Nanostructural transformations during the reduction of hollow and porous nickel oxide nanoparticles†
John A.
Medford
,
Aaron C.
Johnston-Peck‡
and
Joseph B.
Tracy
*
Department of Materials Science and Engineering, North Carolina State University, Raleigh, NC 27695, USA. E-mail: jbtracy@ncsu.edu
First published on 20th November 2012
Abstract
Size-dependent nanostructural transformations occurring during the H2-mediated reduction of hollow and porous NiO nanoparticles were investigated for controlled nanoparticle sizes of ∼10 to 100 nm. Transmission electron microscopy reveals that the location and number of reduction sites strongly depend on the nanoparticle size and structure.
Numerous studies since 2004 have investigated void formation in metal nanoparticles (NPs) during oxidation1–13 and their conversion into sulfides, selenides, and phosphides1,6,13–23 through the Kirkendall effect. The Kirkendall effect has also been demonstrated in multicomponent metal NPs.24 During oxidation, a thin oxide shell first forms, followed by diffusion of metal atoms through the oxide shell toward the NP surface, where oxidation continues. Outward diffusion of metal atoms drives void formation, resulting in hollow or porous NPs. Here, we identify NPs as “hollow” if transmission electron microscopy (TEM) images show a shell that lacks obvious pores, but it is known that the oxide shells generally have nanoscale porosity.1 NPs are called “porous” if TEM images show readily observable gaps in the shells (usually for larger NP sizes, where crystallization of distinct grains is more obvious than for smaller sizes).
During the reverse process, when hollow metal oxide NPs are chemically reduced, “shrinkage” to solid metal NPs has been theoretically predicted because the surface energy is reduced when the void collapses.25–28 There have been limited experimental studies of the shrinkage process during reduction: Nakamura et al. reported the shrinkage of Cu2O and NiO NPs under vacuum in a TEM at elevated temperatures.29 Their study focused primarily on the temperature dependence of the shrinkage process and demonstrated a mechanism, where reduction occurs through the formation and growth of a reduced metal NP inside the void. Chenna et al. have performed studies of the reduction of NiO NPs having sizes smaller than ∼20 nm with CH4 and reported essentially the same nanostructural changes during reduction.30,31 Here, we report the size-dependent nanostructural changes that occur during the reduction of hollow NiO NPs with sizes of ∼10 to 100 nm to solid Ni NPs with H2 at elevated temperatures. Reduction with H2 is chemically more direct than with CH4, where several intermediate species can be present. We report that the NP size and porosity determine whether reduction proceeds from a single site or multiple sites and the arrangement of the sites. Knowledge of the structures of partially reduced Ni NPs and the mechanism of reduction will also be useful for the design and application of Ni NP-based catalysts, particularly for hydrogenation reactions.32 For example, some of the structures obtained at intermediate stages of reduction are small Ni NPs embedded within a nanoporous NiO matrix. Such structures are not observed during the oxidation of Ni9 and may have high catalytic activity owing to the small size of the Ni NPs. The porous shell could also potentially impart selectivity.1
Ni NPs were synthesized and purified as reported previously,9,20,33 based on modifications of a method first reported by Hyeon and coworkers.34 For all sizes of Ni NPs, varying amounts of nickel acetylacetonate hydrate (Ni(acac)2, 98%, TCI America), 2.0 mL (6.1 mmol) of oleylamine (80–90%, Acros), and 5.0 g of trioctylphosphine oxide (TOPO, 99%, Strem) or 5.0 mL of 1-octadecene (ODE, 90%, Sigma-Aldrich) were combined with vigorous stirring and heated to 60 °C in a three-necked, round-bottomed flask for 1.5 h under vacuum before backfilling with nitrogen. For the 12 nm and 24 nm samples, trioctylphosphine (TOP, 97%, Strem) was added to the mixture. For all sizes, the mixture was rapidly heated to 240 °C (∼10 °C min−1 ramp rate) for 30 min and then cooled to room temperature. For the 12 nm Ni NPs, 0.200 g (0.778 mmol) Ni(acac)2 and 0.30 mL (0.67 mmol) of TOP were used, with TOPO as the solvent. The 24 nm Ni NPs were synthesized using the same parameters as the 12 nm NPs, except the amount of TOP was reduced to 0.20 mL (0.45 mmol). No TOP was used for the 40 nm and 96 nm sizes. For the 40 nm Ni NPs, 0.500 g (1.95 mmol) Ni(acac)2 was used, with TOPO as the solvent. For the 96 nm Ni NPs, 0.200 g (0.778 mmol) Ni(acac)2 was used, with ODE as the solvent. After allowing the product to cool to room temperature, methanol was added to flocculate the NPs, followed by centrifugation to separate the NPs from the solvent and excess ligands. The NPs were repeatedly (one to three times) redispersed in hexanes, flocculated by adding methanol, and isolated by centrifugation. The purified NPs were stored in hexanes.
