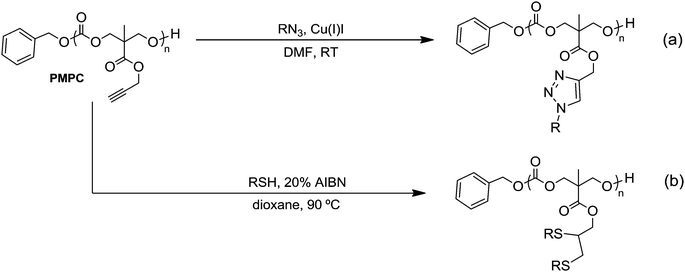
DOI:
10.1039/C2PY20718D
(Paper)
Polym. Chem., 2013,
4, 174-183
Organocatalytic synthesis and post-polymerization functionalization of propargyl-functional poly(carbonate)s†
Received
6th September 2012
, Accepted 15th September 2012
First published on 17th September 2012
Abstract
The synthesis of well-defined propargyl-functional poly(carbonate)s was achieved via the organocatalytic ring-opening polymerization of 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC) using the dual catalyst system of 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (TU) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). The resulting homopolymers showed low dispersities and high end-group fidelity, with the versatility of the system being demonstrated by the synthesis of telechelic copolymers and block copolymers. The synthesized homopolymers with varying degree of polymerization were functionalized with a range of azides via copper-catalyzed Huisgen-1,3-dipolar addition or thiols via radical thiylation, to produce functional aliphatic poly(carbonate)s from a single polymeric scaffold.
Introduction
The ability to readily tune the properties of synthetic biodegradable polymers makes them attractive materials for pharmaceutical applications.1–7 Whilst the synthesis of functional poly(ester)s is a particularly challenging area,8 a number of reported facile preparations of cyclic carbonate monomers from 1,3-diols have led to a poly(carbonate)s with a large range of pendant functionalities being reported.9–15 With many synthetic routes available from which to prepare poly(carbonate)s with specific functional groups, the desire to apply post-polymerization functionalization chemistry presents an attractive method for producing materials with a wide range of properties from a limited monomer pool, as well as enabling the incorporation of reactive groups that are incompatible with the ROP process.
To date, there have been limited reports of the synthesis and post-polymerization functionalizations of such polymers, with allyl-,16,17 maleimide-,18 vinyl sulfone-19 and (meth)acrylate-pendant polymers20 being demonstrated to readily react with thiol-containing molecules. Other examples have reported the synthesis of azide-functional poly(carbonate)s and their modification using copper catalysed-(CuAAC) and strain-promoted (SPAAC) alkyne–azide cycloadditions.21,22
The alkyne functional group is utilized in many organic reactions as a consequence of its versatile reactivity. Alkynes have been widely studied in a range of coupling reactions (Sonogashira, Glaser and Eglinton),23–25 cycloaddition reactions (Diels–Alder with 1,3-dienes, Huisgen 1,3-dipolar)26,27 and in reactions such as hydrogenation, hydroboration, halogenation and the radical thiylation (or ‘thiol-yne’) in which two equivalents of the corresponding reagent can be added to one alkyne group as a result of its double unsaturation.28 In particular the copper-catalyzed derivative Huisgen 1,3-dipolar cycloaddition of alkynes and azides (CuAAC) has been extensively applied in materials chemistry as a fast and quantitative ‘click’ method of functionalization.29,30 The introduction of a pendant propargyl functionality in a poly(carbonate) backbone has previously been reported via the ring-opening polymerization of a propargyl ester functional cyclic carbonate, 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC).31–34 Studies have focused on its copolymerization with lactide in toluene at 120 °C (ref. 34) or at 100 °C in bulk31 using diethyl zinc as a catalyst, with the resulting statistical and block copolymers being used in the immobilization of proteins to biodegradable polymer fibres and in the preparation of protein-grafted polymer microspheres.31–33 Surprisingly however, to date its homopolymerization and the investigation of the versatility of the resulting poly(MPC), (PMPC), for post-polymerization functionalization have not been previously reported.
Herein, the concept of synthesizing a single functional poly(carbonate) scaffold in a controlled manner is extended to those with a pendant propargyl functionality by the ring-opening polymerization of MPC as well as post-polymerization functionalization of the resulting homopolymers via the copper catalyzed addition of azides (CuAAC) or the radical addition of thiols (‘thiol-yne’) to the pendant propargyl groups, giving rise to a range of functional poly(carbonate)s.
Results and discussion
The monomer, 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC), was synthesized in a two-step procedure as previously reported (Scheme 1a).31,32 Initial studies into the ring-opening polymerization (ROP) of MPC were carried out with the dual catalyst system of 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea, TU, (10 mol%) in combination with (−)-sparteine (5 mol%) which had previously shown excellent control in the ROP of the analogous allyl-functional derivative, 5-methyl-5-allyloxycarbonyl-1,3-dioxan-2-one (MAC).16 The resultant polymers revealed narrow, monomodal distributions by gel permeation chromatography (GPC) analysis (e.g.: [M]0/[I]0 = 20; Mn = 6750 g mol−1, ĐM = 1.18) however, the long polymerization times (ca. 24 h for a DP20 homopolymer) and the limited availability of (−)-sparteine presented significant drawbacks.16,35 As such, our attention was turned to the dual catalyst system of TU in combination with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), which had previously shown good control and reduced reaction times in the ROP of other functional poly(carbonate)s.7,36 Polymerizations with [M]0/[I]0 = 100 carried out in CDCl3 ([MPC]0 = 0.5 M) at 25 °C using 5 mol% TU and 1 mol% DBU with benzyl alcohol as initiator achieved >90% monomer conversion within 6 hours. Polymerizations were monitored using 1H NMR spectroscopy by the disappearance of the methylene resonances in the carbonate ring at δ = 4.22 and 4.72 ppm and the appearance of a multiplet at δ = 4.27–4.40 ppm for the corresponding protons on the polymer backbone. Upon completion, pure polymers were obtained by precipitation into cold hexanes and removal of residual catalyst by column chromatography.
