 Open Access Article
Open Access ArticleAccumulation of the cyclobutane thymine dimer in defined sequences of free and nucleosomal DNA†
Amethist S.
Finch‡
a,
William B.
Davis
b and
Steven E.
Rokita§
*a
aDepartment of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA. E-mail: Rokita@jhu.edu
bSchool of Molecular Biosciences, Washington State University, Pullman, WA 99164, USA
First published on 17th June 2013
Abstract
Photochemical cyclobutane dimerization of adjacent thymines generates the major lesion in DNA caused by exposure to sunlight. Not all nucleotide sequences and structures are equally susceptible to this reaction or its potential to create mutations. Photostationary levels of the cyclobutane thymine dimer have now been quantified in homogenous samples of DNA reconstituted into nucleosome core particles to examine the basis for previous observations that such structures could induce a periodicity in dimer yield when libraries of heterogeneous sequences were used. Initial rate studies did not reveal a similar periodicity when a homogenous core particle was analyzed, but this approach examined only formation of this photochemically reversible cyclobutane dimer. Photostationary levels result from competition between dimerization and reversion and, as described in this study, still express none of the periodicity within two alternative core particles that was evident in heterogeneous samples. Such periodicity likely arises from only a limited set of sequences and structural environments that are not present in the homogeneous and well-characterized assemblies available to date.
Introduction
Covalent dimerization of adjacent thymines through a photochemical [2 + 2] reaction was first described over 50 years ago.1 The resulting thymine–thymine cyclobutane dimer product was soon identified as the major DNA lesion formed by UV irradiation of cells and was associated with the mutagenic and carcinogenic effects of solar irradiation.2,3 However, direct connection between thymine dimerization and photomutation is complicated by many factors including those controlling dimer accumulation and its repair (Scheme 1). Both the distribution of cyclobutane thymine dimers and the genetic changes caused by irradiation are heterogeneous within cellular DNA. A purely statistical correlation between frequency of dimerization, number of adjacent thymines, and mutation rate is not often observed. Instead, dimer formation and repair variably contribute to the mutational profiles. Each process is affected by nucleotide sequence, conformation and higher ordered structures.4–9 The biological consequences of a cyclobutane dimer within a particular sequence have been investigated by incorporating a dimer generated synthetically into defined polynucleotides.10–13 Likewise, relative efficiencies of thymine dimerization have been examined for sequence and structure dependence using oligo- and polynucleotides of defined sequence.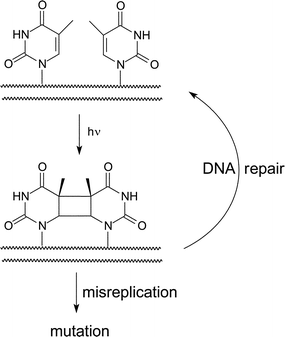 | ||
| Scheme 1 Formation and repair of the cyclobutane thymine dimer. | ||
The effect of neighboring sequences on thymine dimerization was evaluated in a number of laboratories by determining its initial rate of formation within various gene fragments. Although a consensus has not been achieved, thymine dimers are generally more prevalent when flanked by pyrimidines and adenines and less common when flanked by guanine.14–17 Single-stranded DNA also generates more cyclobutane thymine dimers than double-stranded DNA of equivalent sequence,18 and triplex formation further suppresses thymine dimerization in a helix.19,20 Along tracts of (dT)n/(dA)n, dimers seem to accumulate at the 3′-terminal dithymine when irradiated as a duplex in contrast to a uniform distribution of dimers that forms from single-stranded poly(dT).21 Photodimerization within heterogeneous sequences of DNA wrapped in nucleosome core particles (NCP) results in a fascinating periodicity of 10.3 bases and suggest maximum yields of these lesions are generated at sites for which the phosphodiester backbone is farthest from the histones.22–24 A similar distribution of cyclobutane dimers is not evident after irradiation of the same DNA in the absence of NCP. The periodicity of reaction in NCP has been attributed to the differential curvature of its DNA, and a similar periodicity has been observed after a heterogeneous collection of DNA duplexes was constrained by looping enforced by association of Lambda repressor.25
DNA dynamics, conformation and flexibility have all been implicated in the yield of cyclobutane thymine dimerization. For example, dimerization was suppressed near the dyad axis of the NCP, a region known to perturb the helical nature of duplex DNA.22 Model oligonucleotides variably describe the importance of context and suggest that particular conformers may be predisposed to efficient photochemical dimerization.26 Excited-state properties may equally control reaction by affecting the lifetime and population of electronic states required for dimerization.27–30 These properties are similarly dependent on sequence as are subsequent delocalization of excited states through energy and charge transfer.31–35 From a theoretical approach, the helical twist of DNA was proposed as most critical for dimerization.36
To date, the vast majority of studies have focused on the initial rate of cyclobutane dimer formation even though this process is reversible under standard model conditions that include use of the 254 nm emission from a low pressure mercury lamp.37–39 Moreover, the parent thymine pair is favored over its dimer under photostationary conditions.40 The net accumulation of dimers should then be influenced by the rate of dimerization and photoreversion, and together these may create sites known as hotspots for thymine dimerization. The efficiency of photoreversion, similar to photodimerization, is affected by the helical structure and local sequence surrounding the thymine pairs.41,42 This reversion has been described as a process of self-repair and was first observed at sites proximal to guanine quadruplexes.43 Electron donors and acceptors associated with DNA have also demonstrated the ability to repair thymine dimers.44–46 We have since returned to the topic of thymine dimerization from the perspective of its photostationary state, as reported here, to help examine the origins of its accumulation within nucleosomal DNA.
