 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinyl sulfone-based ferrocenylation reagents: applications in conjugation and bioconjugation†
Alicia
Megia-Fernandez
,
Fernando
Hernandez-Mateo
and
Francisco
Santoyo-Gonzalez
*
Departamento de Q. Orgánica, Facultad de Ciencia, Instituto de Biotecnologia, Universidad de Granada, Granada, 18071 Spain. E-mail: fsantoyo@ugr.es; Fax: (+34)-958243186; Tel: (+34)-95824808
First published on 30th January 2013
Abstract
The easy vinyl sulfone derivatization of ferrocene allows the preparation of some effective, versatile and valuable ferrocenylation reagents. The applicability of such compounds in conjugation and bioconjugation of amine and/or thiol containing molecules and biomolecules through Michael-type addition under mild conditions that preserve the biological function of the latter is described. The feasibility of the methodology is demonstrated by the preparation of a variety of conjugates and bioconjugates (ferrocenyl terminated dendrimers and ferrocene–sugar, ferrocene–cyclodextrin, ferrocene–peptide and ferrocene–protein conjugates).
Introduction
Ferrocene (Fc) conjugation and bioconjugation1 has raised considerable interest in many fields owing to the outstanding properties of this prototypic metallocene compound. The well-established and rich chemistry of this sandwich structure allows rapid and easy access to a large number of reagents2 that have great possibilities in terms of organometallic functionalization and molecular engineering. In fact, Fc can be considered nowadays to be one of the most important structural motifs with applications in organometallic and bioorganometallic chemistry,3 electrochemistry,4 asymmetric catalysis,5 materials science,1,6 preparation of functional biomaterials1 and biomedical sciences.7 The academic milieu and also numerous industries (petroleum, plastics, textiles and metallurgy) have benefited from the outstanding characteristics of Fc. In addition to its unique structure, Fc has a panoply of ideal properties such as low price, stability under thermal, aqueous and aerobic conditions, together with a small size and a relative lipophilicity. Among these properties, the favorable electrochemical behavior of Fc is perhaps the most valuable and exploited characteristic that has been widely used in the design and construction of Fc-based systems such as redox molecular switches and sensors. These organometallic compounds have emerged as one of the most important classes of materials for the direct observation of molecular interactions and also as electron mediators, on the basis of their electrochemical detection capabilities.8 Particularly, ferrocenylated peptides and proteins are nowadays the most important ferrocenylated biomolecules with great significance in biological as well as non-biological systems. Among the uses and applications they have found, anticancer activity and cytotoxicity, antimalarial activity, antimycobacterial activity, antiproliferative activity, bacterial infection detector, detection of metal ions, drug carriers, nanobiosensors, nuclease activity, plasmin activity, proteolytic activity, antiviral potency for HIV-1, DNA detection, electrochemical assay of antigens, detection of proteins, synthesis of new water-soluble electrochemically active probes for biomolecules, detection of single-nucleotide polymorphisms, biochip device, DNA cleavage and gene detection have been reported.9,10In Fc-conjugates and bioconjugates, the Fc moiety present plays a variety of roles: molecular scaffold, sensitive probe, chromophore, biological marker, redox-active site, catalytic active site and others. To attain the goal of chemical incorporation of Fc into such systems, a large variety of mono- and disubstituted ferrocenyl derivatizing agents have been developed to perform their covalent conjugation by exploiting their reactivity with different functional groups.2 The majority of ferrocenylation reagents are applied to the conjugation to amino functionalities present in small organic molecules, amino acids, peptides or proteins, which correlates well with the strong demand for powerful analytical methods for the latter biomolecules.9 Traditional amide coupling is the most commonly used strategy for attaching Fc derivatives to amino functionalities either by means of Fc-carboxylic acid derivatives, with the assistance of coupling reagents, or by using activated Fc-carboxylic derivatives (chlorides, activated esters, and anhydrides).2,7b,9,10 In addition, Schiff base chemistry and Fc-derivatives bearing aldehyde,11 isothiocyanate2,9 or carboxyimidate groups12 have also been exploited to generate amino linked Fc-conjugates.
Iodoacetamide13 and maleimide derivatized ferrocenes14 have also been successfully coupled with compounds containing thiol groups, a function that is particularly important for its presence in cysteine residues, one of the most convenient targets for selective modification of proteins owing to the low natural abundance of this proteogenic amino acid. In addition, the alkylthiolation of glycosyl thiols with ferrocenemethanol derivatives has been described as an efficient methodology for the preparation of Fc–carbohydrate conjugates.15 Other functional groups (alcohols, diols, carboxylic acids, alkenes, dienes and imino groups) have been also used for the construction of Fc-conjugates and bioconjugates through well-established synthetic methodologies based on Fc reagents bearing reactive functionalities complementary to those of the target compounds. Furthermore, ferrocenylation has also benefited from the advances made in some contemporary organic reactions and methodologies, such as the Sonogashira and the Suzuki coupling and, particularly, click-chemistry.16
In this context, the excellent capability of vinyl sulfones (VS) to act as Michael acceptors toward amine and thiol groups has been used but not fully exploited up to the present in conjugation and bioconjugation, despite the stability and easy accessibility of this functional group by a broad variety of traditional synthetic methods and other contemporary reactions.17 In fact, VS are productive and widely used intermediates in organic synthesis that also have remarkable biomedical significance18 and have found applications in modern proteomics, particularly in the development of labeling and immobilization techniques.19 In addition, VS have also been used in the conjugation of proteins with other biomolecules to yield post-translational modifications.19 Attractive characteristics offered by the methodology based on the conjugate addition of nucleophiles to VS are the water stability of this function for extended periods, particularly at neutral pH where they are resistant to hydrolysis, the lack of by-products, the lack of need for organometallic catalysts, and the stability of the formed linkages.19 These remarkable characteristics are especially valuable to guarantee the integrity and biological functionality of labile biomolecules.
In some recent contributions, we have demonstrated the feasibility of VS in bioconjugation of proteins to tackle their fluorescent labeling, biotinylation and glycosylation, and also in the immobilization of proteins and carbohydrates to solid supports.20 In a proof of concept we have pointed out the synthetic possibilities of the VS function in ferrocenylation of proteins.20a Considering the wide potential of VS, we report herein the validity of the vinyl sulfone-based ferrocenylation as a general methodology for conjugation and bioconjugation of amine and/or thiol containing molecules and biomolecules.
Results and discussion
1. Synthesis of vinyl sulfone ferrocenylation reagents (Fc-VS)
In order to prove the possibilities of the VS function in the development of a general ferrocenylation methodology for the preparation of Fc-conjugates and bioconjugates, the vinyl sulfone derivatized ferrocenes (Fc-VS) {[2 (ferrocenylmethoxy)ethyl]sulfonyl}ethene 2 (Fc-O-VS)20a and ferrocenylmethyl{2-[2-(vinylsulfonyl)ethoxy]ethyl}sulfane 5 (Fc-SO-VS) were envisioned as valuable reagents (Scheme 1). | ||
| Scheme 1 Synthesis of vinyl sulfone ferrocenylation reagents. | ||
Both compounds are easily accessible from commercially available ferrocene methanol 1 and the rationale behind its design is the introduction of structural variability in the connecting tether between the cyclopentadienyl core and the VS group. In this way, the influence of this parameter on the electrochemical properties of the ferrocenylation reagents and, as a consequence, on the resulting ferrocenylated conjugates could be studied. As reported, Fc-O-VS220a is easily prepared by reaction of 1 with a two-fold molar excess of divinyl sulfone (DVS). By contrast, synthesis of the homologous compound 5 (Fc-SO-VS) is performed in a two-step strategy based on the preparation of the Fc sulfur 3 as a hydroxylated intermediate. Alkylthiolation of 1 with mercaptoethanol in acidic media of trifluoroacetic acid allows easy formation of sulfur derivative 3 that is obtained together with the corresponding acylated compound 4 as a minor product. VS derivatization of 3 was then performed in a similar way to that implemented for accessing compound 2, i.e. reaction with excess of DVS in basic media.