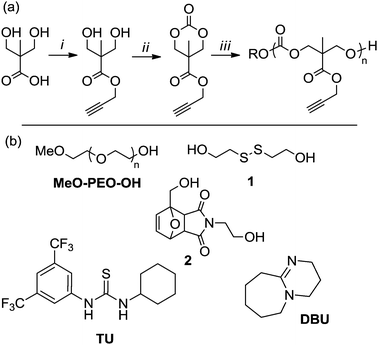 |
| | Scheme 1 (a) Synthesis and ring-opening polymerization (ROP) of 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one, MPC. Conditions: (i) propargyl bromide, KOH, acetone, reflux, 16 h; (ii) ethyl chloroformate, NEt3, THF, 0 °C; (iii) ROH, catalyst, CDCl3, RT; (b) alcohol initiators and organic catalysts used in the ROP of MPC. | |
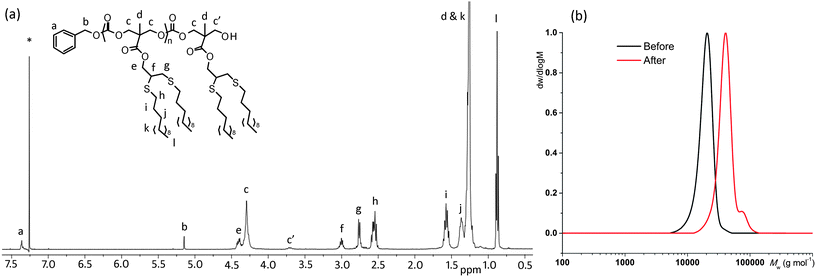
GPC analysis of the resultant BnO-PMPC100-OH polymers revealed a monomodal polymer chain distribution with a number-average molecular weight, Mn = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 540 g mol−1, close to that predicted from the [M]0/[I]0 ratio, and a low dispersity (Fig. 1a). Further investigation of the living characteristics of the polymerization catalyzed by TU and DBU revealed linear correlations between Mn against monomer conversion (Fig. 1b) and initial monomer-to-initiator ratio ([M]0/[I]0) (Fig. 1c) while retaining low dispersity values throughout the polymerization. Analysis of the polymers by 1H NMR spectroscopy revealed resonances expected for polymers with a benzyl alcohol end group (δ = 7.37 (ArH) and 5.15 (CH2) ppm) which demonstrates the end group fidelity of the polymerization (Fig. 2a). Further analysis of a low molecular weight polymer (DP = 12 by 1H NMR spectroscopy) by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF MS) revealed a single distribution with a spacing of 198 m/z, equal to that of a monomer unit, and a main peak at m/z = 2715 corresponding to a sodium charged polymer chain of DP13 with a benzyl alcohol end group; further confirming the living nature of the polymerization (Fig. 2b). Interestingly, 1H NMR spectroscopic analysis of polymers that were synthesized in CDCl3 showed a decrease in intensity of the propargyl proton resonance at δ = 2.50 ppm. MALDI-ToF MS analysis of the corresponding polymers revealed distributions with a repeat unit of ∼199 m/z instead of the expected 198 m/z and an isotope pattern that is consistent with ca. 20% proton-deuterium exchange of the acidic acetylenic proton with the solvent during polymerization (see ESI, Fig. S1†).
540 g mol−1, close to that predicted from the [M]0/[I]0 ratio, and a low dispersity (Fig. 1a). Further investigation of the living characteristics of the polymerization catalyzed by TU and DBU revealed linear correlations between Mn against monomer conversion (Fig. 1b) and initial monomer-to-initiator ratio ([M]0/[I]0) (Fig. 1c) while retaining low dispersity values throughout the polymerization. Analysis of the polymers by 1H NMR spectroscopy revealed resonances expected for polymers with a benzyl alcohol end group (δ = 7.37 (ArH) and 5.15 (CH2) ppm) which demonstrates the end group fidelity of the polymerization (Fig. 2a). Further analysis of a low molecular weight polymer (DP = 12 by 1H NMR spectroscopy) by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF MS) revealed a single distribution with a spacing of 198 m/z, equal to that of a monomer unit, and a main peak at m/z = 2715 corresponding to a sodium charged polymer chain of DP13 with a benzyl alcohol end group; further confirming the living nature of the polymerization (Fig. 2b). Interestingly, 1H NMR spectroscopic analysis of polymers that were synthesized in CDCl3 showed a decrease in intensity of the propargyl proton resonance at δ = 2.50 ppm. MALDI-ToF MS analysis of the corresponding polymers revealed distributions with a repeat unit of ∼199 m/z instead of the expected 198 m/z and an isotope pattern that is consistent with ca. 20% proton-deuterium exchange of the acidic acetylenic proton with the solvent during polymerization (see ESI, Fig. S1†).
![(a) GPC chromatogram of BnO-PMPC100-OH (Mn = 16 540 g mol−1, ĐM = 1.11) prepared by the ROP of MPC; (b) Plot of number-average molecular weight (Mn; ■) and dispersity (ĐM = Mw/Mn; □) against % monomer conversion in the ROP of MPC; [M]0/[I]0 = 45; (c) Plot of number-average molecular weight (Mn; ■) and dispersity (ĐM = Mw/Mn; □) against initial monomer-to-initiator ratio, [M]0/[I]0, in the ROP of MPC. Conditions: [MPC]0 = 0.5 M CDCl3 at 25 °C, 1 mol% DBU, 5 mol% TU, using benzyl alcohol as initiator.](/image/article/2013/PY/c2py20718d/c2py20718d-f1.gif) |
| | Fig. 1 (a) GPC chromatogram of BnO-PMPC100-OH (Mn = 16540 g mol−1, ĐM = 1.11) prepared by the ROP of MPC; (b) Plot of number-average molecular weight (Mn; ■) and dispersity (ĐM = Mw/Mn; □) against % monomer conversion in the ROP of MPC; [M]0/[I]0 = 45; (c) Plot of number-average molecular weight (Mn; ■) and dispersity (ĐM = Mw/Mn; □) against initial monomer-to-initiator ratio, [M]0/[I]0, in the ROP of MPC. Conditions: [MPC]0 = 0.5 M CDCl3 at 25 °C, 1 mol% DBU, 5 mol% TU, using benzyl alcohol as initiator. | |
![(a) 1H NMR spectrum (CDCl3 500 MHz, 298 K; * = residual CHCl3, ** = H2O) and (b) MALDI-ToF MS of PMPC (DP = 12) prepared by the ROP of MPC ([MPC]0 = 0.5 M) catalyzed by 1 mol% DBU and 5 mol% TU.](/image/article/2013/PY/c2py20718d/c2py20718d-f2.gif) |
| | Fig. 2 (a) 1H NMR spectrum (CDCl3 500 MHz, 298 K; * = residual CHCl3, ** = H2O) and (b) MALDI-ToF MS of PMPC (DP = 12) prepared by the ROP of MPC ([MPC]0 = 0.5 M) catalyzed by 1 mol% DBU and 5 mol% TU. | |
Further initiator versatility was demonstrated by the synthesis of amphiphilic block copolymers by initiation of MPC polymerization from commercially available poly(ethylene oxide) monomethyl ethers (Table 1, entries 3 and 4). In each case, chain growth was confirmed by GPC analysis in THF against poly(styrene) standards with the block copolymers revealing shifts to shorter retention times in both cases with narrow dispersities maintained. Furthermore, the synthesis of block copolymers of PMPC with poly(L-lactide) (PLLA) and PMAC further supported that all initiating sites remain active after formation of the first block (Table 1, entries 5–7). For example, the synthesis of a dual functional poly(carbonate) block copolymer was prepared by chain growth of PMAC from a PMPC block. The presence of both functional groups in the BnO-PMPC17-b-PMAC19-OH block copolymer was confirmed by the observation of the resonances at δ = 5.89, 5.33–5.23 and 4.63 ppm corresponding to the pendant allyl ester and at δ = 2.54 ppm for the acetylenic proton. GPC analysis revealed a shift to lower retention from Mn = 4010 g mol−1 for PMPC to Mn = 8110 g mol−1 for the block copolymer (see ESI, Fig. S2†). Such a block copolymer may be further functionalized using orthogonal chemistries.