Experimental section
General materials
Chemicals were purchased from Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO) and were enzymatic grade or the highest grade available. All aqueous solutions were prepared with purified water (Barnstead NANOpure II purifier, 17.8 MΩ). Oligonucleotide primers were purified by polyacrylamide gel electrophoresis (PAGE) and purchased from IDT DNA (Coralville, IA) and Gibco Bioresearch Labs (Carlsbad, California). Washed pooled chicken blood (30%) in Alsevers buffer was purchased from Lampire Biological Laboratories (Pipersville, PA), frozen in liquid N2, and stored at −80 °C until use. Plasmid XP10 (pXP 10) containing the 5S RNA gene of Xenopus borealis was obtained from the late Dr A. Wolffe. Plasmid GEM-3z (pGEM) containing the 601 positioning sequence was obtained from the late Dr J. Widom.General methods
The 154 bp 5S gene was generated by standard PCR (100 μL) of pXP10 (100 pg) with primers 5′-AAT TCG AGC TCG CCC GGG GAT CCG-3′ and 5′-ACT AAC CAG GCC CGA CCC TGC TTC-3′ (50 pmol each) and Vent DNA polymerase (New England Biolabs, Ipswich, MA). Radiolabeled samples were generated analogously with 5′-[32P]-ACT AAC CAG GCC CGA CCC TGC TTC-3′. A 162 bp sequence containing the 601 sequence for nucleosome orientation was similarly generated by PCR of pGEM with primers NH2-(CH2)6-5′-TAT ACG CGG CCG CCC TGG-3′ and 5′-CAC AGG ATG TAT ATA TCT GAC AC-3′ as described previously.47 The presence of an aminolinker at one 5′-terminus allowed for selective radiolabeling of the other 5′-terminus with γ-[32P]-ATP and T4 polynucleotide kinase (New England Biolabs) following manufacturer protocols. Histone isolation and chromatin reconstitution to form the nucleosome core particles (NCP) followed published procedures32,48,49 and was confirmed by native PAGE (6%) on a MiniProtean3 apparatus (Bio-Rad, Hercules, CA).Irradiation of DNA and detection of the cyclobutane thymine dimer
DNA alone and reconstituted in the NCPs (200 kdpm) was irradiated in 10 mM potassium phosphate (pH 7.0) and 100 mM NaCl (10 μL) using a low pressure Hg pencil lamp (6035 Hg-Argon) (Oriel, Stratford, CT). The height of the pen lamp was varied from 6–11 cm to create a light flux from 0.016–0.16 mW cm−2 as needed. Histones were removed from NCP after irradiation by standard chloroform-phenol extraction, and DNA was treated with T4 endonuclease V (endo V) (EpiCentre, Madison, WI) according to manufacturer conditions at 37 °C for 5 h. Samples were analyzed by denaturing PAGE (8%) and quantified by phosphoimagery and ImageQuant software v.5.2 (GE Healthcare, Pittsburgh, PA).Results
The influence of helical structure on the propensity of duplex DNA to form cyclobutane thymine dimers was perhaps best demonstrated by the periodicity of its distribution within DNA bent around the core histones or looped between other DNA-binding proteins.22–25 A detailed correlation between local sequence, structure and dimer accumulation was not initially possible since the data represented a composite of reactivity for heterogeneous sequences bound to the NCP in numerous translational and rotational orientations. More detailed analysis requires a target of defined sequence that forms a uniform assembly. This was first accomplished with the relatively small gene encoding 5S rRNA that forms a NCP of homogeneous structure (Fig. 1).50 The same gene and its NCP were consequently selected to initiate the current study focusing on the photostationary levels of thymine dimer.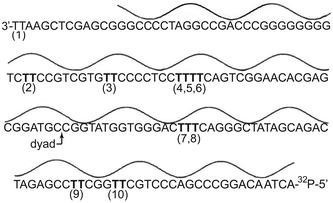 | ||
| Fig. 1 Translational and rotational orientation of the 5S rRNA gene (X. borealis) within the nucleosome core particle. Nucleobases are closest to the histone octamer at the minima and farthest at the maxima of the line above the DNA sequence. Sites of potential thymine dimerization are indicated as (1) through (10). | ||
The 5S gene (X. borealis) was irradiated using a mercury pen lamp (254 nm), and formation of the cyclobutane thymine dimer was detected by subsequent fragmentation of the DNA by T4 endonuclease V, an enzyme specific for this type of pyrimidine dimer.51 The resulting DNA was separated by denaturing polyacrylamide gel electrophoresis, and each fragment was quantified by phosphoimagery (Fig. 2). As expected, the major fragments increased with exposure to UV irradiation, and their gel mobility was consistent with the expected sites of thymine dimers. Irradiation was continued beyond that used for measuring initial rates in order to approach the photostationary levels of dimer accumulation. However, further irradiation degraded the parent DNA and its dimer-containing fragments non-selectively. DNA was also treated with heat and piperidine before and after the maximum irradiation time (30 min) to detect base labile products. The lack of significant fragmentation of the DNA under these conditions indicated that nonspecific degradation was not competitive with thymine dimerization. Analysis of the two sites closest to the 3′ terminus was not included in these investigations. Dimer 1 is beyond the region wrapped within the NCP, and the fragment size associated with dimerization at site 2 was obscured by a fraction of the parent sequence that migrated anomalously during electrophoresis.
