NMR spectrum presented in Fig. 4 shows the disappearance of the characteristic signals for the protons of the TLa ring after the cyclization treatment with propyl amine (a, b, c and e labeled signals). Moreover, the appearance of h and i signals in the spectrum of a c-PS (7a) and their integration values further suggest successful ring closure via the proposed mechanism.
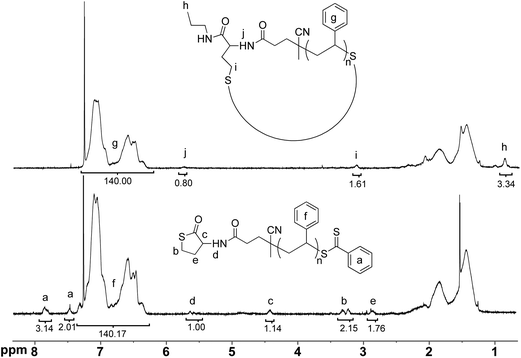 | ||
| Fig. 4 1H-NMR (500 MHz, CDCl3) spectra of linear PS (l-PS, 6, bottom) and cyclic PS (c-PS, 7a, top) with corresponding signal integration values. | ||
In contrast to the results presented above, when using a higher polymer concentration (e.g. 0.25 mM and 50 mM compared to the optimal 0.05 mM), a significant fraction of non-controlled intermolecular disulfide formation was observed in the SEC traces (Fig. S5†), yet lacking the apparent shift toward longer retentions typically observed with the ring creation. Additionally, when no amine was employed in a blank reaction, no cyclization occurred, further confirming the key role of both high dilution and the presence of amine as the only reactant to the additive-free thiolactone–disulfide synthetic strategy for the preparation of cyclic polymers.
Ring opening of disulfide-containing c-PS
MALDI-TOF MS confirmed the formation of disulfides on both polymer chain-ends with the peaks separated by 104 mass units corresponding to the molecular weight of a single styrene unit (Fig. 6). Moreover, each peak of the distribution matched the expected linear structure, yet differing for 289.02 mass units from the starting c-PS (7a) (inset of Fig. 6), which corresponds to the addition of two octanethiyl units during the thiol–disulfide exchange.
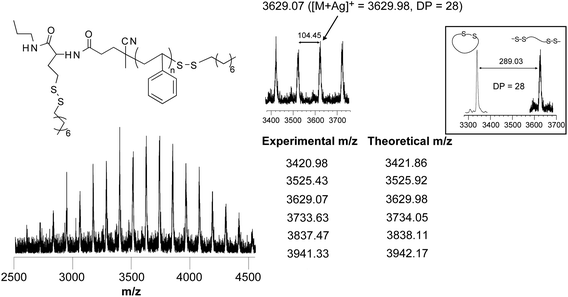 | ||
| Fig. 6 MALDI-TOF mass spectra of linear PS after the thiol–disulfide exchange reaction of c-PS (7a) and octanethiol. Inset: comparison of the characteristic peak distribution of c-PS (before reaction) and linear PS (after reaction). | ||
 | ||
| Fig. 7 MALDI-TOF mass spectra of c-PS (Scheme 2, 7b, R = –OH). | ||
Conclusion
A series of high purity cyclic PS (c-PS, 7) was synthesized in high yield using mild conditions, starting from a linear precursor l-PS-TLa (6) containing the TLa functional group at the α and dithiobenzoate group at the ω terminus. The l-PS-TLa hetero-telechelic was prepared via RAFT polymerization of St mediated by TLa-CTA (5). The nucleophilic reaction of primary amines with both the TLa ring and dithiobenzoate group was utilized to afford the α,ω-thiol-telechelic and its derived one-pot disulfide-promoted cyclization product. SEC, MALDI-TOF MS and 1H-NMR confirmed successful cyclization via a disulfide formation. Moreover, employing ethanolamine, hydroxyl-functionalized c-PS was obtained, demonstrating the opportunity for the preparation of cyclic polymers with the pendent functionality of choice. We have further demonstrated the disulfide ring opening in the presence of a reducing agent or through a thiol–disulfide exchange reaction, re-establishing a parent linear polymer topology. Given the unique character of the synthetic strategy presented here, which enables the fabrication of single cyclic polymers with a desired functional group, the thiolactone–disulfide cyclization offers a key opportunity over other approaches to tailor unusual cyclic polymer topologies. An additional advantage of this system is that the mild reaction conditions are suitable for incorporating a wide range of biomolecules of interest, including systems that are temperature sensitive, such as oligo- and polypeptides. Those and other possibilities ultimately serve as a platform for accessing multi-cyclic complex topological constructions and assemblies via a mild and powerful thiolactone–disulfide strategy, currently being further investigated in our laboratories.Acknowledgements
M.M.S., P.E. and F.E.D.P. acknowledge the Belgian Program on Interuniversity Attraction Poles initiated by the Belgian State, Prime Minister's office (Program P7/05). This work was supported by the cooperation agreement between UGent and TokyoTech, Integrated Doctoral Education Program at TokyoTech (E.B.), The Kurata Memorial Hitachi Science and Technology Foundation (T.Y.) and KAKENHI (23106709 T.Y., 23685022 T.Y. and 23350050 Y.T.).References
- J. A. Semlyen, Cyclic Polymers, Kluwer, Dordrecht, 2nd edn, 2000 Search PubMed.
- T. Yamamoto and Y. Tezuka, Polym. Chem., 2011, 2, 1930 RSC.