| Entry |
Polymer |
DPb |
M
n (NMR)c (g mol−1) |
M
n (GPC)d (g mol−1) |
Đ
M
d
|
|
Reactions were performed in CDCl3 at 25 °C, [MPC]0 = 0.5 M, [M]0/[I]0 = 20 using 1 mol% DBU, 5 mol% TU.
Experimental degree of polymerization (DP) measured by 1H NMR spectroscopy per OH group.
Determined by 1H NMR spectroscopy.
Determined by GPC analysis in THF.
Poly(ethylene oxide) macroinitiator (DP = 114, determined as Mn = 6790 g mol−1, ĐM = 1.03 by GPC analysis in THF against poly(styrene) standards).
Poly(ethylene oxide) macroinitiator (DP = 114, determined as Mn = 14820 g mol−1, ĐM = 1.06 by GPC analysis in THF against poly(styrene) standards).
DP of PMPC block.
DP of PLLA block.
DP of PMAC block.
|
| 1 |
HO-PMPC-O-(C11H10NO3)-O-PMPC-OH |
20g |
8170 |
8100 |
1.08 |
| 2 |
HO-PMPC-O(CH2)2S-S(CH2)2O-PMPC-OH |
24g |
8480 |
8410 |
1.10 |
| 3 |
MeO-PEO114-b-PMPC-OHe |
18g |
8610 |
11020 |
1.06 |
| 4 |
MeO-PEO216-b-PMPC-OHf |
12g |
11930 |
17870 |
1.06 |
| 5 |
BnO-PLLA18-OH |
18h |
2700 |
4200 |
1.09 |
| 5a |
BnO-PLLA18-b-PMPC-OH |
20g |
6670 |
8740 |
1.08 |
| 6 |
BnO-PMPC20-OH |
20g |
4070 |
4540 |
1.12 |
| 6a |
BnO-PMPC20-b-PLLA-OH |
24h |
7530 |
10350 |
1.09 |
| 7 |
BnO-PMPC17-OH |
17g |
3480 |
4010 |
1.11 |
| 7a |
BnO-PMPC17-b-PMAC-OH |
19i |
5280 |
8110 |
1.09 |
Propargyl functional poly(carbonate)s were further functionalized by CuAAC (Scheme 2a) using conditions analogous to those reported for CuAAC functionalizations of (co)polymers from functional δ-valerolactone,41–43 ε-caprolactone44–46 and cyclic carbonate22 monomers. Functionalization of DP10, DP20 and DP72 PMPC homopolymers with 1-azidooctane using 0.4 equivalent CuI and 0.8 eq. diisopropylethylamine (DIPEA), led to >99% conversion of the propargyl groups to the octyl triazole being observed by 1H NMR spectroscopy as evidenced by the disappearance of the resonance attributed to the acetylenic proton at δ = 2.53 ppm and the appearance of new resonances consistent with the triazole proton at δ = 7.73 ppm and the octyl resonances at δ = 4.38, 1.92, 1.36–1.18 and 0.86 ppm (Fig. 3a). GPC analysis of the polymers revealed a shift to lower retention time while maintaining narrow distributions with dispersities similar to that of the unmodified PMPC (DP72, pre: Mn = 9350 g mol−1, ĐM = 1.11; post: Mn = 12740 g mol−1, ĐM = 1.15); Fig. 3b. Interestingly, analysis of the functionalized polymer by MALDI-ToF MS revealed two distributions, both with a repeat unit of 353 m/z. While the primary distribution displayed the expected m/z = 4725 that corresponds to a sodium charged, fully functionalized poly(carbonate) chain after the addition of 1-azidooctane with a DP of 13, the second distribution was observed with a copper counter ion in place of the expected sodium counter ion. This identification was confirmed by the preparation of the MALDI-ToF MS sample in the absence of added sodium trifluoroacetate as a cationization agent resulted in the observation of a single distribution with a repeat unit of 353 m/z and a main peak at m/z = 4765 corresponding to a copper charged, fully functionalized polymer chain of DP13 (see ESI, Fig. S3†).