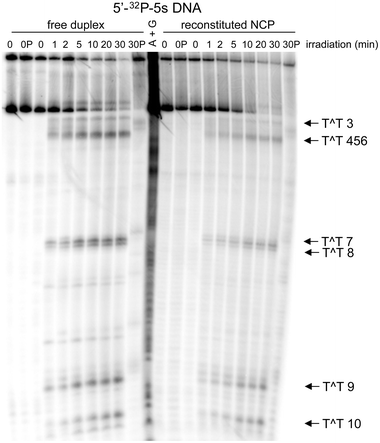 | ||
| Fig. 2 Formation of the cyclobutane thymine dimer within the 5S rRNA gene (X. borealis). Yields and distribution of the cyclobutane photoproduct were determined by strand fragmentation using T4 endonuclease V after irradiation, polyacrylamide gel electrophoresis (8%) and phosphoimage analysis. The radiolabeled 5S gene as the free duplex and reconstituted in a NCP as illustrated in Fig. 1 were analyzed after the indicated time of irradiation (254 nm). Equivalent samples before (OP) and after irradiation (30 min) (30P) were heated with piperidine (1 M) for 30 min at 90 °C. | ||
The collective rate of thymine dimerization and the total accumulation of thymine dimers within the 5S gene did not significantly vary in the presence or absence of the core histones or when reconstituted to form the NCP (Fig. 3). Under the experimental conditions, a photostationary level of ca. 0.5 cyclobutane dimer per duplex was established within 20 min of irradiation. The distribution of dimers among the individual sites was also monitored over time and was not affected by association of the DNA with the core histones (Fig. 4). Dimer 3 (Fig. 1) formed in lowest yield (ca. 4%) under all conditions and reached its photostationary state within 5 min. In contrast, dimer 9 accumulated to a yield of approximately 8% after 20 min. At this position, and to a lesser extent at position 7, more dimer forms in the NCP than in the free duplex. Additionally, a photostationary state may not be achieved after 30 min of irradiation, but the scatter in the data limits further interpretation. Regardless of this constraint, the results are still not reminiscent of the dramatic effect of the NCP on dimer distribution observed previously for a heterogeneous array of DNA sequences.22–25
![Time-dependent formation of the cyclobutane thymine dimer in the 5S rRNA gene. Total incorporation of cyclobutane dimers (T^T) within the 5S gene sequence was determined by the fraction of [32P]-label associated with the fragments representing dimerization relative to the total [32P]-label of each sample as quantified after separation (for example, see Fig. 2). Experimental details are described in the Methods Section for this DNA in the absence (■) and presence of the histone octamer (△) and after reconstitution to form NCP (□).](/image/article/2013/PP/c3pp50147g/c3pp50147g-f3.gif) | ||
| Fig. 3 Time-dependent formation of the cyclobutane thymine dimer in the 5S rRNA gene. Total incorporation of cyclobutane dimers (T^T) within the 5S gene sequence was determined by the fraction of [32P]-label associated with the fragments representing dimerization relative to the total [32P]-label of each sample as quantified after separation (for example, see Fig. 2). Experimental details are described in the Methods Section for this DNA in the absence (■) and presence of the histone octamer (△) and after reconstitution to form NCP (□). | ||
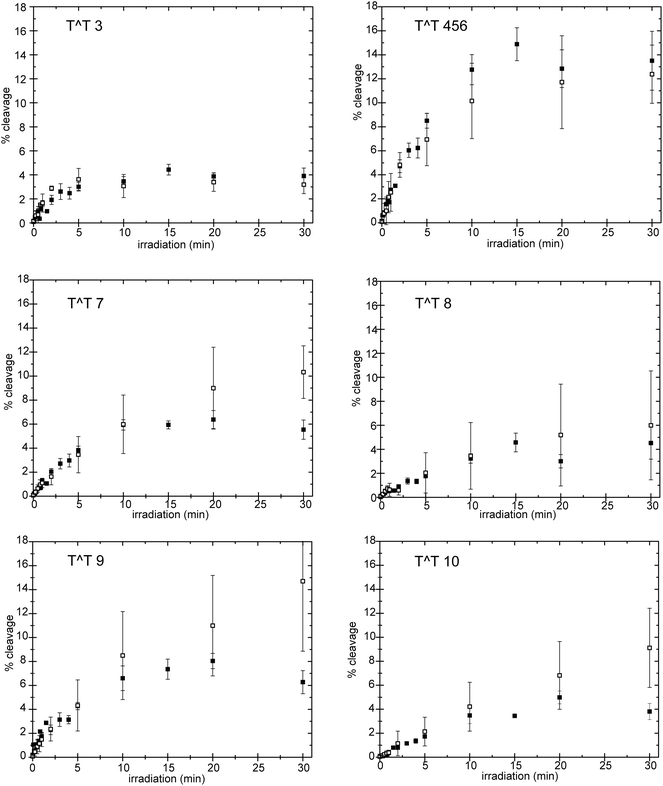 | ||
| Fig. 4 Accumulation of individual cyclobutane thymine dimers (T^T) within the 5S rRNA gene (X. borealis). Fragmentation at each potential cyclobutane dimer site in the absence (■) of the core histones and after reconstitution to form NCP (□) was monitored over time after endo V treatment and separation as illustrated in Fig. 2. Yields are presented relative to total DNA. Individual measurements were averaged when determined repeatedly and error bars indicate the experimental range of the data. Sites of dimerization are illustrated in Fig. 1. | ||
In contrast to the NCP formed collectively by the previous array of sequences, the NCP formed by the 5S gene alone provided only limited-TT-positions for detecting a possible correlation between duplex structure and photostationary levels of the cyclobutane thymine dimer. A second NCP based on a duplex sequence with stronger rotational and translational positioning than the 5S gene was next investigated to expand the range of sites available for dimer formation. The DNA used for this was discovered by the Widom laboratory as “sequence 601” through a systematic selection of strong nucleosome positioning sequences.52 Again, thymine–thymine sites are limited, but at least two (sites 2 and 5) represent regions in which the nucleobases are proximal to the histone octamer, and two (sites 1 and 6) represent regions in which the nucleobases are relatively distal to the histone octamer (Fig. 5).
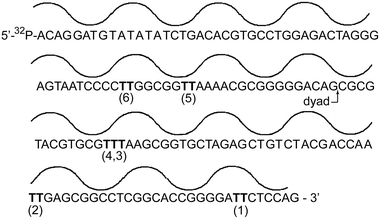 | ||
| Fig. 5 Translational and rotational orientation of the 601 DNA sequence within the nucleosome core particle. Nucleobases are closest to the histone octamer at the minima and farthest at the maxima of the line above the DNA sequence. Sites of potential thymine dimerization are indicated as (1) through (6). | ||
