M1.3 – a small scaffold for DNA origami †
Hassan Saida, Verena J. Schüllerb, Fabian J. Eberc, Christina Wegec, Tim Liedlb and Clemens Richert*a
aInstitute for Organic Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: lehrstuhl-2@oc.uni-stuttgart.de; Fax: +49 711 685 64321; Tel: +49 711 685 64311
bFakultät für Physik and Center for Nanoscience, Geschwister-Scholl-Platz 1, 80539 München, Germany. E-mail: Tim.Liedl@lmu.de; Fax: +49 89 2180 3182
cDepartment of Molecular Biology and Plant Virology, Institute of Biology, University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany. E-mail: christina.wege@bio.uni-stuttgart.de; Fax: +49 711 685-65096
First published on 30th October 2012
Abstract
The DNA origami method produces programmable nanoscale objects that form when one long scaffold strand hybridizes to numerous oligonucleotide staple strands. One scaffold strand is dominating the field: M13mp18, a bacteriophage-derived vector 7249 nucleotides in length. The full-length M13 is typically folded by using over 200 staple oligonucleotides. Here we report the convenient preparation of a 704 nt fragment dubbed “M1.3” as a linear or cyclic scaffold and the assembly of small origami structures with just 15–24 staple strands. A typical M1.3 origami is large enough to be visualized by TEM, but small enough to show a cooperativity in its assembly and thermal denaturation that is reminiscent of oligonucleotide duplexes. Due to its medium size, M1.3 origami with globally modified staples is affordable. As a proof of principle, two origami structures with globally 5′-capped staples were prepared and were shown to give higher UV-melting points than the corresponding assembly with unmodified DNA. M1.3 has the size of a gene, not a genome, and may function as a model for gene-based nanostructures. Small origami with M1.3 as a scaffold may serve as a workbench for chemical, physical, and biological experiments.
Introduction
Deoxyribonucleic acid (DNA) has become a versatile material for the construction of nanoscale objects.1–3 The introduction of the scaffold-based DNA origami method has been an important step in DNA nanostructuring towards larger and more complex assemblies.4 The use of a long single-stranded scaffold strand, folded into the desired geometries through shorter, synthetic ‘staple strands’ is a robust, high-yielding method for generating two- and three-dimensional shapes.5 DNA origami structures have been used to create patterns of proteins,6 or nanoparticles,7–9 and applications include origami-based alignment media for nuclear magnetic resonance structure elucidation,10,11 production of nanocarriers that deliver antibody fragments,12 or immune-stimulating nucleic acid sequences to cells.13 Further, origami can act as a scaffold or a workbench for single molecule analyses or chemical reactions.14–18 Even nanoscale assembly lines have been realized on DNA origami platforms.19 Despite these exciting opportunities to set up spatially controlled experiments at a length scale on the order of 1–100 nm, the DNA origami approach has not yet become a mainstream method for chemists.Most current origami structures are large. Single-stranded, circular M13mp18, a commercial bacteriophage-derived vector of 7249 nucleotides in length,20 is most frequently used as a scaffold. Several reports exist on scaled-up versions of DNA origami with an increased number of addressable positions.21–28 But, DNA origami based on M13mp18 also has drawbacks. This very long scaffold strand increases the likelihood of strand cleavage and makes analysis at a molecular level challenging. Furthermore, known hairpin-forming regions in the M13mp18 sequence can impede folding of parts of the structures. As a consequence of the scaffold length, a large number of staple sequences, typically 120–200 synthetic oligonucleotide sequences, has to be ordered to construct a full origami. If not, a long loop of the unpaired scaffold remains that might interfere with the desired functions of the DNA origami structure. Very recently, a method for assembling large nanoscale shapes without a template strand has been reported.29,30 The full set of strands used to construct the different shapes from the basic ‘canvas’ design includes 362 ‘internal’ oligonucleotides and 1344 ‘edge protectors’, again calling for a very substantial investment in synthetic oligonucleotides prior to generating the desired structures. A large number of staple strands leads to significant costs that can dampen the enthusiasm of experimentalist wishing to set up origami structures, e.g. as chemical work benches. Much smaller origami than established sheets should suffice for many chemical experiments, while still being easy to detect by TEM and/or AFM.