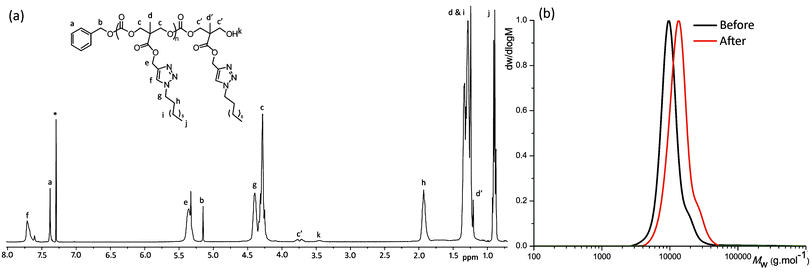 |
| | Fig. 3 (a) 1H NMR in CDCl3 of BnO-PMPC10-OH after functionalization with 1-azidooctane (400 MHz, 298 K; * = residual CHCl3); (b) GPC traces of BnO-PMAC72-OH before (Mn = 9350 g mol−1, ĐM = 1.11) and after post-polymerization functionalization with 1-azidooctane (Mn = 12740 g mol−1, ĐM = 1.15). | |
Complete removal of copper from the polymers was facilitated by stirring a solution of the functionalized polymer with Cuprisorb™ beads. MALDI-ToF analysis of polymers after this process in the presence of NaTFA cationization agent only revealed peaks attributed to sodium charged, fully functionalized poly(carbonate) chains (Fig. 4 and ESI, Fig. S3†).
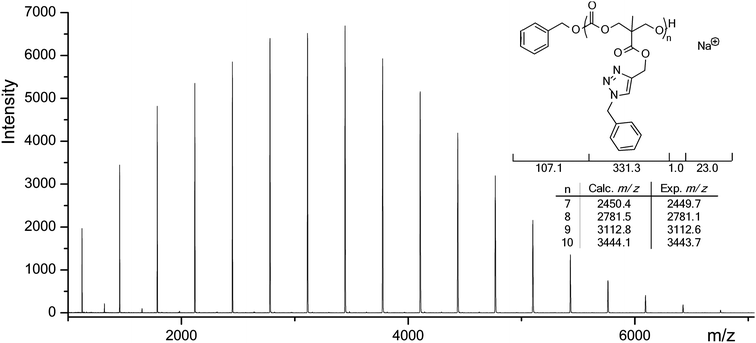 |
| | Fig. 4 MALDI-ToF MS of PMPC (DP = 12) after functionalization with benzyl azide. | |
In order to demonstrate the versatility of this system, additional functionalizations of PMPC homopolymers (DP10 and DP72) were performed with benzyl azide, 2-(2-(2-azidoethoxy)ethoxy)ethanol (TEG azide) and pro-fluorophore 3-azido-7-hydroxylcoumarin (Table 2). 1H NMR spectroscopic analysis of polymers modified with benzyl azide and TEG azide demonstrated that addition of the azide to the propargyl groups was occurring to >99% with data consistent with those expected for the modified side chain groups as well as a resonance corresponding to the triazole proton at δ = 7.6–7.8 ppm. Functionalizations with the profluorophore 3-azido-7-hydroxylcoumarin revealed 85% functionalization of the propargyl groups by 1H NMR spectroscopy and a shift to lower retention time by GPC. Incomplete conversion in this case was ascribed to the bulky nature of the functional group added, thereby sterically blocking the remaining propargyl groups. Interestingly, the GPC traces of polymers functionalized with benzyl azide and TEG azide showed only small shifts to lower retention time or a slight shift to higher retention time compared to that of PMPC (Table 2). These apparent small changes or decrease in Mn compared to the original polymers can be attributed to changes in their hydrodynamic volumes or Cu2+ complexation to the triazole upon functionalization via CuAAC.22 The small change in apparent Mn observed in the addition of benzyl azide is consistent with the observed small change in apparent Mn of allyl-functional poly(carbonate)s upon functionalization with benzyl mercaptan as reported in our previous work,16 and hence the effects of Cu2+ are considered to be minimal. MALDI-ToF MS analysis of the functionalized PMPC10 provided further proof of polymer functionalization with both benzyl azide and 3-azido-7-hydroxylcoumarin with the former showing only a single distribution confirming full functionalization (Fig. 4 and ESI, Fig. S4 and S5†). Strong fluorescence was observed upon irradiation of polymer solutions of 3-azido-7-hydroxylcoumarin functionalized polymers with a UV lamp at 365 nm as a result of the inversion of the 3(n,π*)–1(π–π*) excited states upon triazole formation.47 In contrast, solutions of the starting polymer, the azide or a mixture of both did not show any fluorescence upon irradiation (see ESI, Fig. S6†).
Table 2 CuAAC functionalization of poly(carbonates)a
| Azide |
M
n
b (g mol−1) |
Đ
M
b
|
|
[PMPC]72 = ca. 0.34 M in THF or DMF, 1.1 eq. azide, 25 °C, 24 h.
Measured by GPC analysis using.
THF as eluent.
DMF as eluent.
|
| — |
9350c/15700d |
1.11c/1.13d |
| 1-Azidooctane |
12740c |
1.15c |
| Benzyl azide |
9700c |
1.12c |
| TEG azide |
7000c |
1.21c |
| 3-Azido-7-hydroxylcoumarin |
13200d |
1.30d |
Functionalization of homopolymers by radical thiol-yne addition
Investigation of the post-polymerization functionalization of PMPC using radical ‘thiol-yne’ chemistry was undertaken as an alternative post-polymerization technique for the functionalization of the pendant propargyl esters in PMPC homopolymers (Scheme 2b).48,49 Optimizations of a thermally initiated model system with propargyl acetate (AIBN, dioxane, 90 °C, 24 h, [PMPC]0 = 0.15 M) revealed that 10 equivalents of thiol were required for full conversion to the doubly substituted product to occur. Under these conditions using 10 eq. of 1-dodecanethiol, polymer functionalizations led to complete disappearance of the acetylenic proton at δ = 2.53 ppm being observed by 1H NMR spectroscopic analysis, suggesting full conversion to the new polymer with repeat units substituted with two thiol molecules (Fig. 5a). However, while GPC analysis (Fig. 5b) of the post-polymerization functionalized polymer displayed a shift to lower retention time upon polymer modification, a high molecular weight shoulder was observed that most likely resulted from either coupling of the polymer chains due to the reactive nature of the radical alkynes and alkenes formed during the reaction in combination with their high local concentration,16,49 or from the amplification of water/self-initiated polymer chains that would be present at twice the molecular weight of the benzyl alcohol-initiated polymer. In order to eliminate the latter possibility, synthesis of a polymer initiated from 1,4-butanediol to yield a polymer possessing a homogeneous molecular weight distribution and its subsequent post-polymerization under ‘thiol-yne’ conditions with dodecanethiol was investigated. GPC analysis of the resulting polymeric species revealed the same high molecular weight shoulder, which indicates that this high molecular weight shoulder is indeed the product of coupling between polymer chains that occurs in the reaction. Several attempts to overcome these coupling reactions were attempted including increased thiol equivalence (up to 20 eq.), decreased [PMPC]0 (0.05 M) and milder UV-initiated functionalization using 1 wt% 2-benzyl-2-(dimethylamino)-4′-morpholinobutyrophenone photoinitiator however, consistent with the observations reported by Koo et al.,50 suppression of the coupling was not possible.