The free 601 sequence and its reconstituted NCP were irradiated under the same conditions as described above for the 5S gene. Again, no significant differences in the collective level of the cyclobutane thymine dimers were detected under photostationary conditions in the presence or absence of the associated histone octamer (Fig. 6). Both yielded ca. 0.4 dimer per duplex (Fig. 6). The short time needed to establish this yield versus that needed for the 5S gene was not a function of DNA sequence but rather the replacement of an aging lamp. As in the analysis of the 5S gene, the levels of dimer in the 601 sequence did not include data from position 1 due to the lack of resolution between its fragment formed by the endonuclease and the parent DNA after denaturing gel electrophoresis (ESI†). Further investigation to measure the dimerization of thymine at individual sites was not pursued since reconstitution to the NCP had no apparent effect on its overall yield. This represents the second example of an individual DNA sequence that did not replicate the variable formation of dimer in a heterogeneous population of sequences.
![Time-dependent formation of the cyclobutane thymine dimer in the 601 DNA sequence. Total incorporation of dimers (T^T) was determined by the fraction of [32P]-label associated with the fragments representing dimerization relative to the total [32P]-label of each sample as quantified after separation (see ESI, Fig. S1). Experimental details are described in the Methods Section for this DNA in the absence (■) of the histone octamer and after reconstitution to form NCP (□). The lines are added only to highlight the trends of dimer formation.](/image/article/2013/PP/c3pp50147g/c3pp50147g-f6.gif) | ||
| Fig. 6 Time-dependent formation of the cyclobutane thymine dimer in the 601 DNA sequence. Total incorporation of dimers (T^T) was determined by the fraction of [32P]-label associated with the fragments representing dimerization relative to the total [32P]-label of each sample as quantified after separation (see ESI, Fig. S1†). Experimental details are described in the Methods Section for this DNA in the absence (■) of the histone octamer and after reconstitution to form NCP (□). The lines are added only to highlight the trends of dimer formation. | ||
Discussion
The use of defined sequences of DNA was crucial when first measuring the influence of neighboring nucleobases on the accumulation of cyclobutane thymine dimers.14–17,41 In contrast, large populations of diverse sequences were necessary to detect the effects of conformational constraints on dimerization within duplex DNA such as those enforced by association with proteins.22–25 A previous attempt to identify the interplay between individual sequences and conformation did not recapitulate the earlier studies based on the heterogeneous mixture of sequences. No significant modulation was observed in the initial rates of thymine dimerization when measured in the 5S gene alone and after reconstitution with the core histone proteins.50 However, this kinetic approach focused on only half of the reversible processes. While dimer formation is dominated initially by the forward reaction, ultimately a photostationary level of dimer should form that reflects the relative efficiencies of reversion vs. formation. A second set of sequence and conformational variables combine to control thymine dimer reversion, and thus, photostationary levels of this dimer were determined as an alternative basis for the periodicity of dimerization in heterogeneous samples.After irradiation for 10–30 min, the population of cyclobutane dimers at most positions became constant as expected for a photostationary state (Fig. 3, 4 and 6). However, alternative explanations for the limited yield of dimer were possible. The data might also have been consistent with full consumption of a highly reactive subpopulation of DNA sequences or conformers. This possibility was minimized by selecting systems that form highly uniform NCP.50,52 The duplex DNA in the presence and absence of protein was also expected to be sufficiently dynamic over the time of irradiation to equilibrate between subtle differences in conformation. The accumulation of undetected photochemical products such as the Dewar and (6-4) dimers might have additionally suppressed full conversion to cyclobutane dimer, but this too was not likely. The dominant product generated by irradiation at 254 nm is the cyclobutane thymine–thymine dimer followed by its related thymine–cytosine dimer, the thymine–cytosine 6-4 product and the cyclobutane cytosine–thymine dimer.53 T4 endonuclease V was used to focus on the cyclobutane dimers since only these are recognized as substrates.54 Once again, the cyclobutane thymine–thymine dimer dominated the product profiles