2. Synthesis of ferrocenyl-terminated dendrimers
The capabilities of the new ferrocenylation reagents for conjugation were first tested in the preparation of ferrocenyl-terminated dendrimers.21 These compounds are one of the early examples of metallodendrimers,22 a class of well-organized macromolecules that find multifaceted applications in the fields of molecular electronics21b and encapsulation, and also as sensors,23 reusable catalysts24 and drug carriers to targeted sites. On the basis of the interest of these dendrimers, TREN (tris(2-aminoethyl) amine) 6 was chosen as a model compound to act as the core for the construction of first generation TREN-based dendrimers functionalized in the boundary with Fc units. Michael-type addition of Fc-VS2 or 5 to the primary amino groups present in 6 allowed the easy and effective coupling of both compounds. The corresponding Fc-dendrimers 7 and 8 comprising three Fc derivatized branches departing from a central tertiary amine unit were obtained under mild conditions (rt and 18 h) (Scheme 2). Considering that after the conjugation the primary amine groups are transformed into secondary ones, which are also susceptible to react with VS, the reactions were next performed under reflux in the presence of an excess of 2 or 5 using an extended reaction time (72 h) to ensure the derivatization of the less reactive secondary amine functions. Under these conditions a compound containing five Fc moieties per molecule was isolated in high yields as the only detectable reaction product being identified as the Fc-dendrimer 9, as deduced by its spectroscopic data. Attempts to fully functionalize the remaining amino group by using longer reaction times under identical reaction conditions were unsuccessful, a fact that can be attributed to steric hindrance reasons. However, the formation of the N-acetylated derivative 10 in almost quantitative yield was possible. The isolation of this compound further confirms chemically the structure of the Fc-dendrimer 9 and also suggests the potential of the buried amino group of this multivalent ferrocenyl compound in coupling strategies directed towards its covalent linkage. In particular, azide derivatization of 9 by a classical two step one-pot procedure through its acylation with chloroacetic anhydride followed by treatment with sodium azide was easily performed. In this way, the azide Fc-dendrimer 11 was obtained in high yields, a transformation that endows click capabilities to those dendrimers to be exploited in any click-chemistry scenario.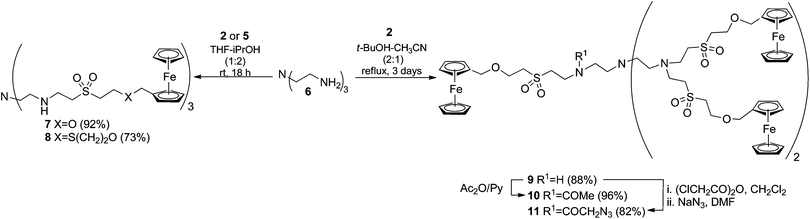 | ||
| Scheme 2 Synthesis of ferrocenyl-terminated dendrimers. | ||
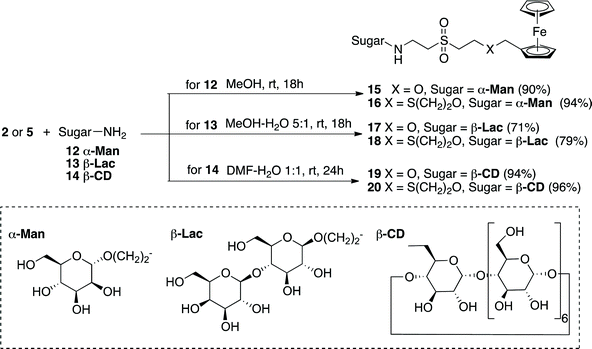
3. Synthesis of Fc–sugar and Fc–cyclodextrin conjugates
The evaluation of the vinyl sulfone-derivatization of Fc in bioconjugation was next undertaken. Amine-containing carbohydrates were first chosen for this aim considering that these are one of the most ubiquitous biomolecules. Decoration of Fc with sugar motifs has been widely used mainly with the purpose of conferring water solubility and biocompatibility on the resulting ferrocenyl carbohydrate hybrids. These principles are the basis of the panoply of applications that those compounds have found. Thus, Fc–sugar conjugates are attractive chiral auxiliaries for asymmetric synthesis owing to the multiple stereocenters present on the carbohydrate units.25 In addition, these conjugates have also been studied for their biological applications in the development of biosensors,26 for the electrochemical detection of carbohydrate–protein27 and carbohydrate–carbohydrate interactions,28 in the search for bacterial adhesion inhibitors29 and to explore their medicinal applications as therapeutics for iron-deficiency disorders30 and as antimalarial31 and anticancer agents.32 Conjugation between the carbohydrate and the ferrocene moieties has been usually performed by means of the different synthetic methodologies mentioned above, with a particular predominance of O, S and N-acylation reactions, giving rise to functionalized derivatives in which the Fc unit is mainly attached covalently to the anomeric position but also to other positions of the carbohydrate ring.With this background, the 2-aminoethyl-O-glycosides derived from the monosaccharide mannose 1233 and the disaccharide lactose 1334 were selected as model carbohydrates and coupled with the VS derivatized reagents 2 and 5. The reactions were performed again under mild conditions using pure or aqueous methanol as a solvent leading to the expected Fc–sugar conjugates 15–18 in a simple manner and with good to high yields (Scheme 3).
 | ||
| Scheme 3 Synthesis of Fc–sugar and Fc–cyclodextrin conjugates. | ||
In order to expand the good results obtained in the preparation of Fc–sugars, the ferrocenylation of cyclodextrins (CD) was next investigated taking into account both the oligosaccharidic nature and the importance of these cyclic compounds. CDs have been widely explored for their host–guest supramolecular encapsulation and carrier capabilities as well as for their scaffold role in the preparation of multivalent systems and artificial enzymes, among other applications. In particular, Fc–CD conjugates have been already prepared with sensing purposes in the construction of redox switches and sensors for organic guests in aqueous solution,35,36 artificial redox enzymes,37 mediators of enzyme-based biosensors38 and redox-controllable amphiphilic molecules.39 With this perspective, the easily accessible mono-6-amino-6-deoxy-β-CD 1440 was conjugated with the VS ferrocenylation reagents 2 and 5 using aqueous DMF as a solvent to facilitate the solubilization of the reagents and that of the resulting Fc–CD conjugates 19 and 20. In these reactions, the protocol of the reaction was slightly modified in order to simplify the isolation and purification procedure by the addition of aminopropyl silica41 to exploit the scavenger capabilities of this hybrid material to remove the excess of the VS reagents.
4. Synthesis of Fc–peptide and Fc–protein conjugates
The ferrocenylation of peptides was next investigated. In the current state of the art a wide variety of Fc–peptide hybrids having different architectures have been reported. These compounds are prepared with two main purposes: (i) the construction of chemical models of protein with secondary structures highly ordered where the Fc serves as a reliable organometallic scaffold and (ii) the preparation of platforms for the design of chemical and biological sensors where the peptide component serves as the recognition element whereas the Fc group acts as the redox probe.9,10b,c,42The intracellular tripeptide glutathione (GSH), composed of glutamine, cysteine and glycine, is an especially important compound owing to the role it plays in biological reactions. Thus, this compound is involved in the reduction of the disulfide bonds formed within cytoplasmic proteins to cysteines, a process in which GSH is converted into its oxidized form glutathione disulfide (GSSG).43 The labelling of GSH with Fc has been, up to the present, reported in a limited number of contributions13,44 both at the C-terminal, in order to keep the sulfhydryl group in the reduced state,44a,b and at the sulfhydryl group of the cysteine residue, by means of Fc-based selective labels.13,44c–f,45 Considering the simultaneous presence in GSH of two different heteroatomic nucleophiles (the thiol and the primary amino group), it was thought that this tripeptide is an ideal prototype to test the selective ferrocenylation by the Fc–VS reagents 2 and 5. On the basis of the different nucleophilicity of the thiol and amino group, it is generally assumed that VS are selective in the reactions with thiol groups relative to amino groups, provided that the reaction is not carried out at alkaline pH. This principle has been experimentally corroborated and is the rationale behind numerous chemoselective modifications of cysteine-containing peptides by vinyl sulfones.19
However, given the multifunctional character and complexity of proteins, the preference of VS for thiol groups should be considered with precaution, as recent findings have demonstrated.19 When GSH21 was reacted with 2 and 5 in the presence of NaBH4 in order to prevent its oxidation to GSSG, almost quantitative Fc labelling of GSH was observed and isolation of the corresponding structurally pure compounds 23 and 24 was performed. In these derivatives the Fc moiety is attached exclusively to GSH through the sulfur atom of the cysteine residue, but a participation of the primary amino group of the glutamic residue was not observed (Scheme 4). This finding is in accordance with the reported chemoselective conjugation capabilities of VS towards the β-sulfhydryl group of cysteine that were mentioned above. In spite of this result, the aza Michael-based Fc derivatization by means of reagents 2 and 5 was next assayed in GSSG22, taking advantage of the masking of the thiol group through the formation of a disulfide bridge. In this case, the conjugation of those reagents was also highly efficient and the N-linked Fc derivatives 25 and 26 were obtained (Scheme 4). This result proves the capabilities of the vinyl sulfone Fc reagents in the conjugation of amine-containing amino acids.