Successful attempts have been made to produce shorter scaffolds. Woolley and co-workers employed the polymerase chain reaction (PCR) with a biotinylated primer, followed by purification over streptavidin-coated magnetic beads31 to produce single-stranded linear scaffolds 756 to 4808 nucleotides in length.31 Högberg et al. used a double-stranded PCR product of 1.3 kb to generate an origami by fast temperature drop and gradual removal of formamide in the presence of staple strands.32 Compared to approaches based on biotechnologically generated M13mp18, PCR is very costly and difficult to scale up. Furthermore, the need for a biotinylated primer, combined with purification over streptavidin-coated magnetic beads,31 makes PCR-based processes more costly than a process using unmodified synthetic oligonucleotides, combined with inexpensive enzymes and conventional gel electrophoresis.
We sought a DNA origami construct as a nanoscale chemical workbench. This bench was to fulfil several criteria: (i) reliable in its folding properties, (ii) inexpensive, (iii) assembling with a small number of staple oligonucleotides and (iv) of sufficient size to be analyzed with the established methodologies for DNA origami structures. It was critical that no more than a small number of staple strands were required, because we were interested in employing custom-modified synthetic oligonucleotides that are costly. For many chemical applications, even unmodified oligonucleotides are costly, and we expected that a robust, small origami, using much fewer staple strands, would more readily open up the field to synthetic organic chemists. Hence, we sought a scaffold strand derived from M13 approx. one order of magnitude smaller than the full vector. Here we report the convenient preparation of a M13 fragment as a linear or cyclic scaffold and the assembly of small origami. Because we were aiming at reducing the size by approx. one order of magnitude, our scaffold was dubbed “M1.3”, with the number “1.3” derived from dividing the number 13 by ten.
Results and discussion
We wished to use a fraction of the M13 sequence because the sequence is well behaved, without a strong propensity to fold intramolecularly or to form aggregates. Further, M13 is commercially available and can be easily produced, even in academic settings by straightforward biotechnological culture.10 A calculation of the material costs for 0.1–1 mg of this DNA is presented in the ESI.†Fig. 1 shows the method developed to excise the desired sequence with the help of restriction endonucleases. The use of restriction enzymes in generating scaffolds is not without precedent.33 In our case, short double-stranded regions were generated by hybridizing two cleavage-inducing oligonucleotides (CIOs) to complementary sites in the scaffold. Type II restriction enzymes were used. Different restriction endonucleases and CIO sequences and lengths (8, 12, 20 and 40 nucleotides) were tested, as well as a range of oligonucleotide concentrations (1 to 1000 equiv.), and the concentration of the enzymes and reaction times were varied. A combination of EcoRI and BglII was selected, together with two CIO 20mers (Fig. 1a), producing M1.3 with a length of 704 nucleotides. Enzyme combinations for other fragments of M13 can be found in Chapter 3 of the ESI.† Both of the selected enzymes are inexpensive, with current total costs below 10 € for a large scale run with both enzymes (250 units each), and both are active in the same restriction buffer. The cleavage fragments of the CIOs are short enough to dissociate from the termini of M1.3 upon mild heating. The remaining long fragments of M13 and M1.3 were easily separable by agarose gel electrophoresis (Fig. 1c). The desired band was excised, the DNA was extracted, and desalted to obtain linear, single-stranded M1.3.