observed. Generation of competing products was checked with a complementary assay based on the 3′–5′ exonuclease activity of T4 DNA polymerase. This approach had previously been used to detect a broad array of DNA lesions including the thymine–cytosine 6-4 photoproduct.50,55,56 Again, the major photochemical product was dimerization at adjacent thymines to form the cyclobutane product (ESI†). Finally, the photostationary state necessitates a product level that can be achieved independently from any initial ratio of starting material and product. This property was confirmed using a DNA duplex of 47 base pairs that contained only a single set of adjacent thymines. Within 40 min of irradiation, cyclobutane thymine dimer yields remained a constant 5–10% whether starting from the parent duplex or its analog containing 100% dimer (ESI†). Consequently, the levels of dimer should reflect the composite variables affecting both its formation and reversion under the conditions of these experiments.
Neither the defined NCP generated with the 5S gene nor the 601 sequence significantly affected dimer levels as illustrated in Fig. 3, 4 and 6. Each defined duplex provided an individual set of thymine pairs in specific locations within the NCP. Such a narrow selection of DNA is a requirement for maintaining consistent translational and rotational orientation of the duplex, but this also severely restricts the variations available for screening. The two sequences of this investigation are negligible in number when compared to the 7 × 1019 possible sequences that were tested while surveying a randomized region of 33 nucleotides for the effect of DNA looping on thymine dimerization.25 Additionally, neither the 5S gene nor the 601 sequence offered potential thymine dimer sites near the dyad axis of NCP where distortion of the duplex is greatest and dimerization is significantly suppressed in the heterogeneous array of sequences.23,24
Only the most reactive sequences of a mixed population are characterized by product detection, and only a fraction of those might respond to structural variation in DNA by a confluence of a particular nucleotide sequence and its associated conformation, dynamics, excitation and relaxation. Access to productive geometries appears key to efficient cyclobutane dimerization as suggested by investigations using systems ranging from simple model oligonucleotides to complex protein DNA assemblies.22,25,57,58 Conversion of single-stranded to double-stranded DNA diminishes the available range of possible nucleobase orientations as well as restricts access to the most productive orientations for dimerization.18 Minor changes to the duplex structure then have the potential to shift the population towards conformers with an enhanced (or suppressed) ability to form cyclobutane thymine dimers as evident from their periodic increase and decrease in duplex DNA held in a loop.25
Neighboring sequences are known to modulate the efficiency of thymine dimerization and reversion as well, and the magnitude of their effect is dependent on structure or at least requires duplex formation to exert its influence.41,42 Little diversity is evident in the nucleotides neighboring the thymine pairs within the 5S gene (Fig. 1). Most are surrounded by pyrimidines. The 601 sequence contains more thymine pairs surrounded by purines (Fig. 5), but still the local sequence in these examples may only set their basal level of dimerization and not necessarily respond to structural perturbations. This is evident from the 5-fold difference in cyclobutane dimerization of thymines between complementary strands of a 175 base pair fragment of the 5S gene despite the similar number of thymine pairs in each strand.50 Still, these differences in reactivity were maintained whether the duplex was free in solution or associated with the core histones.
The lack of NCP-dependent variation in dimer accumulation for the 5S gene and 601 sequence may also reflect a minimal change in helix structure when free in solution versus assembled into a NCP. This possibility is consistent with previous studies based on circular dichroism of the 601 sequence.59 These sequences avidly bind and orient along the histone surface which implies a predisposition to adopt their protein-bound conformation even in the absence of protein. Even a pyrimidine tract inserted in the 5S gene at the dyad axis, the region of greatest distortion, exhibited only minimal change in thymine dimerization in the presence and absence of the NCP.50 Perhaps the weaker binding of other sequences to core histones indicates their need for a much greater distortion in conformational before accommodation into the NCP. The magnitude of these perturbations may then modulate the response of thymine dimerization to binding and looping around proteins.