Despite its limitations, the radical ‘thiol-yne’ process was repeated for a range of thiol-containing molecules including those with alcohol groups which are incompatible with the investigated ring-opening polymerization (Table 3). 1H NMR spectroscopic analysis of the modified polymers all demonstrated that full conversion of the propargyl and allyl intermediates had occurred with data consistent with those expected for the modified side-chain groups. GPC analysis of the polymers revealed a shift to lower retention times with a shoulder due to cross-linking being observed in all cases. Further analysis using MALDI-ToF MS of DP10 polymers functionalized with 1-dodecanethiol and benzyl mercaptan revealed a main distribution consistent with full addition of two thiol molecules per repeat units in both cases. For example, functionalization with 1-dodecanethiol led to an increase of the repeat unit from 198 m/z to 603 m/z (+2 × 202 m/z) with a main peak corresponding to a sodium charged, fully functionalized polymer chain of DP4 at m/z = 2542 (Fig. 6). In both cases a second distribution was observed showing a difference of −135 Da in comparison to the main distribution; this is an apparent loss of a benzyl ester and was ascribed to mid-chain cleavage followed by loss of CO occurring during the ionization of the sample. The benzyl mercaptan-functional polymer displays a smaller third distribution corresponding to the loss of 4 benzyl mercaptan units from the polymer chain and is most likely due to reaction of two propargyl groups (see ESI, Fig. S8†).
Table 3 Radical ‘thiol-yne’ functionalization of BnO-PMPC72-OHa
| Thiol |
M
n
b (g mol−1) |
Đ
M
b
|
|
[PMPC]72 = ca. 0.15 M in dioxane, 10 eq. thiol, 25 °C, 24 h.
Measured by GPC analysis using.
THF as eluent.
DMF as eluent.
|
| — |
9350c/15700d |
1.11c/1.13d |
| 1-Dodecanethiol |
18140c |
1.17c |
| Benzyl mercaptan |
14020c |
1.21c |
| 1-Thioglycerol |
11550d |
1.57d |
| Mercaptoethanol |
24830d |
1.16d |
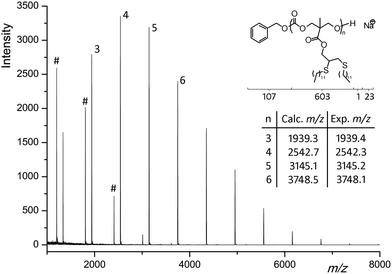 |
| | Fig. 6 MALDI-ToF-MS spectra of PMPC12 after post-polymerization functionalization with 1-dodecanethiol (secondary distribution – #). | |
Conclusions
In conclusion, the controlled organocatalytic ring-opening polymerization of 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one, MPC, was achieved using the dual 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (TU)/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) catalyst system. Polymers with predictable molecular weights (based on [M]0/[I]0) and narrow dispersities were able to be prepared with the polymerization occurring in a controlled manner. Notably, the post-polymerization functionalization of the pendant propargyl groups in the polymer backbone via CuAAC was shown to be highly efficient and occurring without observable polymer degradation leading to the synthesis of a range of new functional aliphatic poly(carbonate)s. Although the functionalization of pendant propargyl groups via the radical ‘thiol-yne’ reactions resulted in low levels of crosslinking being observed due to unavoidable side reactions of the reactive intermediates, reactions proceed with quantitative conversion of the propargyl groups and without degradation of the materials. The versatility of the functionalization of the pendant propargyl groups make these polymers scaffold essential materials for the facile preparation of new functional biodegradable and biocompatible materials and for the preparation of complex degradable materials for bioconjugation and drug delivery applications.
Experimental section
Materials
L-Lactide was purified from dry methylene chloride and sublimed twice before use and stored under inert atmosphere. Poly(ethylene oxide) methyl ethers were purchased from Sigma-Aldrich, dried in a desiccator over P2O5 and stored under inert atmosphere. CDCl3, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and (−)-sparteine were dried over CaH2, distilled, degassed, and stored under inert atmosphere. Benzyl alcohol was dried and stored over 4 Å molecular sieves under inert atmosphere. 2-Hydroxyethyl disulfide was purchased from Aldrich and dried and purified by distillation from CaH2. Methylene chloride was purified over an Innovative Technology SPS alumina solvent column and degassed before use. 5-Methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC) was synthesized as reported previously31,32 recrystallized several times before use, and dried 4 Å molecular sieves under inert atmosphere in dry methylene chloride. Silica gel (pore size = 40 Å) was obtained from Fisher Scientific and used as received. The thiourea catalyst (TU), and maleimide diol (2) were synthesized as reported previously,37,38,51 then dried over calcium hydride in dry tetrahydrofuran and recrystallized from dry methylene chloride. Azides were synthesized as previously reported.47,52 All other solvents and chemicals were obtained from Sigma-Aldrich or Fisher Scientific and used as received.