 | ||
| Scheme 4 Synthesis of Fc–GSH and Fc–GSSG conjugates. | ||
Having studied the conjugation of thiol and amine-containing peptides, we next addressed the Fc bioconjugation of proteins. The covalent attachment of Fc to proteins is a well-established post-translational modification of interest in chemical biology owing to the very well behaved redox chemistry of Fc that allows multiple applications particularly in the field of biological sensing.3,46 Among other archetypal proteins, bovine serum albumin (BSA) has been the object of ferrocenylation in different studies by means fundamentally of a variety of N-labelling reagents that exploit the high ratio of nucleophilic lysine residues present in this commercial protein.45,47 As indicated, we have recently communicated the ability of diverse proteins, including BSA, to react via the amine groups of the Lys and His residues with VS derivatized biological, functional and bifunctional tags including the vinyl sulfone Fc 2.20a The reported results indicate the validity of the VS-based labelling of proteins in mild conditions that preserve their biological functionality, conjugations that take place regardless of the isoelectric point, the number of potential nucleophiles, or the presence of free Cys residues in those biomolecules.
On the basis of the preliminary results on the ferrocenylation of BSA with 2, we now extended the bioconjugation studies to the reaction of BSA with 5 in order to study the plausible influence of the differences in the chemical structure of the connecting tether. In addition, the reaction was performed by using different Fc-VS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BSA stoichiometric ratios to also determine the effect of this parameter on the extent of labelling (Table 1). The reactions were performed by directly mixing the reagents in aqueous DMSO at room temperature, pH 8.5 and for reaction times of 24 h (Scheme 5). The Fc:BSA ratio of the resulting conjugates 27 (Fc-O-BSA)20a and 28 (Fc-SO-BSA) was determined through the evaluation of the BSA content using the Bradford method48 and simultaneously the Fe content by spectrophotometry using the method reported by Badia et al.47f
:BSA stoichiometric ratios to also determine the effect of this parameter on the extent of labelling (Table 1). The reactions were performed by directly mixing the reagents in aqueous DMSO at room temperature, pH 8.5 and for reaction times of 24 h (Scheme 5). The Fc:BSA ratio of the resulting conjugates 27 (Fc-O-BSA)20a and 28 (Fc-SO-BSA) was determined through the evaluation of the BSA content using the Bradford method48 and simultaneously the Fe content by spectrophotometry using the method reported by Badia et al.47f
 | ||
| Scheme 5 Synthesis of Fc–BSA conjugates. | ||
| Conjugate |
Fc-VS:BSA (mol) |
Fc/BSA (mol) (Experimental) |
|---|---|---|
| 27 (Fc-O-BSA)20a | 10:1 |
9.28 |
| 50:1 |
25.03 | |
| 100:1 |
28.75 | |
| 28 (Fc-SO-BSA) | 10:1 |
7.02 |
| 50:1 |
13.20 | |
| 100:1 |
17.00 |
The results showed that BSA is labeled very efficiently in all cases as demonstrated by the fact that an Fc-VS:BSA ratio as unfavourable as 10:1 yielded between 7 and 9 labels per molecule of BSA. In addition, it was also observed that the extent of labelling concomitantly increased as the Fc-VS:BSA ratio increased to reach a maximum that corresponded to less than 30 labels in the case of the Fc-BSA conjugate 27. However, a comparative analysis showed paradoxically that the ferrocenylation reagent 2 is more efficient than its homologous 5 in spite of the higher length of the connecting tether of this last compound that could be anticipated as a factor that should facilitate the accessibility to the reactive functional groups of BSA. This result can be tentatively attributed to the observed lower solubility in the reaction media of 5 with respect to that of 2. The resulting labeled proteins were analysed by MALDI-TOF (Fig. 1), which confirmed that the reaction takes place, and that variations of stoichiometry exert a clear direct effect on the number of labels coupled to BSA. Finally, a closer analysis of the overall results based on the existence in BSA of a single free Cys and 77 reactive groups demonstrates the good reactivity of amine groups present in proteins towards vinyl sulfones, and the existence of reactive groups that do not react, in accordance with our previous results.
 | ||
| Fig. 1 MALDI spectrum of Fc–BSA conjugates 27 (left) and 28 (right). (A) BSA; (B) Fc-VS:BSA 10:1; (C) Fc-VS:BSA 50:1; (D) Fc-VS:BSA 100:1. | ||
5. Electrochemical behaviour of ferrocene conjugates and bioconjugates
The electrochemical properties of Fc conjugates and bioconjugates 7–8, 15–20 and 23–26 were studied by cyclic voltammetry (CV), the experiments being carried out in water and/or acetonitrile solutions depending on their solubility. Thus, the cyclic voltammograms showed reversible redox couples of ferrocene/ferrocenium (see ESI, Fig. S1–S5†). The associated values of the half-wave potential (E1/2) for the oxidation of the Fc appendages and the diffusion coefficient (D0) are collected in Table 2. From these data two primary conclusions can be extracted. First, the E1/2 value of the Fc moiety is dependent on the nature of the tether decreasing on moving from the conjugates derived from 2 (conjugates 7, 15, 1719, 23 and 25) to those obtained in the coupling with 5 (conjugates 8, 16, 18, 20 and 24), in concordance with the higher electron-withdrawing nature of the oxygenated tether of the former with respect to the sulfur tether of the latter.35a Second, the E1/2 value is also determined by the solvent in such a way that superior values are observed when the less polar acetonitrile is used instead of water, an effect that can be attributed to the higher solvation of the ferrocenium ion in aqueous solution that facilitates oxidation of the iron atom.| Compound | E 1/2 (V) | D 0 × 106 (cm2 s−1) | ||
|---|---|---|---|---|
| H2O | CH3CN | H2O | CH3CN | |
| 1 | 0.222 | 0.376 | 11.7 | 27.0 |
| 2 | 0.253 | 0.411 | 8.6 | 6.6 |
| 5 | 0.232 | 0.395 | 6.0 | 7.2 |
| 7 | 0.421 | 6.0 | ||
| 8 | 0.403 | 9.4 | ||
| 15 | 0.262 | 0.416 | 7.5 | 30.3 |
| 16 | 0.246 | 0.404 | 3.7 | 7.7 |
| 17 | 0.262 | 0.410 | 4.5 | 18.3 |
| 18 | 0.236 | 0.399 | 5.2 | 26.4 |
| 19 | 0.368 | 1.2 | ||
| 20 | 0.390 | 1.0 | ||
| 23 | 0.249 | 4.7 | ||
| 24 | 0.218 | 5.8 | ||
| 25 | 0.251 | 7.1 | ||
On the other hand, it is also remarkable that the ferrocenyl-terminated dendrimers 7 and 8 show a unique wave in their voltammograms. This result indicates that all the Fc moieties present have identical half-wave oxidation potential and, therefore, the absence of any interaction between the different metal centers. In addition, for the ferrocene–sugar conjugates the carbohydrate moiety barely influences the E1/2, since no significant differences are observed for conjugates with the same tether and different carbohydrates (mannose and lactose). However, in the particular case of the Fc–cyclodextrin conjugates 19 and 20 the CV curve shows a positive shift in the redox potential that can be attributed to the inclusion of the ferrocene in the CD cavity and to a decomplexation process prior to the oxidation of the ferrocene, in concordance with an identical behaviour observed in similar conjugates.35a
The D0 values listed in Table 2 are consistent with the degree of substitution of Fc. According to the Stokes–Einstein equation, the D0 parameter is inversely proportional to the hydrodynamic radius of the molecule. Therefore, a higher molecular mass of the glycoconjugate is expected to diffuse more slowly. In accordance with that, cyclodextrin conjugates 19 and 20 show the lowest D0 values.