The structural perturbations generated by cyclobutane thymine dimers had the additional potential to influence the net accumulation of product under photostationary conditions without affecting its initial rate of formation. The helical kink generated by a thymine dimer is not expected to be equally tolerated in all rotational and translational settings of the NCP and could therefore contribute to the periodic increase and decrease of thymine dimers. However, the presence of a dimer in the 5S gene only slightly reduced its affinity for histones relative to that of the undamaged gene as published previously.60 Destabilization by the dimer was also not sufficient to alter the orientation of this DNA with respect to the histones.50,61 Only the rate of DNA release from a NCP based on the 5S gene appeared to increase in the presence of the dimer.62 Thus, the insensitivity of the photostationary level of cyclobutane thymine dimer to NCPs formed by the 5S gene and 601 sequence is consistent with this lack of an observed reorganization or distortion around the dimer. Identifying the sequences that express the greatest range of photochemical reactivity when incorporated into a NCP will require a future strategy that combines the variable reactivity of heterogeneous populations of DNA with the structural details of individual constituents.
Acknowledgements
This research was supported in part by the National Science Foundation (SER) a SMART Fellowship from the Department of Defense (ASF), and a NSF CAREER Award (WBD). We thank Prof. John-Stephen Taylor and Dr Matthew Ray Holman for preparing and characterizing the duplex DNA containing a single cyclobutane thymine dimer within a defined sequence.References
- R. Beukers and W. Berends, Isolation and identification of the irradiation product of thymine, Biochim. Biophys. Acta, 1960, 41, 550–551 CrossRef CAS.
- Photochemistry and Photobiology of Nucleic Acids, ed. S. Y. Wang, Academic Press, New York, 1976 Search PubMed.
- J.-S. Taylor, Unraveling the molecular pathway from sunlight to skin cancer, Acc. Chem. Res., 1994, 27, 76–82 CrossRef CAS.
- D. E. Brash and W. A. Haseltine, UV-induced mutation hotspots occur at DNA damage hotspots, Nature, 1982, 298, 189–192 CrossRef CAS.
- P. A. Todd and B. W. Glickman, Mutational specificity of UV light in Escherichia coli: indications for a role of DNA secondary structure, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4123–4127 CrossRef CAS.
- S. Gao, R. Drouin and G. P. Holmquist, DNA repair rates mapped along the Human PGK1 gene at nucleotide resolution, Science, 1994, 263, 1438–1440 CAS.
- D. Chandrasekhar and B. Van Houten, High resolution mapping of UV-induced photoproducts in the Escherichia coli lacI gene, J. Mol. Biol., 1994, 238, 319–332 CrossRef CAS.
- S. Tornaletti and G. P. Pfeifer, Slow repair of pyrimidine dimers at P53 mutation hotspots in skin cancer, Science, 1994, 263, 1436–1438 CAS.
- R. Drouin and J.-P. Therrien, UVB-induced cyclobutane pyrimidine dimer frequency correlates with skin cancer mutational hotspots in p53, Photochem. Photobiol., 1997, 66, 719–726 CrossRef CAS.
- S. K. Banerjee, R. B. Christensen, C. W. Lawrence and J. E. LeClerc, Frequency and spectrum of mutations produced by a single cis–syn thymine–thymine cyclobutane dimer in a single-stranded vector, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 8141–8145 CrossRef CAS.
- C. A. Smith, M. Wang, N. Jiang, L. Che, X. Zhao and J.-S. Taylor, Mutation spectra of M13 vectors containing site-specific cis–syn, trans–syn-I, (6-4), and Dewar pyrimidone photoproducts of thymidylyl-(3′–5′)-thymidine in Escherichia coli under SOS conditions, Biochemistry, 1996, 35, 4146–4154 CrossRef CAS.
- I. Ensch-Simon, P. M. J. Burgers and J.-S. Taylor, Bypass of a site-specific cis–syn thymine dimer in a SV40 vector during in vitro replication by HeLa and XPV cell-free extracts, Biochemistry, 1998, 37, 8218–8226 CrossRef CAS.
- H. Kamiya, N. Murata, T. Murata, S. Iwai, A. Matsukage, C. Masutani, F. Hanaoka and E. Ohtsuka, Cyclobutane thymine dimers in a ras proto-oncogene hot spot activate the gene by point mutation, Nucleic Acids Res., 1993, 21, 2355–2361 CrossRef CAS.
- W. A. Haseltine, L. K. Gordon, C. P. Lindan, R. H. Grafstrom, N. L. Shaper and L. Grossman, Cleavage of pyrimidine dimers in specific DNA sequences by a pyrimidine dimer DNA-glycosylase of M. luteus, Nature, 1980, 285, 634–641 CrossRef CAS.
- F. Bourre, G. Renault, P. C. Seawell and A. Sarasin, Distribution of ultraviolet-induced lesions in Simian Virus 40 DNA, Biochimie, 1985, 67, 293–299 CrossRef CAS.
- D. L. Mitchell, J. Jen and J. E. Cleaver, Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation, Nucleic Acids Res., 1992, 20, 225–229 CrossRef CAS.
- J.-H. Yoon, C.-S. Lee, T. R. O'Connor, A. Yasui and G. P. Pfeifer, The DNA damage spectrum produced by simulated sunlight, J. Mol. Biol., 2000, 299, 681–693 CrossRef CAS.
- J. L. Hosszu and R. O. Rahn, Thymine dimer formation in DNA between 25 °C and 100 °C, Biochem. Biophys. Res. Commun., 1967, 29, 327–330 CrossRef CAS.