General considerations
Polymerizations were performed under inert atmosphere in a glovebox. Polymer functionalizations were carried out under oxygen-free conditions using standard Schlenk-line techniques. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX-300, DPX-400, DRX-500, AV II-600 or AV II-700 spectrometer at 293 K unless stated otherwise. Chemical shifts are reported as δ in parts per million (ppm) and referenced to the residual solvent signal (CDCl3: 1H, δ = 7.26 ppm; 13C, δ = 77.16 ppm; (CD3)2SO: 1H, δ = 2.50 ppm; 13C, δ = 39.52 ppm). Mass spectra were acquired by MALDI-ToF (matrix-assisted laser desorption ionization-time of flight) mass spectrometry using a Bruker Daltonics Ultraflex II MALDI-ToF mass spectrometer, equipped with a nitrogen laser delivering 2 ns laser pulses at 337 nm with positive ion ToF detection performed using an accelerating voltage of 25 kV. Solutions of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propylidene]malonitrile (DCTB) as a matrix (0.3 μL of a 10 g L−1 solution in methylene chloride or tetrahydrofuran), sodium trifluoroacetate as a cationisation agent (0.3 μL of a 10 g L−1 solution in methylene chloride or tetrahydrofuran), and analyte (0.3 μL of a 1 g L−1 solution in methylene chloride or tetrahydrofuran) were applied sequentially to the target followed by solvent evaporation to prepare a thin matrix/analyte film. The samples were measured in linear and reflectron ion mode and calibrated by comparison to 2 × 103 and 5 × 103 g mol−1 poly(ethylene oxide) monomethyl ether standards. Gel-permeation chromatography (GPC) was used to determine the molecular weights and polydispersities of the synthesized polymers. GPC in THF was conducted on a system composed of a Varian 390-LC-Multi detector suite fitted with differential refractive index (DRI), light scattering (LS), and ultraviolet (UV) detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 μM, 50 × 7.5 mm) and two mixed D columns (Varian Polymer Laboratories PLGel 5 μM, 300 × 7.5 mm). The mobile phase was either tetrahydrofuran eluent or tetrahydrofuran with 5% triethylamine eluent at a flow rate of 1.0 mL min−1, and samples were calibrated against Varian Polymer Laboratories Easi-Vials linear poly(styrene) standards (162 to 3.7 × 105 g mol−1) using Cirrus v3.3. GPC in DMF was conducted on a system composed of a Varian 390-LCMulti detector suite fitted with differential refractive index (DRI) and ultraviolet (UV) detectors equipped with a guard column (Varian Polymer PLGel 5 μM, 50 × 7.5 mm) and two mixed D columns (Varian Polymer Laboratories PLGel 5 μM, 300 × 7.5 mm). The mobile phase was DMF with 5% LiBr eluent at a flow rate of 1.0 mL min−1, and samples were calibrated against Varian Polymer Laboratories Easi-Vials linear poly(methyl methacrylate) standards (690 to 1.9 × 106 g mol−1) using Cirrus v3.3.
Typical procedure for the organocatalytic ROP of 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC)
A solution of MPC ([MPC]0 = 0.5 M) in dry CDCl3 or dry CH2Cl2 was added to a stirred solution of alcohol initiator, DBU (1 mol% to monomer), and 1-(3,5-bis(trifluoromethyl) phenyl)-3-cyclohexylthiourea (5 mol% to monomer) in the same solvent. After the desired time, the polymerizations were quenched by the addition of Amberlyst® 16 (acidic) ion exchange resin and precipitated into hexanes. Polymers with degrees of polymerization of 40 or higher were purified by repeated precipitation in hexanes, whereas 1-(3,5- bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea and DBU impurities were removed by column chromatography on silica gel in hexanes–ethyl acetate (4:1) for polymers with DP < 40. The polymer containing fractions were directly precipitated into hexanes. 1H NMR (600 MHz, CDCl3, 298 K): δ 7.37 (m, OBn–ArH), 5.15 (s, OBn–CH2), 4.73 (m, OCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) 4.44–4.24 (m, OC(O)OCH2), 3.73 (m, OC(O)OCH2), 2.53 (s, CH2CCH), 2.40 (br s, OH), 1.29 (s, CH3), 1.24 (s, 3H, C(CH3)CH2OH). 13C NMR (151 MHz, CDCl3): δ 171.5 (CC(O)O), 154.4 (OC(O)O), 135.1, 128.8, 128.6 (ArC), 75.7 (CH2CCH), 70.1 (CH2CCH), 68.6 (OC(O)OCH2), 64.7 (C(O)OCH2), 53.0 (CH2CCH), 46.7 (CCH3), 17.5 (CH3). GPC (THF, RI, DP = 72): Mn (ĐM) = 9350 g mol−1 (1.11). GPC (DMF, RI, DP = 72): Mn (ĐM) = 15700 g mol−1 (1.13).
CH) 4.44–4.24 (m, OC(O)OCH2), 3.73 (m, OC(O)OCH2), 2.53 (s, CH2CCH), 2.40 (br s, OH), 1.29 (s, CH3), 1.24 (s, 3H, C(CH3)CH2OH). 13C NMR (151 MHz, CDCl3): δ 171.5 (CC(O)O), 154.4 (OC(O)O), 135.1, 128.8, 128.6 (ArC), 75.7 (CH2CCH), 70.1 (CH2CCH), 68.6 (OC(O)OCH2), 64.7 (C(O)OCH2), 53.0 (CH2CCH), 46.7 (CCH3), 17.5 (CH3). GPC (THF, RI, DP = 72): Mn (ĐM) = 9350 g mol−1 (1.11). GPC (DMF, RI, DP = 72): Mn (ĐM) = 15700 g mol−1 (1.13).