Conclusions
In summary, the reported results demonstrate that the vinyl sulfone functionalization of ferrocene derivatives allows the implementation of a general strategy to easily perform the ferrocenylation of amine and thiol containing molecules and biomolecules through their effective Michael-type addition. The preparation of the vinyl sulfone-based ferrocene reagents is straightforward. The mild conditions required are a remarkable characteristic particularly in the case of applications in bioconjugation as they preserve the biological functionalities of the labeled biomolecules. The developed methodology expands the repertoire of protocols available in conjugation and bioconjugation for the efficient electroactive labeling with Fc.Experimental
General methods
Unless otherwise noted, commercially available reagents and solvents were used as purchased without further purification. TLCs were performed on Silica Gel 60 F254 aluminium sheets. Reagents used for developing plates include ethanolic sulfuric acid (10% w/v), potassium permanganate (1% w/v) and UV light is used where applicable. Flash column chromatography was performed on Silica Gel (230–400 mesh, ASTM). Melting points are uncorrected. FAB mass spectra were recorded using m-nitrobenzyl alcohol or thioglycerol as a matrix. MALDI-TOF and MALDI mass spectra were recorded using HCCA as a matrix. Ferrocenyl(2-methoxyethyl)sulfonyl)ethene 2 (Fc-O-VS) was prepared following the procedure reported in the literature.20aSynthesis of vinyl sulfone ferrocenylation reagent 5
:5 to 1:2). Eluted first was compound 4 (71 mg, 9%) as a yellow syrup: νmax(film)/cm−1: 3093, 2923, 1785, 1221, 1151 and 821; 1H-NMR (CDCl3, 400 MHz): δ 4.40 (t, 2H, J = 6.9 Hz), 4.20–4.15 (m, 9H), 3.57 (s, 2H), 2.76 (t, 2H, J = 6.9 Hz); 13C-NMR (CDCl3, 100 MHz): δ 158.3, 157.7, 157.2, 156.6, 120.3, 116.6, 112.8, 109.0, 84.8, 69.3, 69.2, 68.8, 67.1, 32.4, 29.4. Eluted second was compound 3 (482 mg, 82%) as a yellow solid: νmax(film)/cm−1: 3421, 3093, 2921, 1703, 1410, 1041 and 819; 1H-NMR (CDCl3, 400 MHz): δ 4.18–4.14 (m, 9H), 3.66 (t, 2H, J = 6.0 Hz), 3.52 (s, 2H), 2.66 (t, 2H, J = 5.9 Hz), 2.22 (s, 1H); 13C-NMR (CDCl3, 100 MHz): δ 84.9, 68.8, 68.7, 68.2, 60.4, 35.0, 31.5. HRMS m/z (FAB+): calcd for C13H16OSFe [M]+ 276.0272, found 276.0271.
Synthesis of ferrocenylmethyl{2-[2-(vinylsulfonyl)ethoxy]ethyl]sulfane 5 (Fc-SO-VS)
t-BuOK (20 mg, 0.18 mmol) and DVS (0.27 mL, 2.71 mmol) were added to a solution of 3 (500 mg, 1.81 mmol) in THF (20 mL). After 1 h at rt, the solvent was evaporated under reduced pressure and the resulting crude product purified by column chromatography (EtOAc–hexane 1:7 to 1:1) giving compound 5 (600 mg, 84%) as a yellow syrup: νmax(film)/cm−1: 2921, 1639, 1314, 1126, 820 and 754; 1H-NMR (CDCl3, 400 MHz): δ 6.78 (dd, 1H, J = 16.0, 10.0 Hz), 6.39 (d, 1H, J = 16.4 Hz), 6.07 (d, 1H, J = 10.0 Hz), 4.24–4.13 (m, 9H), 3.84 (t, 2H, J = 5.6 Hz), 3.55 (t, 2H, J = 6.5 Hz), 3.52 (s, 2H), 3.22 (t, 2H, J = 5.6 Hz), 2.65 (t, 2H, J = 6.5 Hz); 13C-NMR (CDCl3, 75 MHz): δ 138.0, 128.9, 85.3, 70.6, 69.2, 69.0, 68.5, 64.5, 55.1, 32.4, 31.2; HRMS m/z (FAB+): calcd for C17H22O3S2Fe [M]+ 394.0357, found 394.0360.
Synthesis of ferrocenyl-terminated dendrimers
:1, 20 mL). The reaction mixture was magnetically stirred at rt for 18 h. Evaporation of the solvent under reduced pressure yielded a crude product that was purified by column chromatography giving the ferrocene–dendrimers conjugates 7–8.
Compound 7: Column chromatography (MeOH–CH2Cl2 1:20 to 1:10) gave 7 (330 mg, 96%) as an orange solid: νmax(KBr)/cm−1: 3420, 3096, 2922, 2852, 1638, 1463, 1312, 1124, 820 and 732; 1H-NMR (CDCl3, 300 MHz): δ 4.33 (s, 6H), 4.22 (t, 6H, J = 1.6 Hz), 4.16 (t, 6H, J = 1.6 Hz), 4.13 (s, 15H), 3.83 (t, 6H, J = 5.4 Hz), 3.34 (t, 6H, J = 6.1 Hz), 3.27 (t, 6H, J = 5.4 Hz), 3.12 (t, 6H, J = 6.2 Hz), 2.86 (brs, 3H), 2.64 (t, 6H, J = 5.0 Hz), 2.54 (t, 6H, J = 5.1 Hz); 13C-NMR (CDCl3, 75 MHz) δ 82.5, 70.0, 69.8, 69.0, 68.8, 63.5, 54.6, 54.0, 54.0, 47.2, 42.6; HRMS m/z (MALDI): calcd for C51H73N4O9S3Fe3 [M]+ 1149.2582, found 1149.2583.
Compound 8: Column chromatography (MeOH–CH2Cl2 1:10) gave 8 (291 mg, 73%) as an orange syrup: νmax(KBr)/cm−1: 3584, 3317, 3090, 2920, 2855, 1638, 1464, 1311, 1285, 1123, 1103, 820 and 734; 1H-NMR (CDCl3, 300 MHz): δ 4.18–4.12 (m, 27 H), 3.85 (t, 6 H, J = 5.4 Hz), 3.57 (t, 6H, J = 6.6 Hz), 3.55 (s, 6H), 3.44–3.40 (m, 6H), 3.34 (t, 6H, J = 5.4 Hz), 3.18 (t, 6H, J = 5.8 Hz), 2.74–2.70 (m, 6H), 2.66 (t, 6H, J = 6.6 Hz), 2.63–2.59 (m, 6H); 13C-NMR (CDCl3, 75 MHz): δ 85.0, 70.4, 68.9, 68.8, 68.2, 64.4, 54.4, 53.9, 53.9, 47.1, 42.5, 32.3, 31.3; HRMS m/z (MALDI): calcd for C57H85N4O9S6Fe3 [M]+ 1329.2679, found 1329.2684.