- M.-S. Tang, H. Htun, Y. Cheng and J. E. Dahlberg, Suppression of cyclobutane and <6-4> dipyrimidines formation in triple-stranded H-DNA, Biochemistry, 1991, 30, 7021–7026 CrossRef CAS.
- V. A. Malkov, V. N. Soyfer and M. D. Frank-Kamenetskii, Effect of intermolecular triplex formation on the yield of cyclobutane photodimers in DNA, Nucleic Acids Res., 1992, 20, 4889–4895 CrossRef CAS.
- V. Lyamichev, Unusual conformation of (dA)n·(dT)n-tracts as revealed by cyclobutane thymine–thymine dimer formation, Nucleic Acids Res., 1991, 19, 4491–4496 CrossRef CAS.
- J. M. Gale, K. A. Nissen and M. J. Smerdon, UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6644–6648 CrossRef CAS.
- J. M. Gale and M. J. Smerdon, Photofootprint of nucleosome core DNA in intact chromatin having different structural states, J. Mol. Biol., 1988, 204, 949–958 CrossRef CAS.
- J. R. Pehrson, Thymine dimer formation as a probe of the path of DNA in and between nucleosomes in intact chromatin, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9149–9153 CrossRef CAS.
- J. R. Pehrson and L. H. Cohen, Effects of DNA looping on pyrimidine dimer formation, Nucleic Acids Res., 1992, 20, 1321–1324 CrossRef CAS.
- S. Breeger, U. Hennecke and T. Carell, Excess electron-transfer-based repair of a cis–syn thymine dimer in DNA is not sequence dependent, J. Am. Chem. Soc., 2004, 126, 1302–1303 CrossRef CAS.
- C. E. Crespo-Hernández, B. Cohn and B. Kohler, Base stacking controls excite-state dynamics in A-T DNA, Nature, 2005, 436, 1141–1144 CrossRef.
- N. K. Schwalb and F. Temps, Base sequence and higher-order structure induce the complex excited-state dynamics in DNA, Science, 2008, 322, 243–245 CrossRef CAS.
- A. Banyasz, I. Vayá, P. Changenet-Barret, T. Gustavsson, T. Douki and D. Markovitsi, Base pairing enhances fluorescence and favors cyclobutane dimer formation induced upon absorption of UVA radiation by DNA, J. Am. Chem. Soc., 2011, 133, 5163–5165 CrossRef CAS.
- A. Banyasz, T. Douki, R. Improta, T. Gustavsson, D. Onidas, I. Vayá, M. Perron and D. Markovitsi, Electronic excited states responsible for dimer formation upon UV absorption directly by thymine strands: joint experimental and theoretical study, J. Am. Chem. Soc., 2012, 134, 14834–14845 CrossRef CAS.
- S. Delaney and J. K. Barton, Long-range DNA charge transport, J. Org. Chem., 2003, 68, 6475–6483 CrossRef CAS.
- C. C. Bjorklund and W. B. Davis, Attenuation of DNA charge transport by compaction into a nucleosome core particle, Nucleic Acids Res., 2006, 34, 1836–1846 CrossRef CAS.
- T. M. Nordlund, Sequence, structure and energy transfer in DNA, Photochem. Photobiol., 2007, 83, 625–636 CrossRef CAS.
- I. Buchvarov, Q. Wang, M. Raytchev, A. Trifonov and T. Fiebig, Electronic energy delocalization and dissipation in single- and double-stranded DNA, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4794–4797 CrossRef CAS.
- I. Vayá, T. Gustavsson, T. Douki, Y. Berlin and D. Markovitsi, Electronic excitation energy transfer between nucleobases of natural DNA, J. Am. Chem. Soc., 2012, 134, 11366–11368 CrossRef.
- Y. K. Law, J. Azadi, C. E. Crespo-Hernández, E. Olmon and B. Kohler, Predicting thymine dimerization yields from molecular dynamics simulations, Biophys. J., 2008, 94, 3590–3600 CrossRef CAS.
- E. Sztumpf and D. Shugar, Photochemistry of model oligo- and polynucleotides, Biochim. Biophys. Acta, 1962, 61, 555–566 CAS.
- H. E. Johns, M. L. Pearson, J. C. LeBlanc and C. W. Helleiner, The ultraviolet photochemistry of thymidylyl-(3′–>5′)-thymidine, J. Mol. Biol., 1964, 9, 503–524 CrossRef CAS.
- J. Cadet and P. Vigny, The photochemistry of nucleic acids, in Bioorganic Photochemistry: Photochemistry and the Nucleic Acids, ed. H. Morrison, John Wiley & Sons, New York, 1990, pp. 1–272 Search PubMed.
- D. L. Wulff, Kinetics of thymine photodimerization in DNA, Biophys. J., 1963, 3, 355–362 CrossRef CAS.
- M. R. Holman, T. Ito and S. E. Rokita, Self-repair of thymine dimer in duplex DNA, J. Am. Chem. Soc., 2007, 129, 6–7 CrossRef CAS.
- Z. Pan, J. Chen, W. J. Schreier, B. Kohler and F. D. Lewis, Thymine dimer photoreversal in purine-containing trinucleotides, J. Phys. Chem. B, 2012, 116, 698–704 CrossRef CAS.