To prepare samples for oxidation and subsequent reduction, NPs with average diameters (measured by TEM) of 12, 24, 40, and 96 nm dispersed in hexanes were sonicated for one minute to improve their dispersion, followed by drop casting onto 50 nm thick SiN membranes from Protochips, Inc. After the solvent completely evaporated, each sample was exposed to ultraviolet light with ozone treatment for five minutes to remove the ligands.9 A separate SiN membrane was used for each reduction time investigated. Oxidation treatments were performed under ambient atmosphere in a preheated Protherm PC422 tube furnace at 500 °C for one hour. Each sample was promptly removed after one hour.
Reduction experiments were performed in the same tube furnace under a non-flammable atmosphere of forming gas (5% H2 in 95% N2) with the tube sealed and vented via a bubbler. Immediately after introducing the samples at 350 °C, the furnace was purged with forming gas, followed by a constant flow of 0.5 cubic foot per hour. This relatively slow flow rate was chosen in order to minimize thermal perturbations. The samples were reduced for one, two, or four hours. At the end of the reaction, special precautions were taken to avoid oxidation of the reduced NPs while they were still hot. Under a forming gas flow exceeding 5 cubic feet per hour, the sample substrates were moved out of the hot zone in the furnace while remaining in the tube. The samples cooled quickly under the faster flow of forming gas and were removed at room temperature. We have previously shown that the native oxides formed on Ni NPs at room temperature are usually thinner than 2 nm.33 The reduction reaction was initially performed at temperatures of 300–400 °C. For this study, 350 °C was selected because it allows for reduction on the time scale of a few hours, which enables simple observation of partially reduced intermediates without requiring in situ TEM. After reduction, the specimens were stored in a glovebox under inert atmosphere to minimize oxidation.
Bright-field TEM images and selected-area electron diffraction (SAED) were acquired using a JEOL 2000FX microscope. High-resolution TEM (HRTEM) was performed using a JEOL 2010F microscope. The colored layers in the HRTEM images were obtained from fast Fourier transforms (FFTs) of the images. Symmetric, annular (i.e. bandpass) masks of width ∼1.3 nm−1 were separately overlaid on the points corresponding to the Ni {200} and NiO {111} plane spacings. Inverse FFTs for each set of masked areas were calculated, while discarding information from the unmasked area. Color-coded composites were prepared by overlaying the colored layers on the original image.