:1, 20 mL). The reaction mixture was magnetically stirred under reflux for 72 h. Evaporation of the solvent under reduced pressure yielded a crude product that was purified by column chromatography (CH2Cl2 to CH2Cl2–MeOH 20:1) giving the ferrocene–dendrimer conjugate 9 (221 mg, 88%) as a syrup: νmax(film)/cm−1: 3091, 2954, 2919, 2851, 1461, 1287, 1098, 820 and 734; 1H-NMR (CDCl3, 400 MHz): δ 4.33 (s, 10H), 4.25–4.10 (m, 45H), 3.81 (t, 10H, J = 5.3 Hz), 3.30–3.10 (m, 22H), 2.87 (t, 8H, J = 6.7 Hz), 2.59 (brs, 2H), 2.44 (s, 10H); 13C-NMR (CDCl3, 100 MHz): δ 82.4, 70.0, 69.9, 69.0, 68.8, 63.5, 54.7, 52.8, 51.9, 51.8, 46.9; HRMS m/z (MALDI): calcd for C81H108N4O15S5Fe5Na [M + Na]+ 1839.3043, found 1839.3054.
:30) to give 10 as a syrup (49 mg, 96%): νmax(film)/cm−1: 3091, 2923, 2854, 1640 and 1126; 1H-NMR (CDCl3, 500 MHz): δ 4.33 (s, 10H), 4.24–4.11 (m, 45H), 3.81 (t, 10H, J = 5.2 Hz), 3.69 (t, 2H, J = 6.6 Hz), 3.32 (t, 2H, J = 6.9 Hz), 3.27 (m, 2H), 3.21–3.11 (m, 16H), 2.86 (t, 8H, J = 6.4 Hz), 2.49–2.37 (m, 8H), 2.04 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 170.9, 82.1, 69.8, 69.7, 69.5, 68.8, 68.6, 68.5, 63.4, 63.3, 63.0, 54.5, 54.3, 52.8, 52.6, 51.6, 46.8, 46.7, 42.6, 40.3, 21.6; HRMS m/z (MALDI): calcd for C83H110N4O16S5Fe5Na [M + Na]+ 1881.3164, found 1881.3160.
:30) giving 11 (90 mg, 82%) as a syrup: νmax(film)/cm−1: 3091, 2923, 2858, 2104, 1655, 1288, 1126, 1097, 819 and 751; 1H-NMR (CDCl3, 400 MHz): δ 4.27 (s, 10H), 4.19–4.04 (m, 45H), 3.86 (s, 2H), 3.75 (t, 10H, J = 5.0 Hz), 3.66 (t, 2H, J = 6.4 Hz), 3.28 (t, 2H, J = 6.4 Hz), 3.20 (brs, 2H), 3.12 (t, 8H, J = 5.0 Hz), 3.06 (t, 8H, J = 6.4 Hz), 2.88 (brs, 2H), 2.83–2.76 (m, 10H), 2.42–2.29 (m, 8H); 13C-NMR (CDCl3, 100 MHz): δ 167.1, 81.1, 68.8, 68.7, 68.6, 67.9, 67.6, 62.4, 62.3, 62.0, 53.5, 53.4, 52.6, 51.8, 51.6, 51.3, 50.5, 50.4, 49.3, 45.7, 45.6, 39.5. HRMS m/z (MALDI): calcd for C83H109N7O16S5Fe5Na 1922.3181, found 1922.3179.
Synthesis of ferrocene–sugar conjugates
Compound 15: Column chromatography (CH3CN–H2O 10:1) gave 15 (250 mg, 90%) as a yellow syrup: [α]D +21.2 (c 0.5, MeOH); νmax(film)/cm−1: 3549, 2920, 1657, 1460 and 1045; 1H-NMR (CD3OD, 400 MHz): δ 4.76 (d, 1H, J = 1.5 Hz), 4.36 (s, 2H), 4.26 (t, 2H, J = 1.8 Hz), 4.18 (t, 2H, J = 1.8 Hz), 4.15 (s, 5H), 3.86–3.79 (m, 5H), 3.75–3.66 (m, 2H), 3.60 (t, 1H, J = 9.4 Hz), 3.54–3.50 (m, 2H), 3.36–3.32 (m, 4H), 3.11 (t, 2H, J = 6.7 Hz), 2.83–2.76 (m, 2H); 13C-NMR (CD3OD, 75 MHz): δ 101.8, 83.8, 74.8, 72.5, 72.0, 70.7, 70.6, 69.7, 69.5, 68.7, 67.3, 64.5, 62.9, 55.1, 54.7, 49.1, 43.2; HRMS m/z (MALDI): calcd for C23H35NO9SFeNa [M + Na]+ 580.1273, found 580.1274.
Compound 16: Column chromatography (CH3CN–H2O 5:1) gave 16 (290 mg, 94%) as a yellow syrup: [α]D +11.2 (c 0.5, MeOH); νmax(film)/cm−1: 3504, 2920, 1656, 1460, 1070 and 811; 1H-NMR (CD3OD, 500 MHz): δ 4.77 (d, 1H, J = 1.1 Hz), 4.20 (t, 2H, J = 1.6 Hz), 4.14 (s, 5H), 4.12 (t, 2H, J = 1.7 Hz), 3.87–3.81 (m, 5H), 3.74–3.68 (m, 2H), 3.63–3.56 (m, 5H), 3.56–3.50 (m, 2H), 3.40 (t, 2H, J = 6.8 Hz), 3.38 (t, 2H, J = 5.1 Hz), 3.12 (t, 2H, J = 6.6 Hz), 2.86–2.81 (m, 2H), 2.68 (t, 2H, J = 6.4 Hz); 13C-NMR (CD3OD, 100 MHz): δ 101.8, 86.6, 74.8, 72.6, 72.1, 71.4, 69.9, 69.8, 69.0, 68.7, 67.4, 65.6, 63.0, 55.1, 54.9, 49.3, 43.3, 32.9, 32.3; HRMS m/z (MALDI): calcd for C25H39NO9S2FeNa [M + Na]+ 640.1307, found 640.1308.
Compound
1717
: Column chromatography (CH3CN–H2O 5:1) gave 17 (255 mg, 71%) as a yellow syrup: νmax(KBr)/cm−1: 3407, 2924, 1653, 1291, 1121 and 619; 1H-NMR (DMSO-d6, 400 MHz): δ 4.36 (s, 2H), 4.35 (d, 1H, J = 8.7 Hz), 4.31 (d, 1H, J = 7.7 Hz), 4.26 (s, 2H), 4.18 (s, 2H), 4.15 (s, 4H), 3.95 (m, 1H), 3.91 (dd, 1H, J = 12.1, 2.3 Hz), 3.86–3.80 (m, 4H), 3.78 (dd, 1H, J = 11.4 and 7.5 Hz), 3.69 (dd, 2H, J = 11.3 and 4.4 Hz), 3.60–3.51 (m, 4H), 3.48 (dd, 1H, J = 9.7 and 3.2 Hz), 3.44–3.39 (m, 1H), 3.37–3.32 (m, 4H), 3.27 (d, 1H, J = 8.3 Hz), 3.11 (m, 2H), 2.82 (m, 2H); 13C-NMR (DMSO-d6, 100 MHz): δ 105.9, 105.1, 82.3, 81.4, 77.9, 77.3, 77.1, 75.6, 75.5, 73.4, 71.5, 71.1, 70.6, 70.4, 69.7, 65.2, 63.3, 62.7, 55.8, 55.2; HRMS m/z (MALDI): calcd for C29H45NO14SFeNa [M + Na]+ 742.1795, found 742.1802.
Compound 18: Column chromatography (CH3CN–H2O 5:1) gave 18 (308 mg, 79%) as a yellow syrup: νmax(film)/cm−1: 3381, 2925, 1654, 1284, 1123 and 619; 1H-NMR (DMSO-d6, 400 MHz): δ 5.04 (brs, 1H), 4.22–4.10 (m, 11H), 3.79 (m, 1H), 3.74 (t, 3H, J = 5.5 Hz), 3.61 (brs, 2H), 3.55 (s, 2H), 3.53–3.39 (m, 7H), 3.35–3.20 (m, 7H), 3.03 (t, 1H, J = 7.9 Hz), 2.98 (brs, 2H), 2.75 (brs, 2H), 2.60 (t, 2H, J = 6.7 Hz); 13C-NMR (DMSO-d6, 100 MHz): δ 103.8, 102.7, 85.0, 80.7, 75.4, 74.8, 74.5, 73.2, 73.1, 70.5, 69.7, 68.5, 68.4, 68.1, 67.6, 63.7, 60.5, 60.3, 53.2, 48.0, 41.9, 31.1, 30.4; HRMS m/z (MALDI): calcd for C31H50NO14S2Fe [M]+ 780.2016; found 780.2017.