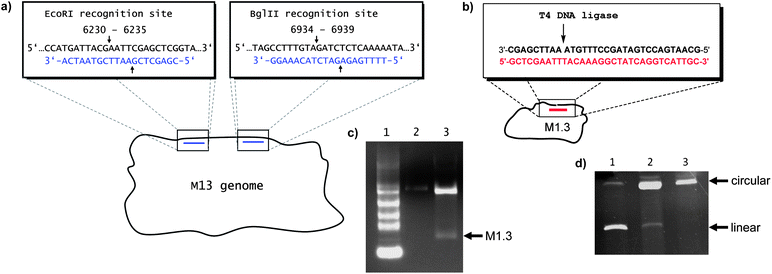 | ||
| Fig. 1 Preparation of M1.3. (a) Excising M1.3 from the single-stranded M13mp18 vector DNA with the aid of two cleavage-inducing oligonucleotides (blue) and restriction endonucleases EcoRI and BglII. The cleavage sites are marked with arrows, and positions in the M13 sequence (GenBank accession no: X02513) are given numerically in the blow-up boxes. (b) Ligating linear M1.3 to cyclicM1.3 (cM1.3) with the aid of a template oligonucleotide of asymmetric coverage (red) and T4 DNA ligase. (c) Agarose electrophoresis gel showing the digestion of M13 to M1.3. Lane 1: 0.24–9.5 kb RNA ladder, lane 2: 0.2 pmol M13, lane 3: 0.8 pmol M13 after digestion, showing the fast-migrating band of M1.3. (d) PAGE (8% denaturing) of the products of the enzymatic cyclization of M1.3 with T4 DNA ligase, using a template strand with symmetric (lane 1) or asymmetric coverage of the termini of the linear scaffold (lane 2). Lane 3 shows purified circular M1.3. | ||
Most origami structures can be readily produced with linear scaffold strands, but circular scaffolds offer an additional entropic benefit and fold more readily. Therefore, a protocol for cyclizing the 704mer linear M1.3 with the aid of a template strand and an inexpensive ligase (T4 DNA ligase) was developed. For efficient ligation, the template strand and linear scaffold need to be at an equimolar concentration. For very long DNA, stoichiometries are difficult to adjust accurately. Initial ligation attempts, using conventional template strands that cover the ligation site symmetrically, were unsatisfactory (Fig. 1d, lane 1). To overcome this problem, an asymmetric template strand was applied at a 5-fold excess. After annealing, the mixture was heated to 40 °C, a temperature at which the shorter double-stranded region at the 5′-terminus of the scaffold melts, and excess template strand was removed by ultrafiltration. The desired duplex with a template to scaffold ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was cooled to 16 °C and ligation was induced through addition of ATP and T4 DNA ligase, leading to circular M1.3 (cM1.3) in >80% yield (Fig. 1d, lane 2).
:1 was cooled to 16 °C and ligation was induced through addition of ATP and T4 DNA ligase, leading to circular M1.3 (cM1.3) in >80% yield (Fig. 1d, lane 2).
Starting from linear or circular M1.3, a first small origami sheet was assembled, using 24 unique staple strands (see ESI† for sequences). The sheet was designed using caDNAno.34 It is non-symmetrical, with one receding corner to facilitate the assignment of the two faces. It is of sufficient size (approx. 20 × 30 nm) to set up a ‘chemical workbench’ with both small molecules and small proteins and to detect its shape by AFM. Due to its remote resemblance to features of a human hand, it was dubbed ‘four finger sheet’ or ‘4F sheet’. Fig. 2 shows a cartoon of the sheet, a gel of assembly mixtures for linear and circular M1.3, and a TEM micrograph. Images of partly frayed structures, as in the blow-up in Fig. 2c, further confirmed the formation of the desired shape, but more compact forms were more abundant in the TEM images, as expected for the design. In addition, alternating laser excitation (ALEX)35 measurements showed that two fluorescently labeled staple strands are efficiently integrated into the 4F sheet, confirming the successful formation of the full DNA origami structure (see Fig. S4, ESI†).