We identify each sample by the average diameter (12, 24, 40, or 96 nm) of the initial Ni NPs prior to oxidation to NiO, which is the starting point for this study. For convenience, we name these as the Ni12, Ni24, Ni40, and Ni96 samples. (The hollow oxide NPs are larger than these sizes due to the increase in volume that accompanies void formation and lattice expansion during oxidation.) According to SAED, NPs are completely oxidized after the oxidation treatment, though small Ni-rich regions could still remain. Based on our previous study, however, oxidation at 500 °C for one hour is more than sufficient to completely oxidize the NPs.9 TEM images of different selected single NPs for all sizes and reduction times are shown in Fig. 1, along with NPs before and after oxidation but prior to reduction. After complete reduction, the NPs return to their original sizes, but minor size differences are apparent in Fig. 1 due to the size distribution of each sample. Additional TEM images for each sample and SAED are presented in the ESI, Fig. S1–22,† which show that many of the NPs agglomerated during the oxidation and reduction treatments. It is important to note that while most NPs within the same sample (provided they have not agglomerated) undergo the same general structural changes during the reduction process, some NPs appear to be reduced more quickly or slowly than others. The speckled contrast across individual NPs in the TEM images confirms their polycrystalline structures, which are also observed in HRTEM images (Fig. 2 and S23†). Analysis of the lattice spacings enables unique identification of the regions of reduced Ni.
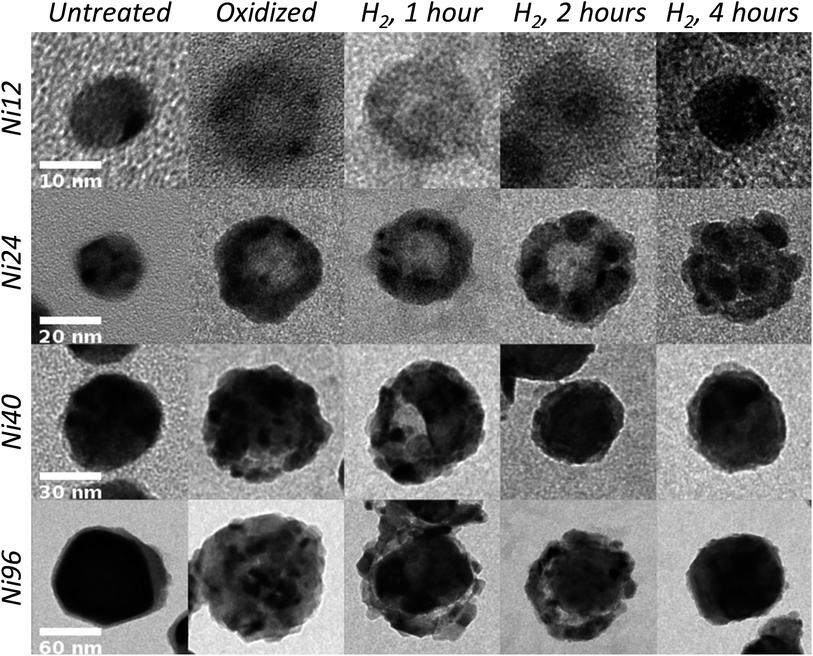 | ||
| Fig. 1 TEM images of 12, 24, 40, and 96 nm diameter Ni nanoparticles before oxidation, after oxidation, and after reduction for 1, 2, or 4 hours. Each row has a common scale bar. | ||
 | ||
| Fig. 2 HRTEM images of (a) a Ni24 nanoparticle after reduction for two hours at 350 °C and (b) a Ni40 nanoparticle after reduction for one hour at 350 °C. Overlaid colors indicate regions where the lattice spacings uniquely match (red) Ni {200} and (green) NiO {111}. | ||
In the Ni12 and Ni24 samples, formation of Ni grains during reduction can be discerned through the darker contrast of Ni because it is more electron dense than NiO. In the Ni40 and Ni96 samples, the larger size of the crystalline NiO grains results in more distinct lightly and darkly shaded regions in the completely oxidized NPs, and we are less certain about exactly where the Ni nucleates and grows until the grain size significantly exceeds that of the NiO grains. In each sample, reduction occurred from within the NP first, followed by reduction of the outside of the NP, but the number of sites for nucleation and growth of reduced Ni and their arrangement depend strongly on size. The smallest NPs (Ni12) have only one reduction site, and larger NPs have multiple reduction sites. Growth of the reduced Ni from NiO grains is consistent with the known autocatalytic mechanism of reduction, in which reduction proceeds most favorably at Ni–NiO interfaces.35 The grains of reduced Ni are polycrystalline, though with sufficient reduction time, single-crystalline NPs might be achieved. Completely reduced Ni NPs free of oxide were not observed, however, due to the thin native oxide layer that quickly forms upon exposure to ambient atmosphere.