Synthesis of ferrocene–cyclodextrin conjugates
:1, 10 mL). The reaction mixture was magnetically stirred at rt for 24 h. After this time, aminopropyl silica41 (40 mg) was added and the resulting suspension was heated at 60 °C until complete disappearance of the excess of 220a or 5 was observed by TLC (4 h). After filtration and washing of the filtrate with water (10 mL), evaporation of the resulting solution under reduced pressure was followed by lyophilization giving the corresponding ferrocene–cyclodextrin conjugates 19–20.
Compound 19: Isolated as a yellow solid (121 mg, 94%). Mp 191–192 °C; [α]D +86.6 (c 1, water); νmax(film)/cm−1: 3398, 2916, 1642, 1410, 1150, 1077 and 1026; 1H-NMR (DMSO-d6, 500 MHz) (selected signals): δ 4.91–4.79 (m, 7H), 4.24–4.08 (m, 11H); 13C-NMR (DMSO-d6, 100 MHz): δ 102.7, 102.6, 102.5, 82.4, 82.3, 82.2, 81.9, 81.7, 73.4, 73.2, 72.9, 72.6, 72.4, 69.7, 69.6, 68.7, 60.4, 60.3, 60.1, 55.9, 55.3, 53.4, 48.9; HRMS m/z (MALDI): calcd for C57H89NO37SFeNa [M + Na]+ 1490.4092, found 1490.4076.
Compound 20: Isolated as a yellow solid (129 mg, 96%): Mp 198–199 °C; [α]D +84.7 (c 1, water), νmax(KBr)/cm−1: 3361, 2925, 1654, 1384, 1155 and 1030; 1H-NMR (DMSO-d6, 400 MHz) (selected signals): δ 4.87 (s, 7H), 3.04 (m, 2H), 2.82 (t, 2H, J = 6.0 Hz), 2.69 (m, 2H), 2.40 (t, 2H, J = 6.0 Hz); 13C-NMR (DMSO-d6, 125 MHz): δ 103.0, 102.9, 102.7, 102.6, 102.3, 85.4, 84.6, 82.4, 82.3, 82.0, 81.9, 81.8, 79.4, 73.3, 73.2, 73.1, 72.8, 72.7, 72.6, 72.4, 71.6, 69.1, 68.9, 68.7, 68.6, 68.3, 68.1, 64.6, 60.3, 60.2, 60.1, 54.3, 53.9, 50.1, 42.7, 32.0, 30.2; HRMS m/z (MALDI): calcd for C59H93NO37S2FeNa [M + Na]+ 1550.4102, found 1550.4109.
Synthesis of ferrocene–GSH conjugates
:1, 15 mL) under magnetic stirring. The solution was kept at rt for 1 h. AcOH was then added to neutralize the excess of NaBH4. Evaporation and co-evaporation with toluene of the solvent under reduced pressure yielded a crude product that was purified by column chromatography to give the corresponding ferrocene–GSH conjugates 23–24.
Compound 23: Column chromatography (H2O–CH3CN 1:4) gave 23 (209 mg, 100%) as an orange solid: Mp 185–186 °C; [α]D −12.6 (c 1, water); νmax(film)/cm−1: 3420, 3094, 2924, 1651, 1542, 1397, 1315, 1118 and 668; 1H-NMR (D2O, 400 MHz): δ 4.50 (dd, 1H, J = 8.6, 5.0 Hz), 4.40–4.15 (m, 11H), 3.85 (t, 2H, J = 4.8 Hz), 3.72–3.66 (m, 3H), 3.42–3.31 (m, 4H), 2.95 (dd, 1H, J = 14.1, 4.9 Hz), 2.86 (dd, 2H, J = 9.0, 5.8 Hz), 2.77 (dd, 1H, J = 14.1, 8.8 Hz), 2.45 (t, 2H, J = 7.8 Hz), 2.07 (q, 2H, J = 7.3 Hz); 13C-NMR (D2O, 75 MHz): δ 177.0, 175.8, 174.9, 172.5, 82.8, 70.9, 70.3, 69.9, 63.5, 55.1, 54.9, 44.3, 34.0, 32.4, 27.2, 24.2; HRMS m/z (MALDI): calcd C25H35N3O9S2Fe [M]+ 641.1142, found 641.1164.
Compound 24: Column chromatography (H2O–CH3CN 1:4) gave 24 (224 mg, 98%) as an orange syrup: [α]D −13.4 (c 1, water); νmax(film)/cm−1: 3382, 3083, 2922, 1643, 1607, 1544, 1405, 1313, 1113 and 618 cm−1; 1H-NMR (DMSO-d6, 400 MHz): δ 4.50 (m, 1H), 4.18–4.09 (m, 9H), 3.74 (d, 2H, J = 4.5 Hz), 3.69–3.59 (m, 3H), 3.49 (s, 4H), 3.41 (t, 2H, J = 7.3 Hz), 3.32 (m, 2H), 2.99 (m, 2 H), 2.86 (t, 2H, J = 7.7 Hz), 2.84–2.75 (m, 2H), 2.59 (t, 2H, J = 5.1 Hz), .2.41 (t, 2H, J = 7.0 Hz), 2.04 (q, 2H, J = 6.9 Hz); 13C-NMR (D2O, 125 MHz): δ 177.0, 175.7, 175.0, 172.5, 86.4, 71.1, 70.3, 70.1, 69.5, 64.8, 55.3, 55.2, 54.3, 54.1, 44.6, 34.4, 32.8, 32.6, 32.1, 27.4, 24.7; HRMS m/z (MALDI): calcd C27H39N3O9S3FeNa [M + Na]+ 724.1099, found 724.1096.
Synthesis of ferrocene–GSSG conjugates
:1, 10 mL) under magnetic stirring. The solution was kept at rt for 18 h. Aminopropyl silica41 (150 mg) was then added and the suspension heated at 60 °C until complete disappearance of 2 or 5 was detected by TLC (4 h). Filtration and washing with water (10 mL) was followed by evaporation of the solvent giving the corresponding ferrocene–GSSG conjugates 25–26.
Compound 25: Isolated as a yellow syrup (208 mg, 100%): νmax(film)/cm−1: 3273, 3089, 2919, 1734, 1646, 1605, 1544, 1526, 1373, 1271, 1119 and 819; 1H-NMR (DMSO-d6, 400 MHz) (selected signals): δ 8.32 (t, 2H, J = 8.3 Hz), 8.24 (d, 2H, J = 8.3 Hz), 4.56 (m, 2H), 4.27–4.13 (m, 22H), 3.72 (t, 8H, J = 5.7 Hz), 2.25 (t, 4H, J = 6.0 Hz); 13C-NMR (DMSO-d6, 75 MHz): δ 172.0, 171.0, 170.2, 82.6, 69.1, 68.4, 68.2, 68.1, 62.5, 60.2, 52.9, 52.8, 51.8, 41.2, 40.1, 31.6, 27.5, 27.4; HRMS m/z (MALDI): calcd for C50H68Fe2N6O18S4 [M]+ 1280.210, found 1280.2166.
Compound 26: Isolated as a yellow syrup (224 mg, 100%). Mp 180–182 °C; νmax(film)/cm−1: 3285, 3092, 2923, 1649, 1608, 1548, 1529, 1306, 1103 and 822; 1H-NMR (CDCl3, 400 MHz) (selected signals): δ 5.23 (s, 4H), 4.13–4.03 (m, 18H), 3.79 (m, 8H), 2.60 (m, 4H), 2.30 (m, 8H), 1.34 (m, 8H), 1.19 (m, 8H); 13C-NMR (DMSO-d6, 75 MHz): δ 85.1, 69.6, 68.4, 67.5, 63.7, 60.1, 52.9, 52.7, 31.0, 30.2; HRMS m/z (MALDI): calcd for C54H76Fe2N6O18S6 [M + H] 1400.2262, found 1400.2234.