- D. J.-F. Chinnapen and D. Sen, A deoxyribozyme that harnesses light to repair thymine dimers in DNA, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 65–69 CrossRef CAS.
- P. J. Dandliker, R. E. Holmlin and J. K. Barton, Oxidative thymine dimer repair in the DNA helix, Science, 1997, 275, 1465–1468 CrossRef CAS.
- A. Schwögler, L. T. Burgdorf and T. Carell, Self-repairing DNA based on a reductive electron transfer through the base stack, Angew. Chem., Int. Ed., 2000, 39, 3918–3920 CrossRef.
- K. V. Nguyen and C. J. Burrows, A prebiotic role for 8-oxoguanosine as a flavin mimic in pyrimidine dimer photorepair, J. Am. Chem. Soc., 2011, 133, 14586–14589 CrossRef CAS.
- C. C. Bjorklund and W. B. Davis, Stable DNA-protein cross-links are products of DNA charge transport in a nucleosome core particle, Biochemistry, 2007, 46, 10745–10755 CrossRef CAS.
- L. J. Libertini and E. W. Small, Salt-induced transitions of chromatin core particles studied by tyrosine fluorescence anisotropy, Nucleic Acids Res., 1980, 8, 3517–3534 CrossRef CAS.
- K. Luger, T. J. Rechsteiner and T. J. Richmond, Preparation of nucleosome core particle from recombinant histones, Methods Enzymol, 1999, 304, 3–19 CAS.
- X. Liu, D. B. Mann, C. Suquet, D. L. Springer and M. J. Smerdon, Ultraviolet damage and nucleosome folding of the 5S ribosomal RNA gene, Biochemistry, 2000, 39, 557–566 CrossRef CAS.
- A. K. McCullough, O. Schärer, G. L. Verdine and R. S. Lloyd, Structural determinants for specific recognition by T4 endonuclease V, J. Biol. Chem., 1996, 271, 32147–32152 CrossRef CAS.
- P. T. Lowary and J. Widom, New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning, J. Mol. Biol., 1998, 276, 19–42 CrossRef CAS.
- T. Douki and J. Cadet, Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions, Biochemistry, 2001, 40, 2495–2501 CrossRef CAS.
- L. K. Gordon and W. A. Haseltine, Comparison of the cleavage of pyrimidine dimers by the bacteriophage T4 and Micrococcus luteus UV-specific endonuclease, J. Biol. Chem., 1980, 255, 12047–12050 CAS.
- P. W. Doetsch, G. L. Chan and W. A. Haseltine, T4 DNA polymerase (3′–5′) exonuclease, an enzyme for the detection and quantitation of stable DNA lesions: the ultraviolet light examples, Nucleic Acids Res., 1985, 13, 3285–3304 CrossRef CAS.
- G. B. Panigrahi and I. G. Walker, The N2-guanine adduct but not the C8-guanine or N6-adenine adducts formed by 4-nitroquinoline 1-oxide blocks the 3′–5′ exonuclease action of T4 DNA polymerase, Biochemistry, 1990, 29, 2122–2126 CrossRef CAS.
- M. Hariharan and F. D. Lewis, Context-dependent photodimerization in isolated thymine–thymine steps in DNA, J. Am. Chem. Soc., 2008, 130, 11870–11871 CrossRef CAS.
- M. Hariharan, M. McCullagh, G. C. Schatz and F. D. Lewis, Conformational control of thymine photodimerization in single-strand and duplex DNA containing locked nucleic acid TT steps, J. Am. Chem. Soc., 2010, 132, 12856–12858 CrossRef CAS.
- W. B. Davis, C. C. Bjorklund and P. S. Cho, Hole transport dynamics in mixed sequence DNA can vary with salt concentration, J. Phys. Chem. C, 2010, 114, 20821–20833 CAS.
- D. B. Mann, D. L. Springer and M. J. Smerdon, DNA damage can alter the stability of nucleosomes: effects are dependent on damage type, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2215–2220 CrossRef CAS.
- Z. Svedružić, C. Wang, J. V. Kosmoski and M. J. Smerdon, Accommodation and repair of a UV photoproduct in DNA at different rotational settings on the nucleosome surface, J. Biol. Chem., 2005, 280, 40051–40057 CrossRef.
- M.-R. Duan and M. J. Smerdon, UV damage in DNA promotes nucleosome unwrapping, J. Biol. Chem., 2010, 285, 26295–26303 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1, denaturing polyacrylamide gel analysis of thymine dimerization within the 601 sequence alone and reconstituted into a nucleosome core particle; Fig. S2, photoproduct detection using the 3′–5′ exonuclease activity of T4 polymerase; Fig. S3, approach to a photostationary level of thymine dimer from duplex DNA alternatively containing a single thymine pair or its dimer analog. See DOI: 10.1039/c3pp50147g |
| ‡ Current address: US Army Research Laboratory, 2800 Powder Mill Road, Adelphi, MD 20783, USA. |
| § Current address: Department of Chemistry, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218, USA. E-mail: Rokita@jhu.edu |
| This journal is © The Royal Society of Chemistry and Owner Societies 2013 |