The Ni12 NPs consist of symmetrical NiO shells before reduction. Reduction of NiO first occurs at a single nucleation site usually located on the shell–void interface, from which the grain of reduced Ni grows (Fig. 3a). Experimental evidence for a single reduction site in the Ni12 NPs is provided in the image of a Ni12 NP after 1 hour of reduction (Fig. 1). As reduction proceeds, the void fills, and the NiO shell becomes thinner. The Ni24 NPs are asymmetrical hollow shells before reduction.9 Multiple reduction sites form at locations within the shells but generally not on the inner or outer surfaces. The Ni grains continue growing and collapse into the void, usually at times between 2 and 4 hours of reduction treatment (illustrated in Fig. 3b). The individual grains in each Ni24 NP eventually merge together and form a single metal NP, though this process requires more time than the complete reduction of other NP sizes and is not consistently observed after 4 hours of reduction. This longer time for complete reduction of Ni24 NPs may arise from their larger size than Ni12 NPs and from having the reduction sites embedded within the shell, where diffusion may be slower than in the more open, porous Ni40 and Ni96 NPs. We note that reduction from a single site inside the NiO shell has been reported previously,29,30 but we are unaware of previous reports of nucleation of reduced Ni within the NiO shells. Multiple Ni nucleation sites appear to develop throughout the Ni40 and Ni96 NPs, often dominated by a larger single site.
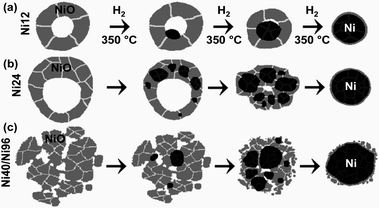 | ||
| Fig. 3 Graphical depictions of the nanostructural transformations that accompany reduction with H2 over a period of several hours at 350 °C for (a) Ni12, (b) Ni24, and (c) Ni40 and Ni96 nanoparticles. Black coloring depicts Ni, and NiO is shaded gray. White and gray lines indicate grain boundaries. | ||
Bulk NiO is known to undergo reduction through the nucleation model of reduction, wherein Ni grains form at the NiO surface, and then the grains grow until reduction is completed.29,36 Removing oxygen from the lattice leaves anion vacancies that are eliminated by atomic rearrangements in the reduced material. The results for the reduction of all four sizes of hollow NiO NPs can be understood using the nucleation model, but there are significant differences in nucleation and growth of the reduced Ni among the different sizes of NPs. Nucleation is most favorable at defects, but diffusion and geometrical factors determine the growth. The preference for the nucleation of reduced Ni grains on the inside of the shell for Ni12 NPs is suggestive that there is a higher defect density on the inner surface of the shell than elsewhere in the NP. This preference may arise from the oxidation process:9 The outer surface of the NP is replenished and constantly restructured during the oxidation process, which could decrease the defect density. In contrast, vacancies accumulated on the inside of the shell as Ni diffused outward through the shell. Therefore the inside of the shell may retain a high defect density after completing oxidation. For all sizes, the exterior surface of the NP was one of the last regions to undergo reduction.
Reduction from the inside can be explained using the porosity1 of the NPs; H2 diffuses through the pores in the shell and into the inner surface of the void. The Ni12 NPs are sufficiently small that a single Ni grain effectively serves as the reduction site for the whole NP; adsorbed H2 travels a short distance before it reduces NiO at the interface with the Ni grain. The larger size of the Ni24 NPs does not permit diffusion of adsorbed H2 over the larger distance before it can react with NiO, and nucleation of Ni occurs at multiple sites. It is not fully clear why the Ni grains in the Ni24 NPs nucleate within the NiO shells, but this is suggestive of a high defect density within the shells. Size-dependent changes in the nanostructure would be expected to cause size-dependent nucleation behavior. For example, the NiO grain size after oxidation strongly depends on the NP size; large grains of NiO are clearly identifiable in the Ni40 and Ni96 samples (Fig. 1). The grains in the Ni24 sample are smaller, and the grains in the Ni12 sample are difficult to discern, presumably due to their small size. The Ni24 NPs could have high defect densities within the pores or at grain boundaries within the shell. Growth of the Ni grains is determined geometrically, both by the location of the Ni–NiO interface and by the thermodynamically driven tendency for hollow shells to collapse and reduce their surface energy.25–28 A remaining question is whether the reduction temperature affects the nanostructural evolution during reduction. For example, since H2 diffuses faster at higher temperatures, would there be fewer nuclei in the Ni24 and larger samples?