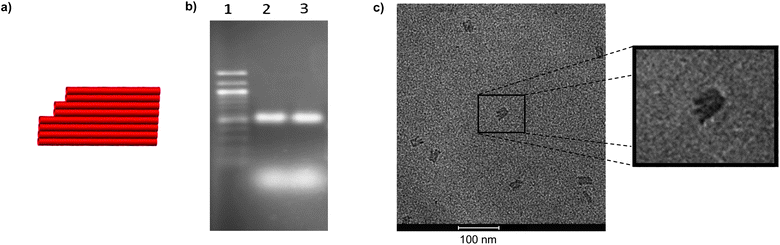 | ||
| Fig. 2 Folding of M1.3 with 24 staple strands into a four finger sheet (4F sheet). (a) Design of 4F sheet, (b) fluorescence image of agarose gel with ethidium bromide staining, lane 1: 100 bp ladder; lane 2: 0.5 pmol linear M1.3, assembled with 10 equivalents of staple strands; lane 3: 0.5 pmol circular M1.3, assembled with 10 equivalents of staple strands, diffuse bands at the bottom are from excess staple strands, (c) transmission electron micrograph. Note the characteristic finger-like substructure of the sheet when viewed perpendicular to the sheet plane. | ||
Encouraged by these results, an additional set of M1.3 origami was prepared, including a flat, ‘two-dimensional’ triangle, a curved six helix bundle,36 and a three-dimensional cube37 (Fig. 3). The latter two are smaller versions of the known M13 origami. The small structures cover a range of different design features, such as deletions and insertions and three-dimensional packing arrangements. They required between 15 and 21 different staple strands, demonstrating that the number of staple oligonucleotides can be scaled with the length of the scaffold.
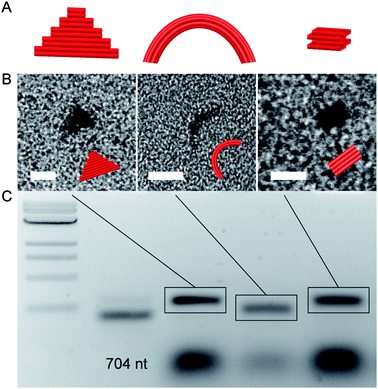 | ||
| Fig. 3 DNA origami structures assembled by using the M1.3 scaffold. (A) Schemes of DNA origami structures: 2D triangle, curved six-helix bundle (6HB), and 3D cube. (B) Electron micrographs of DNA origami structures. (C) Agarose gel of assembled structures. Left to right: 2-log 1 kb DNA ladder, M1.3 704 nt scaffold, 2D triangle, curved six-helix bundle (6HB), and 3D cube (0.2 pmol each). Scale bars: 20 nm. Different granularities are due to different sample settings. | ||
As the long-term focus of our study is on synthetically enforced origami, we then devised a method for generating sets of staple strands consisting entirely of modified strands. We chose staples that each bear a molecular cap at their 5′-terminus. Caps can enhance duplex stability and base-pairing fidelity at the termini of linear duplexes by bridging helices with correctly paired terminal base pairs more strongly than helices with mispaired termini.38Scheme 1 shows the synthesis of modified strands with a pyrenyl-C-nucleoside39 (PyC) or a pyrenylmethyl-pyrrolidinol residue (PyPy) as the 5′-cap.40 Pyrenes can intercalate,41 show weak fluorescence, and can give excimer bands when in close proximity.42 The phosphoramidite of the PyPy cap is commercially available.43
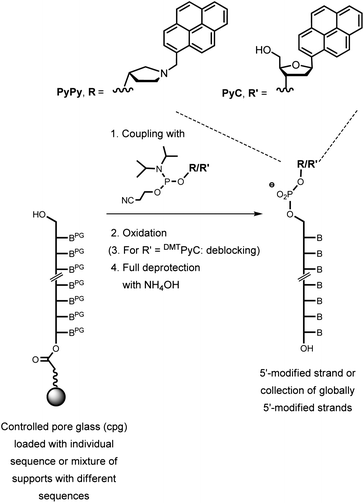 | ||
| Scheme 1 Synthesis of capped staple strands by coupling a cap phosphoramidite on protected oligonucleotides on controlled pore glass (cpg) as a solid support. One synthesis cycle with either the PyPy (R) or the PyC phosphoramidite (R′) yielded a 5′-capped oligonucleotide or a set of 24 globally 5′-capped staple strands. Deprotection was two steps for PyC and one step for PyPy. Crudes were used for the assembly of the M1.3 4F motif when the control synthesis involving only one sequence showed >90% yield. | ||
The two sets of globally 5′-modified staple strands for the 4F sheet were prepared by pooling 2 mg each of the controlled pore glass supports (cpg's) bearing the individual protected staple strand sequences. After thorough mixing, the pooled supports were subjected to one cycle of DNA chain extension using the pyrenyl building blocks. Conventional deprotection procedures then gave the mixture of strands required to assemble the modified origami with a pyrenyl moiety at every 5′-terminus of a staple strand. A control synthesis with a single sequence was also performed to confirm high coupling yields for the reagents used. When annealed to the M1.3 scaffold, either set of staple strands gave high yields of 4F origami. The origami bands showed slightly decreased mobility in agarose gels compared to unmodified 4F origami (Fig. S5, ESI†), as expected for structures with a modest increase in rigidity and size.