- T. Yamamoto and Y. Tezuka, in Complex Macromolecular Architectures: Synthesis, Characterization and Self-assembly, ed. N. Hadjichristidis, A. Hirao, Y. Tezuka and F. Du Prez, John Wiley & Sons (Asia) PTE LTD, Singapore, 2011, p. 3 Search PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 165 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- M. G. Finn, H. C. Kolb, V. V. Fokin and K. B. Sharpless, Prog. Chem., 2008, 20, 1 Search PubMed.
- Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2010, 50, 60 CrossRef.
- N. Sugai, H. Heguri, K. Ohta, Q. Meng, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 14790 CrossRef CAS.
- N. Sugai, H. Heguri, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2011, 133, 19694 CrossRef CAS.
- S. Perrier, Nat. Chem., 2011, 3, 194 CrossRef CAS.
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251 CrossRef CAS.
- H. Oike, T. Mouri and Y. Tezuka, Macromolecules, 2001, 34, 6229 CrossRef CAS.
- K. Miki, Y. Inamoto, S. Inoue, T. Uno, T. Itoh and M. Kubo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5882 CrossRef CAS.
- K. Ishikawa, T. Yamamoto, H. Harada and Y. Tezuka, Macromolecules, 2010, 43, 7062 CrossRef CAS.
- Y. Tezuka and R. Komiya, Macromolecules, 2002, 35, 8667 CrossRef CAS.
- E. Baba, S. Honda, T. Yamamoto and Y. Tezuka, Polym. Chem., 2011, 3, 1903 RSC.
- R. P. Quirk, S.-F. Wang, M. D. Foster, C. Wesdemiotis and A. M. Yol, Macromolecules, 2011, 44, 7538 CrossRef CAS.
- M. Schappacher and A. Deffieux, Macromolecules, 2001, 34, 5827 CrossRef CAS.
- M. Kubo, T. Nishigawa, T. Uno, T. Itoh and H. Sato, Macromolecules, 2003, 36, 9264 CrossRef CAS.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2011, 36, 455 CrossRef CAS.
- M. Glassner, J. P. Blinco and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 724 CrossRef CAS.
- H. Durmaz, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5083 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338 RSC.
- U. Mansfeld, C. Pietsch, R. Hoogenboom, C. Remzi Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238 CrossRef CAS.
- A. S. Goldmann, D. Quémener, P.-E. Millard, T. P. Davis, M. H. Stenzel, C. Barner-Kowollik and A. H. E. Muller, Polymer, 2008, 49, 2274 CrossRef CAS.
- A. Touris and N. Hadjichristidis, Macromolecules, 2011, 44, 1969 CrossRef CAS.
- M. J. Stanford, R. L. Pflughaupt and A. P. Dove, Macromolecules, 2010, 43, 6538 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley – VCH, New York, 2008 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9 CrossRef CAS.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123 CrossRef CAS.
- M. R. Whittaker, Y.-K. Goh, H. Gemici, T. M. Legge, S. Perrier and M. J. Monteiro, Macromolecules, 2006, 39, 9028 CrossRef CAS.
- T. He, G. H. Zheng and C. Y. Pan, Macromolecules, 2003, 36, 5960 CrossRef CAS.
- Md. H. Hossain, D. Valade, Z. Jia and M. Monteiro, Polym. Chem., 2012, 3, 2986 RSC.
- P. Espeel, F. Goethals and F. E. Du Prez, J. Am. Chem. Soc., 2011, 133, 1678 CrossRef CAS.
- R. Benesch and R. E. Benesch, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 848 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolay, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444 CrossRef CAS.
- B. Yameen, M. Ali, M. Alvarez, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Polym. Chem., 2010, 1, 183 RSC.
- K. A. Aamer and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5618 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435 CrossRef CAS.
- P. Espeel, F. Goethals, M. M. Stamenović, L. Petton and F. E. Du Prez, Polym. Chem., 2012, 3, 1007 RSC.
- P. Vana, L. Albertin, L. Barner, T. P. Davis and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4032 CrossRef CAS.
- C. Schilli, M. G. Lanzendorfer and A. H. E. Muller, Macromolecules, 2002, 35, 6819 CrossRef CAS.
- M. M. Stamenovic, P. Espeel, W. Van Camp and F. E. Du Prez, Macromolecules, 2011, 44, 5619 CrossRef CAS.
- H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka, A. Uchibori and Y. Tezuka, J. Am. Chem. Soc., 2000, 122, 9592 CrossRef CAS.
- H. Oike, M. Hamada, S. Eguchi, Y. Danda and Y. Tezuka, Macromolecules, 2001, 34, 2776 CrossRef CAS.
- T. E. Hogen-Esch, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2139 CrossRef CAS.
- G. J. King, A. Jones, B. Kobe, T. Huber, D. Mouvadov, D. L. Hume and I. L. Ross, Anal. Chem., 2008, 80, 5036 CrossRef CAS.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180 CrossRef CAS.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087 CrossRef CAS.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133 CrossRef CAS.
- D.-C. Wu, X. J. Loh, Y.-L. Wu, C. L. Lay and Y. Liu, J. Am. Chem. Soc., 2010, 132, 15140 CrossRef CAS.
- Y. Shen, L. Zhong, S. Markwell and D. Cao, Biochimie, 2010, 92, 530 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2py20751f |
| This journal is © The Royal Society of Chemistry 2013 |