In conclusion, distinct, size-dependent nanostructural changes were observed during the reduction of hollow and porous NiO NPs. Symmetrical Ni12 NP shells reduce from a single location on the shell–void interface. In contrast to previous studies, which examined only smaller sizes30,31 or reported reduction only from a single site,29 we observed multiple reduction sites in the larger NPs. Reduction commenced from multiple sites throughout the asymmetrical shells of Ni24 NPs. In the Ni40 and Ni96 NPs, multiple reduction sites are present but are more difficult to unambiguously identify. The limited number of reduction sites can be described by reduction occurring most favorably at the Ni–NiO interface, which is consistent with the autocatalytic mechanism of reduction of NiO.35 The lack of reduction sites on the exterior surfaces of NPs suggests that locations inside the NPs, such as grain boundaries or the inside of the shell are richer in defects than the outer surface, which may arise from the manner in which the hollow NiO NPs initially formed. These results will facilitate the development of new tools for fabricating heterostructured nanomaterials and the design of improved catalysts.
Acknowledgements
This work was supported by the National Science Foundation (DMR-1056653) and startup funds from North Carolina State University. A.C.J.-P. acknowledges support from a GAANN fellowship. We thank Protochips, Inc. for donating the SiN membranes and Andrew J. Medford for helpful discussions.References
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS.
- J. B. Tracy, D. N. Weiss, D. P. Dinega and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 064404 CrossRef.
- C. M. Wang, D. R. Baer, L. E. Thomas, J. E. Amonette, J. Antony, Y. Qiang and G. Duscher, J. Appl. Phys., 2005, 98, 094308 CrossRef.
- S. Peng and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 4155–4158 CrossRef CAS.
- A. Cabot, V. F. Puntes, E. Shevchenko, Y. Yin, L. Balcells, M. A. Marcus, S. M. Hughes and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 10358–10360 CrossRef CAS.
- H. J. Fan, U. Gösele and M. Zacharias, Small, 2007, 3, 1660–1671 CrossRef CAS.
- Q. Peng, X. Y. Sun, J. C. Spagnola, C. Saquing, S. A. Khan, R. J. Spontak and G. N. Parsons, ACS Nano, 2009, 3, 546–554 CrossRef CAS.
- P. Y. Keng, B. Y. Kim, I. B. Shim, R. Sahoo, P. E. Veneman, N. R. Armstrong, H. Yoo, J. E. Pemberton, M. M. Bull, J. J. Griebel, E. L. Ratcliff, K. G. Nebesny and J. Pyun, ACS Nano, 2009, 3, 3143–3157 CrossRef CAS.
- J. G. Railsback, A. C. Johnston-Peck, J. W. Wang and J. B. Tracy, ACS Nano, 2010, 4, 1913–1920 CrossRef CAS.
- F. Güder, Y. Yang, S. Goetze, A. Berger, N. Ramgir, D. Hesse and M. Zacharias, Small, 2010, 6, 1603–1607 CrossRef.
- Y. Ren, W. K. Chim, S. Y. Chiam, J. Q. Huang, C. Pi and J. S. Pan, Adv. Funct. Mater., 2010, 20, 3336–3342 CrossRef CAS.
- Q. K. Ong, X.-M. Lin and A. Wei, J. Phys. Chem. C, 2011, 115, 2665–2672 CAS.