The 4F origami sheets were freed of excess staple strands by membrane filtration over 100 kDa cut-off spin filters. To study the effect of the caps on the stability of the sheets, UV-melting curves of the origami assemblies, monitored at 260 nm, were measured for the unmodified 4F origami and either of its counterparts with 5′-capped staple strands. All three gave a sharp transition and a Tm typical for M13 origami44 (Fig. 4 and S6 in the ESI†).
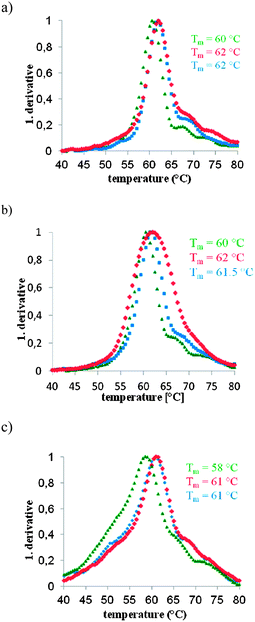 | ||
| Fig. 4 Results from UV-melting curve experiments with unmodified 4F M1.3 origami (green), 4F M1.3 origami with globally 5′-PyPy capped staples (blue) and 4F M1.3 origami with globally 5′-PyC capped staples (red). All curves were measured at 260 nm and 50 nM concentrations of M1.3. The first derivatives of the melting curves are shown. See Fig. S4 (ESI†) for plots of primary data. (a) Heating curves at a rate of 0.2 °C min−1, (b) heating curves at a rate of 1 °C min−1, and (c) cooling curves at a rate of 0.2 °C min−1; Tm = UV-melting point. | ||
Both pyrenyl caps induced an increase in the UV-melting point, as defined by the maximum of the first derivative, by approximately 2 °C (Table S1, ESI†). Perhaps more importantly, 4F origami with the capped staples shows slower kinetics of melting, meaning that a fraction of the duplex regions survives longer under thermal stress, resulting in a broader transition. This effect is most pronounced with the PyC cap at a heating rate of 1 °C min−1 (Fig. 4b). We hypothesize that a bridging effect of the pyrenyl residues that stabilize selected duplex regions causes this effect. The global assembly kinetics appear to be similar for sheets with and without capped staples, though, as judged by similar shapes of transitions observed in cooling curves measured at a cooling rate of 0.2 °C per minute (Fig. 4c). Here, the main peaks observed for the pyrenyl-modified origami are slightly sharper, suggesting that the polycyclic aromatic substituents aid in the cooperative process of origami formation. At the faster cooling rate of 1 °C min−1, only the sheet with PyPy caps shows this sharpening effect (Fig. S7 and Table S2, ESI†).
We note that the current design of the 4F sheet is not optimized for accommodating tetracyclic aromatic hydrocarbons, such as pyrenes. We expect that optimization of caps and their binding sites will lead to stronger effects than what the current, strictly exploratory results show. For example, larger ligands that can bridge two duplexes at a time, placed at strategic positions, are expected to have stronger effects. Both thermal stability and rapid assembly can be favorable for practical applications. Perhaps more importantly, catalytically active moieties,45 recognition motifs,46 or reporter groups,47 properly positioned in space with the aid of modified staple strands, can be expected to lead to new, functional origami. The small M1.3 platform may help to rapidly prototype such origami.