Typical procedures for the synthesis of PMPC block copolymers
PEO-b-PMPC block copolymers were synthesized using the abovementioned method using commercially available poly(ethylene oxide) monomethyl ethers as initiator ([M]/[I] = 20). For the synthesis of PMPC-b-PLLA and PMPC-b-PMAC block copolymers, benzyl alcohol, DBU, and 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea were weighed and dissolved in dry CDCl3. MPC (or L-Lactide) was dissolved separately in CDCl3 and added to the initiator/catalyst solution. After 2 h (>90% conversion), a small aliquot was taken from the mixture and used for GPC analysis, and the reaction mixture was poured in a vial containing L-lactide (or MPC or MAC). The polymerization was carried out until >90% conversion was reached. The mixture was then quenched by addition of Amberlyst® 16 (acidic) ion exchange resin and precipitated in hexanes. MeO-PEO114-b-PMPC18-OH.1H NMR (400 MHz, CDCl3): δ 4.73 (OCH2CCH), 4.39–4.27 (OC(O)OCH2), 3.63 (OCH2CH2), 3.37 (OCH3), 2.53 (m, CCH), 1.28 (s, CH3). GPC (THF, RI): Mn (ĐM) = 11020 g mol−1 (1.06). MeO-PEO216-b-PMPC12-OH.1H NMR (400 MHz, CDCl3): δ 4.73 (OCH2CCH), 4.40–4.25 (OC(O)OCH2), 3.63 (OCH2CH2), 3.36 (OCH3), 2.50 (m, CCH), 1.28 (s, CH3). GPC (THF, RI): Mn (ĐM) = 17870 g mol−1 (1.06). BnO-PLLA18-b-PMPC20-OH.1H NMR (400 MHz, CDCl3): δ 7.35 (OBn–ArH), 5.21–5.11 (PLLA–CH and OBn–CH2), 4.72 (OCH2CCH), 4.45–4.24 (OC(O)OCH2), 3.74 (OC(O)CH2), 2.54 (C CH), 1.57 (PLLA–CH3), 1.29 (PMPC–CH3), 1.24 (C(CH3)CH2OH). GPC (THF, RI): Mn (ĐM) = 8740 g mol−1 (1.08). BnO-PMPC20-b-PLLA24-OH.1H NMR (400 MHz, CDCl3): δ 7.37 (OBn–ArH), 5.22–5.11 (PLLA–CH and OBn–CH2), 4.73 (OCH2CCH), 4.38–4.25 (OC(O)OCH2 and CHOH), 3.74 (OC(O)CH2), 2.54 (CCH), 1.58 (PLLA–CH3), 1.29 (PMPC–CH3). GPC (THF, RI): Mn (ĐM) = 10350 g mol−1 (1.09). BnO-PMPC17-b-PMAC19-OH.1H NMR (400 MHz, CDCl3): δ 7.37 (OBn–ArH), 5.89 (CHvinyl), 5.33–5.23 (CH2-vinyl), 5.15 (OBn–CH2), 4.73 (OCH2CCH), 4.63 (OCH2CHCH2), 4.45–4.27 (OC(O)OCH2), 3.72 (OC(O)CH2), 2.54 (CCH), 1.29 (PMPC–CH3), 1.27 (PMAC–CH3). GPC (THF, RI): Mn (ĐM) = 8110 g mol−1 (1.06).
Post-polymerisation functionalization of PMPC homopolymers via CuAAC
Stock solutions of polymer in dioxane (50 mg polymer mL−1) and stock solutions of diisopropylamine in THF or DMF were prepared and degassed prior to the reactions. In a typical experiment, an ampoule was charged with of polymer stock solution (1 mL of a solution of 50 mg polymer, 3.5 × 10−3 mmol), before the dioxane was removed in vacuo. Under a nitrogen atmosphere, CuI (19.1 mg, 0.10 mmol), azide (0.28 mmol) and degassed diisopropylamine stock solution (0.4 M) were added, and the ampoule stirred for 24 h. Amberlyst® 16 (acidic) ion exchange resin and THF or DMF were added and the mixture was stirred vigorously, then passed over a neutral alumina plug. The mixture was then stirred for 16 h with Cuprisorb™ beads to remove residual copper, filtered and concentrated in vacuo; the residue was dissolved in a minimal amount of methylene chloride and precipitated into hexanes. 1H NMR (600 MHz, CDCl3): δ 7.71 (br s, N3CHC), 7.35 (m, OBn–ArH), 5.27 (s, C(O)OCH2), 5.12 (s, OBn–CH2), 4.37 (m, NCH2), 4.30–4.22 (m, OC(O)OCH2), 1.91 (m, NCH2CH2(CH2)5CH3), 1.36–1.17 (m, N(CH2)2(CH2)5CH3 and CCH3), 0.86 (m, (CH2)7CH3). 13C NMR (151 MHz, CDCl3): δ 172.1 (CC(O)O), 154.4 (OC(O)O), 142.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN3), 131.7 (CHvinyl), 68.7 (OC(O)OCH2), 58.7 (OCH2CCHN3), 50.7 (N3CH2(CH2)6CH3), 46.7 (CCH3), 31.8, 30.4, 29.2, 29.1 and 26.6 ((CH2)5CH2CH3), 22.7 ((CH2)5CH2CH3), 17.5 (m, CH3), 14.2 ((CH2)7CH3)). GPC (THF, RI, DP = 72): Mn (ĐM) = 12690 g mol−1 (1.15).
CHN3), 131.7 (CHvinyl), 68.7 (OC(O)OCH2), 58.7 (OCH2CCHN3), 50.7 (N3CH2(CH2)6CH3), 46.7 (CCH3), 31.8, 30.4, 29.2, 29.1 and 26.6 ((CH2)5CH2CH3), 22.7 ((CH2)5CH2CH3), 17.5 (m, CH3), 14.2 ((CH2)7CH3)). GPC (THF, RI, DP = 72): Mn (ĐM) = 12690 g mol−1 (1.15).
Post-polymerization functionalization of PMPC homopolymers via radical ‘thiol-yne’
Stock solutions of polymer and AIBN (20%) in dioxane (50 mg polymer mL−1) and stock solutions of thiol in dioxane were prepared and degassed prior to the reactions. In a typical experiment, an ampoule was charged with 1 mL of polymer–AIBN stock solution, after which the dioxane was removed in vacuo. Under a nitrogen atmosphere, degassed 1-dodecanethiol stock solution was added, and the ampoule was placed in an oil bath at 90 °C and stirred for 24 h. The mixture was then concentrated in vacuo; the residue was dissolved in a minimal amount of methylene chloride and precipitated cold methanol. 1H NMR (600 MHz, CDCl3): δ 7.36 (m, OBn–ArH), 5.15 (s, OBn–CH2), 4.46–4.22 (m, OC(O)OCH2 and OCH2CH2CH2S), 3.70 (m, CH2OH), 3.00 (m, OCH2CHCH2S), 2.76 (m, OCH2CH2CH2S), 2.61–2.50 (m, SCH2(CH2)10CH3), 1.58 (m, SCH2CH2(CH2)9CH3), 1.37 (m, S(CH2)2CH2(CH2)8CH3), 1.32–1.19 (m, CCH3 and (CH2)8CH3), 0.88 (m, (CH2)8CH3). 13C NMR (151 MHz, CDCl3): δ 171.8 (CC(O)O), 154.5 (OC(O)O), 128.7, 128.5 (ArC), 68.9 (OC(O)OCH2), 65.9 (OCH2CHCH2S), 46.8 (CCH3), 44.6 (OCH2CHCH2S), 34.9 (OCH2CHCH2S), 33.4 (CHSCH2(CH2)10CH3), 32.1 (CH2SCH2(CH2)10CH3), 31.7 (CH2CH2CH3), 30.0, 29.9, 29.8, 29.7, 29.5, 29.1, 29.0 (CH2)8CH2CH2CH3), 22.8 (CH2CH2CH3), 17.6 (CCH3), 14.3 (S(CH2)11CH3). GPC (THF, RI, DP = 72): Mn (ĐM) = 18140 g mol−1 (1.17).