Cyclic voltammetry (CV) assays
CV experiments were carried out in nitrogen-purged (180 s) solutions with an instrument interfaced to a potentiostat connected to a computer running EcoChimie B.V. GPES 4.9 software. The working electrode was a platinum ring electrode (∅︀ 5 mm, effective area 0.410 ± 0.009 cm2). It was immersed in a H2SO4 50% v/v solution for 5 min and sonicated in MilliQ water for 10 min before each experiment. The counter electrode was a glassy carbon electrode (65 mm, ∅︀ 2 mm), which was immersed in a 0.1 M HNO3 solution for 5 min, carefully polished with a basic Al2O3–water slurry and sonicated in a H2O–MeOH–CH3CN 1:1:1 mixture at 40 °C for 10 min prior to use. An Ag/AgCl (3 M KCl) electrode was used as a reference. Electrochemical behaviour of each compound was studied on 0.2 mM solutions in MilliQ water or acetonitrile with 50 mM NaClO4 as a supporting electrolyte. Cyclic voltammograms were obtained measuring five scans at six sweep rates varying from 0.05 to 0.5 V s−1 with a step potential of 0.002 V. The diffusion coefficients were calculated by fitting the anodic peak current versus square root of the sweep rate plots to the Randles–Sevcik equation.
Ferrocene labelling of BSA. Synthesis of conjugates 27 (Fc-BSA(O)) and 28 (Fc-BSA(SO))
The ferrocenylation of BSA with 5 (conjugated Fc-BSA(SO) (28)) was undertaken by following the reported procedure for conjugated Fc-BSA(SO) (27):20a To three aliquots (2 mL) of a BSA solution (2 mg mL−1) in HEPES pH 8.5 was added 5 (0.24, 1.20 and 2.4 mg) dissolved in DMSO (0.2 mL) to get BSA:VS molar ratios of 1:10, 1:50 and 1:100, respectively. The samples were kept at rt for 24 h. After centrifugation, dialysis against phosphate buffer (85 mM, pH 7.0) was performed. The BSA concentration was determined by the Bradford method48 and the Fe content by spectrophotometry.47f An aliquot of each sample was dialyzed against distilled water prior to the MALDI analysis.
Acknowledgements
Financial support was provided by Ministerio de Ciencia e Innovacion (CTQ2011-29299-CO2-01).Notes and references
- Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Stepnicka, John Wiley & Sons Ltd., Chichester, UK, 2008 Search PubMed.
- B. Seiwert and U. Karst, Anal. Bioanal. Chem., 2008, 390, 181 CrossRef CAS.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931 CrossRef CAS.
- W. E. Geiger, Organometallics, 2007, 26, 5738 CrossRef CAS.
- Chiral Ferrocenes in Asymmetric Catalysis: Synthesis and Applications, ed. L.-X. Dai and X.-L. Hou, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- W. A. Amer, L. Wang, A. M. Amin, L. Ma and H. Yu, J. Inorg. Organomet. Polym. Mater., 2010, 20, 605 CrossRef CAS.
- (a) M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613 CrossRef CAS; (b) N. Metzler-Nolte, Top. Organomet. Chem., 2010, 32, 195 CrossRef CAS.
- X. Bin, P. M. Diakowski, K. Kerman and H.-B. Kraatz, in Electrochemisty of Functional Supramolecular Systems, ed. P. Ceroni, A. Credi and M. Venturi, John Wiley & Sons Inc., Hoboken, NJ, USA, 2010, p. 261 Search PubMed.
- S. Martic, M. Labib, P. O. Shipman and H.-B. Kraatz, Dalton Trans., 2011, 40, 7264 RSC.
- (a) B. Lal, A. Badshah, A. A. Altaf, N. Khan and S. Ullah, Appl. Organomet. Chem., 2011, 25, 843 CrossRef CAS; (b) T. Moriuchi and T. Hirao, Top. Organomet. Chem., 2006, 17, 143 CrossRef CAS; (c) H.-B. Kraatz, J. Inorg. Organomet. Polym. Mater., 2005, 15, 83 CrossRef CAS.
- C. Xu, H. Cai, P. He and Y. Fang, Fresenius’ J. Anal. Chem., 2000, 367, 593 CrossRef CAS.
- H. Falk, M. Peterlik and K. Schloegl, Monatsh. Chem., 1969, 100, 787 CrossRef CAS.
- K. Kam-Wing Lo, J. Shing-Yip Lau, D. Chun-Ming Ng and N. Zhu, J. Chem. Soc., Dalton Trans., 2002, 1753 RSC.
- (a) Y. Wang, X. Zhu, Z. Hu, J. Wang and F. Zhou, Electroanalysis, 2005, 17, 2163 CrossRef CAS; (b) K. D. Gleria, H. A. O. Hill and L. L. Wong, FEBS Lett., 1996, 390, 142 CrossRef.
- J. M. Casas-Solvas, E. Ortiz-Salmerón, J. J. Giménez-Martínez, L. García-Fuentes, L. F. Capitán-Vallvey, F. Santoyo-González and A. Vargas-Berenguel, Chem.–Eur. J., 2009, 15, 710 CrossRef CAS.
- V. Ganesh, V. S. Sudhir, T. Kundu and S. Chandrasekaran, Chem.–Asian J., 2011, 6, 2670 CrossRef CAS.
- (a) N. S. Simpkins, Tetrahedron, 1990, 46, 6951 CrossRef CAS; (b) I. Forristal, J. Sulfur Chem., 2005, 26, 163 CrossRef CAS.
- D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 793 CrossRef CAS.
- F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, in Integrative Proteomics, ed. H.-C. Eastwood Leung, InTech, 2012, 301–327 DOI:10.5772/29682.
- (a) J. Morales-Sanfrutos, J. Lopez-Jaramillo, M. Ortega-Muñoz, A. Megia-Fernandez, F. Perez-Balderas, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2010, 8, 667 RSC; (b) J. Morales-Sanfrutos, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2010, 75, 4039 CrossRef CAS; (c) M. Ortega-Muñoz, J. Morales-Sanfrutos, A. Megia-Fernandez, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Mater. Chem., 2010, 20, 7189 RSC.
- (a) D. Astruc, New J. Chem., 2011, 35, 764 RSC; (b) D. Astruc, C. T. Ornelas and J. Ruiz, Acc. Chem. Res., 2008, 41, 841 CrossRef CAS.
- G. R. Newkome, E. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689 CrossRef CAS.
- C. M. Casado, B. Alonso, J. Losada and M. P. Garcia-Armada, in Des. Dendrimers, ed. S. Campagna, P. Ceroni and F. Puntoriero, John Wiley & Sons, Inc., Hoboken, N. J, 2012, p. 219 Search PubMed.
- A. Berger, R. J. M. K. Gebbink and G. van Koten, Top. Organomet. Chem., 2006, 20, 1 CrossRef CAS.
- (a) C. G. Hartinger, A. A. Nazarov, M. Galanski, M. Reithofer and B. K. Keppler, J. Organomet. Chem., 2005, 690, 3301 CrossRef CAS; (b) A. A. Nazarov, C. G. Hartinger, V. B. Arion, G. Giester and B. K. Keppler, Tetrahedron, 2002, 58, 8489 CrossRef CAS; (c) A. Albinati, P. S. Pregosin and K. Wick, Organometallics, 1996, 15, 2419 CrossRef CAS.
- (a) M. H. Chahma, J. S. Lee and H.-B. Kraatz, J. Organomet. Chem., 2002, 648, 81 CrossRef CAS; (b) P. R. Ashton, V. Balzani, M. Clemente-Leon, B. Colonna, A. Credi, N. Jayaraman, F. M. Raymo, J. F. Stoddart and M. Venturi, Chem.–Eur. J., 2002, 8, 673 CrossRef CAS; (c) M. Takeuchi, T. Mizuno, S. Shinkai, S. Shirakami and T. Itoh, Tetrahedron: Asymmetry, 2000, 11, 3311 CrossRef CAS.