Conclusions
The transition from the molecular scale, where structures at or below the 1 nm limit can readily be produced by organic synthesis to the 100 nm scale of full size origami sheets, is quite dramatic. Most chemical experiments that cross the boundaries of traditional synthetic assemblies require spatial control on the 10–30 nm scale, rather than the 100 nm scale. Small scaffolds, such as M1.3, give access to this scale. We are currently starting a spectroscopic study on chromophore arrays on an M1.3 sheet that offers the functionality of larger arrays, based on M13 platforms,18 with a fraction of the number of staple strands. But, the advantage of the reduction in oligonucleotide staples is greatest when many or all of them are chemically modified. Globally capping or derivatizing oligonucleotides requires accurately weighing out individual cpg samples, and completely reacting the mixture batch in a solid-phase apparatus, a task that is difficult to do with conventional synthesizer cartridges for sets of 100 or more different cpg supports. We note that properly derivatized staples allow for covalently locking individual staple strands. We are actively pursuing locking methodologies.48Routine access to both small and large origami broadens the range of functional constructs that can be formed through inter-origami hybridization. But, smaller origami structures have hydrodynamic properties that make it easier to study them spectroscopically with bulk methods in solution, using conventional chemical analysis methods. Our data show how inexpensive ensemble experiments can yield data on the stability of origami structures and the global kinetics of their formation. Together, our data suggest a role for small origami structures that complements that of genome-sized ones, both for asking scientific questions and for developing devices that require a defined three-dimensional structure at the low nanometer scale.
Experimental
Excision of M1.3 from M13mp18 ssDNA
A solution of 20 pmol (50 μg) circular single-stranded M13mp18 DNA and ten equivalents of each of the two cleavage-inducing oligonucleotides (20mers) in 450 μL of restriction buffer (50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 100 mM NaCl, 0.1 mg mL−1 BSA) was heated to 85 °C for 5 min and allowed to cool to 20 °C in 2 h. To the solution were added 25 μL each of stock solutions of EcoRI and BglII (10 U μL−1) to give a final volume of 500 μL and final concentrations of 40 nM M13mp18 DNA, 400 nM oligonucleotides and 5 units of each enzyme per μg of DNA. The solution was incubated at 40 °C for 6 h. The digestion was stopped by adding 25 μL of 0.5 M EDTA (pH 8.0) to give a final concentration of EDTA of 24 mM. The volume of the solution was reduced to 200 μL by lyophilisation, followed by loading into several wells of a 1% agarose gel in 1× TAE buffer (40 mM Tris, 1 mM EDTA, 40 mM acetic acid). After running the gel for 90 min at 60 V and staining with ethidium bromide (0.5 μg mL−1), the desired band was excised with an extraction kit (NucleoSpin gel and PCR clean-up, Macherey-Nagel, Düren, Germany). Then, the DNA was desalted using an Amicon Ultra 0.5 mL centrifugal filter with a molecular weight cut-off of 30000 Da (Millipore, Billerica, MA, USA). The yield of M1.3, as determined by UV absorption at 260 nm, was 62% (12.4 pmol).Cyclization with asymmetric template strand