Acknowledgements
The Research Councils UK (RCUK) are acknowledged for funding a fellowship to A.P.D. We gratefully acknowledge financial support from EPSRC (EP/F068808/1) for funding a studentship (S.T.) and (EP/C007999/1) the purchase of the Bruker Ultraflex MALDI-ToF MS instrument. The GPC equipment used in this research was obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). CIRMAP is grateful to the “Belgian Federal Government Office Policy of Science (SSTC)” for general support in the frame of the PAI-6/27 and thanks the Belgian F.R.S.-FNRS in the frame of the ERA-Chemistry/FRFC 2.4.616.09.F project.
Notes and references
- F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Polym. Chem., 2011, 2, 528–533 RSC.
- W. Zhao, Y. Wang, X. Liu and D. Cui, Chem. Commun., 2012, 48, 4588–4590 RSC.
- M. J. Stanford and A. P. Dove, Macromolecules, 2008, 42, 141–147 CrossRef.
- H. Middleton, S. Tempelaar, D. M. Haddleton and A. P. Dove, Polym. Chem., 2011, 2, 595–600 RSC.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS.
- J. Lu and M. S. Shoichet, Macromolecules, 2010, 43, 4943–4953 CrossRef CAS.
- Y. Qiao, C. Yang, D. J. Coady, Z. Y. Ong, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 1146–1153 CrossRef CAS.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS.
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS.
- L. Mespouille, F. Nederberg, J. L. Hedrick and P. Dubois, Macromolecules, 2009, 42, 6319–6321 CrossRef CAS.
- R. J. Williams, R. K. O'Reilly and A. P. Dove, Polym. Chem., 2012, 3, 2156–2164 RSC.
- A. L. Z. Lee, S. Venkataraman, S. B. M. Sirat, S. Gao, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2012, 33, 1921–1928 CrossRef CAS.
- F. Suriano, R. Pratt, J. P. K. Tan, N. Wiradharma, A. Nelson, Y.-Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637–2645 CrossRef CAS.
- S. Tempelaar, L. Mespouille, P. Dubois and A. P. Dove, Macromolecules, 2011, 44, 2084–2091 CrossRef CAS.
- D. M. Stevens, S. Tempelaar, A. P. Dove and E. Harth, ACS Macro Lett., 2012, 915–918 CrossRef CAS.
- S. Onbulak, S. Tempelaar, R. J. Pounder, O. Gok, R. Sanyal, A. P. Dove and A. Sanyal, Macromolecules, 2012, 45, 1715–1722 CrossRef CAS.
- R. Wang, W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2011, 44, 6009–6016 CrossRef CAS.
- W. Chen, H. Yang, R. Wang, R. Cheng, F. Meng, W. Wei and Z. Zhong, Macromolecules, 2009, 43, 201–207 CrossRef.
- X. Zhang, Z. Zhong and R. Zhuo, Macromolecules, 2011, 44, 1755–1759 CrossRef CAS.
- J. Xu, F. Prifti and J. Song, Macromolecules, 2011, 44, 2660–2667 CrossRef CAS.
- L. B. Sessions, B. R. Cohen and R. B. Grubbs, Macromolecules, 2007, 40, 1926–1933 CrossRef CAS.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267–14273 CrossRef CAS.
- H. Abe, H. Kurokawa, Y. Chida and M. Inouye, J. Org. Chem., 2010, 76, 309–311 CrossRef.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- K. Stumpe, H. Komber and B. I. Voit, Macromol. Chem. Phys., 2006, 207, 1825–1833 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- C. Lu, Q. Shi, X. Chen, T. Lu, Z. Xie, X. Hu, J. Ma and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204–3217 CrossRef CAS.
- Q. Shi, X. Chen, T. Lu and X. Jing, Biomaterials, 2008, 29, 1118–1126 CrossRef CAS.
- Y. Han, Q. Shi, J. Hu, Q. Du, X. Chen and X. Jing, Macromol. Biosci., 2008, 8, 638–644 CrossRef CAS.
- Q. Shi, Y. Huang, X. Chen, M. Wu, J. Sun and X. Jing, Biomaterials, 2009, 30, 5077–5085 CrossRef CAS.
- D. J. Coady, A. C. Engler, H. W. Horn, K. M. Bajjuri, K. Fukushima, G. O. Jones, A. Nelson, J. E. Rice and J. L. Hedrick, ACS Macro Lett., 2011, 1, 19–22 CrossRef.
- These conditions (5 mol% 1 and 1 mol% DBU) also give excellent control in the ROP of MAC.
- K. M. Wiggins, J. A. Syrett, D. M. Haddleton and C. W. Bielawski, J. Am. Chem. Soc., 2011, 133, 7180–7189 CrossRef CAS.
- W. H. Heath, F. Palmieri, J. R. Adams, B. K. Long, J. Chute, T. W. Holcombe, S. Zieren, M. J. Truitt, J. L. White and C. G. Willson, Macromolecules, 2008, 41, 719–726 CrossRef CAS.
- M. J. Stanford, R. L. Pflughaupt and A. P. Dove, Macromolecules, 2010, 43, 6538–6541 CrossRef CAS.
- R. J. Pounder, M. J. Stanford, P. Brooks, S. P. Richards and A. P. Dove, Chem. Commun., 2008, 5158–5160 RSC.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS.
- B. M. Cooper and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7054–7065 CrossRef CAS.
- H. Misaka, R. Kakuchi, C. Zhang, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 5091–5096 CrossRef CAS.
- R. Riva, S. Schmeits, C. Jérôme, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 796–803 CrossRef CAS.
- H. Li, R. Riva, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 824–831 CrossRef CAS.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, Chem. Commun., 2006, 4010–4012 RSC.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2008, 42, 211–217 CrossRef.
- S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- M. H. Ngai, P.-Y. Yang, K. Liu, Y. Shen, M. R. Wenk, S. Q. Yao and M. J. Lear, Chem. Commun., 2010, 46, 8335–8337 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectral and chromatographic data of the (un)functionalized polymers. See DOI: 10.1039/c2py20718d |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.