- (a) J. M. Casas-Solvas, E. Ortiz-Salmeron, L. Garcia-Fuentes and A. Vargas-Berenguel, Org. Biomol. Chem., 2008, 6, 4230 RSC; (b) J. L. Kerr, J. S. Landells, D. S. Larsen, B. H. Robinson and J. Simpson, J. Chem. Soc., Dalton Trans., 2000, 1411 RSC.
- A. Otsuka, K. Sakurai and T. Hasegawa, Chem. Commun., 2009, 5442 RSC.
- (a) M. Kovacevic, L. Barisic, R. Ribic, V. P. Perokovic, S. Tomic and V. Rapic, Appl. Organomet. Chem., 2012, 26, 74 CrossRef CAS; (b) E. Szanti-Pinter, J. Balogh, Z. Csok, L. Kollar, A. Goemoery and R. Skoda-Foeldes, Steroids, 2011, 76, 1377 CAS.
- Y. Chen, G.-f. Han and H.-y. Liu, Jiangsu Gongye Xueyuan Xuebao, 2004, 16, 8 CAS.
- (a) C. Herrmann, P. F. Salas, B. O. Patrick, C. d. Kock, P. J. Smith, M. J. Adam and C. Orvig, Organometallics, 2012, 5748 CrossRef CAS; (b) C. Herrmann, P. F. Salas, J. F. Cawthray, C. de Kock, B. O. Patrick, P. J. Smith, M. J. Adam and C. Orvig, Organometallics, 2012, 5736 CrossRef CAS; (c) C. L. Ferreira, C. B. Ewart, C. A. Barta, S. Little, V. Yardley, C. Martins, E. Polishchuk, P. J. Smith, J. R. Moss, M. Merkel, M. J. Adam and C. Orvig, Inorg. Chem., 2006, 45, 8414 CrossRef CAS; (d) T. Itoh, S. Shirakami, N. Ishida, Y. Yamashita, T. Yoshida, H. S. Kim and Y. Wataya, Bioorg. Med. Chem. Lett., 2000, 10, 1657 CrossRef CAS.
- (a) A. Hottin, F. Dubar, A. Steenackers, P. Delannoy, C. Biot and J.-B. Behr, Org. Biomol. Chem., 2012, 10, 5592 RSC; (b) C. G. Hartinger, A. A. Nazarov, V. B. Arion, G. Giester, M. Jakupec, M. Galanski and B. K. Keppler, New J. Chem., 2002, 26, 671 RSC.
- A. L. Martin, B. Li and E. R. Gillies, J. Am. Chem. Soc., 2008, 131, 734 CrossRef.
- X.-L. Sun, C. A. Haller, X. Wu, V. P. Conticello and E. L. Chaikof, J. Proteome Res., 2005, 4, 2355 CrossRef CAS.
- (a) J. M. Casas-Solvas, E. Ortiz-Salmeron, I. Fernandez, L. Garcia-Fuentes, F. Santoyo-Gonzalez and A. Vargas-Berenguel, Chem.–Eur. J., 2009, 15, 8146 CrossRef CAS; (b) Y. Willener, K. M. Joly, C. J. Moody and J. H. R. Tucker, J. Org. Chem., 2008, 73, 1225 CrossRef CAS; (c) S. Sato, T. Nojima and S. Takenaka, J. Organomet. Chem., 2004, 689, 4722 CrossRef CAS.
- (a) G. Favero, L. Campanella, A. D'Annibale and T. Ferri, Microchem. J., 2004, 76, 77 CrossRef CAS; (b) G. Cooke, Angew. Chem., Int. Ed., 2003, 42, 4860 CrossRef CAS; (c) I. Suzuki, Q. Chen, A. Ueno and T. Osa, Bull. Chem. Soc. Jpn., 1993, 66, 1472 CrossRef CAS; (d) A. Ueno, F. Moriwaki, T. Matsue, T. Osa, F. Hamada and H. Murai, Makromol. Chem., Rapid Commun., 1985, 6, 231 CrossRef CAS.
- S. K. Schreyer and S. R. Mikkelsen, Bioconjugate Chem., 1999, 10, 464 CrossRef CAS.
- R. Kataky, R. Dell and P. K. Senanayake, Analyst, 2001, 126, 2015 RSC.
- (a) Y. Han, K. Cheng, K. A. Simon, Y. Lan, P. Sejwal and Y.-Y. Luk, J. Am. Chem. Soc., 2006, 128, 13913 CrossRef CAS; (b) I. Suzuki, Q. Chen, Y. Kashiwagi, T. Osa and A. Ueno, Chem. Lett., 1993, 1719 CrossRef CAS.
- F. Trotta, K. Martina, B. Robaldo, A. Barge and G. Craotto, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 3 CrossRef CAS.
- M. Zougagh, J. M. Cano Pavón and A. Garcia de Torres, Anal. Bioanal. Chem., 2005, 381, 1103 CrossRef CAS.
- S. Beheshti, S. Martić and H.-B. Kraatz, Chem.–Eur. J., 2012, 18, 9099 CrossRef CAS.
- D. A. Dickinson and H. J. Forman, Biochem. Pharmacol., 2002, 64, 1019 CrossRef CAS.
- (a) Y. Peng, Y.-N. Liu and F. Zhou, Electroanalysis, 2009, 21, 1848 CrossRef CAS; (b) S. Jahn, W. Lohmann, S. Bomke, A. Baumann and U. Karst, Anal. Bioanal. Chem., 2012, 402, 461 CrossRef CAS; (c) M. C. Martos-Maldonado, J. M. Casas-Solvas, R. Téllez-Sanz, C. Mesa-Valle, I. Quesada-Soriano, F. Garcia-Maroto, A. Vargas-Berenguel and L. Garcia-Fuentes, Biochimie, 2012, 94, 541 CrossRef CAS; (d) W. Hyk, M. Karbarz, B. Misterkiewicz and Z. Stojek, J. Phys. Chem. B, 2007, 111, 13090 CrossRef CAS; (e) B. Misterkiewicz, M. Salmain and G. Jaouen, Tetrahedron Lett., 2004, 45, 7511 CrossRef CAS; (f) K. Di Gleria, H. A. O. Hill and L. L. Wong, FEBS Lett., 1996, 390, 142 CrossRef CAS.
- F.-B. Wang, Y. Peng, M.-Y. Fan, Y.-N. Liu and K.-L. Huang, Wuli Huaxue Xuebao, 2009, 25, 1125 CAS.
- N. Metzler-Nolte and M. Salmain, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Willey & Sons, Ltd-, Chichester, UK, 2008, p. 499 Search PubMed.
- (a) V. S. Tripathi, V. B. Kandimalla and H. Ju, Biosens. Bioelectron., 2006, 21, 1529 CrossRef CAS; (b) H. Shinohara, T. Kusaka, E. Yokota, R. Monden and M. Sisido, Sens. Actuators, B, 2000, 65, 144 CrossRef; (c) T. Kohno, S. Asai, Y. Iribe, I. Hosoi, K. Shibata and K. Ishikawa, J. Brain Sci., 1998, 24, 47 CAS; (d) T. Suzawa, Y. Ikariyama and M. Aizawa, Bull. Chem. Soc. Jpn., 1995, 68, 165 CrossRef CAS; (e) S. Kunugi, Y. Murakami, K. Ikeda and N. Itoh, Int. J. Biol. Macromol., 1992, 14, 210 CrossRef CAS; (f) A. Badia, N. H. Huy Thai, A. M. English, S. R. Mikkelsen and R. T. Patterson, Anal. Chim. Acta, 1992, 262, 87 CrossRef CAS; (g) F. Mizutani and M. Asai, Denki Kagaku Oyobi Kogyo Butsuri Kagaku, 1988, 56, 1100 CAS; (h) H. Falk, M. Peterlik and K. Schlögl, Monatsh. Chem., 1969, 100, 787 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Voltammograms and diffusion coefficient D0 (plot of Ipavs. v1/2) of different compounds and NMR spectra for new compounds (3–5, 7–11, 15–20 and 23–26). See DOI: 10.1039/c3ob27209e |
| This journal is © The Royal Society of Chemistry 2013 |