- M. Ibáñez, J. Fan, W. Li, D. Cadavid, R. Nafria, A. Carrete and A. Cabot, Chem. Mater., 2011, 23, 3095–3104 CrossRef.
- J. H. Gao, B. Zhang, X. X. Zhang and B. Xu, Angew. Chem., Int. Ed., 2006, 45, 1220–1223 CrossRef CAS.
- Y. D. Yin, C. K. Erdonmez, A. Cabot, S. Hughes and A. P. Alivisatos, Adv. Funct. Mater., 2006, 16, 1389–1399 CrossRef CAS.
- R. K. Chiang and R. T. Chiang, Inorg. Chem., 2007, 46, 369–371 CrossRef CAS.
- A. E. Henkes, Y. Vasquez and R. E. Schaak, J. Am. Chem. Soc., 2007, 129, 1896–1897 CrossRef CAS.
- A. Cabot, R. K. Smith, Y. Yin, H. Zheng, B. r. M. Reinhard, H. Liu and A. P. Alivisatos, ACS Nano, 2008, 2, 1452–1458 CrossRef CAS.
- A. Cabot, M. Ibáñez, P. Guardia and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 11326–11328 CrossRef CAS.
- J. W. Wang, A. C. Johnston-Peck and J. B. Tracy, Chem. Mater., 2009, 21, 4462–4467 CrossRef CAS.
- E. Muthuswamy, G. H. L. Savithra and S. L. Brock, ACS Nano, 2011, 5, 2402–2411 CrossRef CAS.
- S. Carenco, X. F. Le Goff, J. Shi, L. Roiban, O. Ersen, C. d. Boissière, C. m. Sanchez and N. Mézailles, Chem. Mater., 2011, 23, 2270–2277 CrossRef CAS.
- D.-H. Ha, L. M. Moreau, C. R. Bealing, H. Zhang, R. G. Hennig and R. D. Robinson, J. Mater. Chem., 2011, 21, 11498–11510 RSC.
- E. González, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377–1380 CrossRef.
- A. M. Gusak, T. V. Zaporozhets, K. N. Tu and U. Gösele, Philos. Mag., 2005, 85, 4445–4464 CrossRef CAS.
- F. D. Fischer and J. Svoboda, J. Nanopart. Res., 2008, 10, 255–261 CrossRef.
- A. V. Evteev, E. V. Levchenko, I. V. Belova and G. E. Murch, Philos. Mag., 2008, 88, 1525–1541 CrossRef CAS.
- A. M. Gusak and T. V. Zaporozhets, J. Phys.: Condens. Matter, 2009, 21, 415303 CrossRef CAS.
- R. Nakamura, D. Tokozakura, J. G. Lee, H. Mori and H. Nakajima, Acta Mater., 2008, 56, 5276–5284 CrossRef CAS.
- S. Chenna, R. Banerjee and P. A. Crozier, ChemCatChem, 2011, 3, 1051–1059 CrossRef CAS.
- S. Chenna and P. A. Crozier, Micron, 2012, 43, 1188–1194 CrossRef CAS.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons , 2001 Search PubMed.
- A. C. Johnston-Peck, J. W. Wang and J. B. Tracy, ACS Nano, 2009, 3, 1077–1084 CrossRef CAS.
- I. S. Lee, N. Lee, J. Park, B. H. Kim, Y. W. Yi, T. Kim, T. K. Kim, I. H. Lee, S. R. Paik and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 10658–10659 CrossRef CAS.
- A. F. Benton and P. H. Emmett, J. Am. Chem. Soc., 1924, 46, 2728–2737 CrossRef CAS.
- H. K. Harold, in Stud. Surf. Sci. Catal., Elsevier, 1989, vol. 45, pp. 91–109 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional TEM images, SAED, and HRTEM images without color overlays. See DOI: 10.1039/c2nr33005a |
| ‡ Current address: Center for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA. |
| This journal is © The Royal Society of Chemistry 2013 |