A sample of linear M1.3 (2 pmol) and 10 pmol of the asymmetric template strand in ligase buffer (30 μL, 40 mM Tris–HCl, pH 7.8, 10 mM MgCl2, 10 mM DTT, 5 mM ATP) were heated to 85 °C for 5 min and allowed to cool to 4 °C for 2 h, followed by incubation at 4 °C for 16 h. The solution was diluted with ligase buffer to 500 μL and warmed to 40 °C for 20 min. The excess template strand was removed by filtration at 40 °C, using an Amicon Ultra 0.5 mL centrifugal filter with a molecular weight cut-off of 30000 Da. The filter was washed twice with ligase buffer. The DNA was recovered (20 μL), and the solution was allowed to cool slowly from 40 °C to 16 °C, followed by incubation at 16 °C for 2 h. Then, solutions of ATP (1.5 μL of 10 mM) and T4 DNA ligase (3 μL, 1 Weiss unit per μL), 50% PEG 4000 (3 μL) and 1× ligase buffer were added to a final volume of 30 μL. After 3 h, the ligation was stopped by heating to 70 °C for 10 min. The solution was lyophilized, and the residue was dissolved in sample buffer (20 μL, 50% formamide, 1 mM EDTA, pH 8.0), heated to 90 °C for 2 min, and immediately placed on ice. Successful cyclization was confirmed by 8% denaturing PAGE (7 M urea).Assembly of origami
For the 4F sheet, a solution of 0.5 pmol M1.3 and 5 pmol staple strands in 20 μL of folding buffer (pH 8.0) containing 5 mM Tris–HCl, 1 mM EDTA, and 12 mM MgCl2 was heated to 85 °C for 5 min, and then cooled to 4 °C over the course of 2.5 h, followed by incubation at 4 °C for 12 h. For gel electrophoresis, 0.75 g of agarose in 50 mL of TBE buffer (45 mM Tris borate, 1 mM EDTA, pH 8.3) was boiled, cooled to 60 °C, and treated with MgCl2 solution (2 M, 300 μL) to give a 12 mM MgCl2 concentration, and cast as gel. A loading dye (0.025% xylene cyanol in 30% aqueous glycerol) was added to origami samples, followed by electrophoresis in the gel for 4 h at 50 V, followed by staining with ethidium bromide (0.5 mg mL−1) for 20 min. A similar procedure was employed for the assembly of the other M1.3 origami (Fig. 3), by annealing 10 nM scaffolds and 100 nM staple strands, in 1× TE with 10 mM MgCl2 for 1 h, and purification using a 2% agarose gel, 0.5× TBE, 11 mM MgCl2 running buffer for 2 h at 70 V. Isolation involved the ‘freeze and squeeze’ procedure with 10 min in a freezer and centrifugation for 10 min at 13000 rpm.Synthesis of globally 5′-capped sets of staple strands
For the global modification of 24 staple strands, 2 mg of each cpg loaded with a given sequence (approx. 0.06 μmol loading) and the dimethoxytriyl (DMT)-protected 3′-phosphoramidite of the PyCC-nucleoside (23.6 mg, 28.8 μmol, 20 eq.) or the phosphoramidite of the PyPy nucleoside analog (14.5 mg, 28.8 μmol, 20 eq.) were dried at 0.1 mbar in a polypropylene reaction vessel for 2 h. Then, activator solution (200 μL, 4,5-dicyanoimidazole, 0.25 M in CH3CN) was added under a N2 stream. After 1 h, the cpg was washed with CH3CN and then treated with oxidizer solution (200 μL of 0.02 M iodine in water–pyridine–THF, 2:21:77 v/v/v) for 10 min, followed by washing with CH3CN. In the case of the PyC-capped oligonucleotide, deblock solution (200 μL, trichloroacetic acid in CH2Cl2, 3:97, 200 μL) was added to the cpg, and the mixture was incubated for 20 min, followed by washing with CH3CN. After drying at 0.1 mbar, the cpg was treated with ammonium hydroxide (25% aqueous NH3, 500 μL) for 5 h at 55 °C. (Caution: pressure builds up when heating ammonia solution.) After cooling, excess ammonia was removed by gently blowing a stream of nitrogen onto the surface until the solution was odorless, and the solution was filtered (0.2 μm pore size, Whatman Inc., Chilton, NJ). Solutions were lyophilized and the modified staple strand mixtures were dissolved in water (100 μL) to produce a stock solution.UV-melting curve experiments
After assembly of the 4F origami from 4 pmol M1.3 and 80 pmol (modified) staple strands in 50 μL of folding buffer (pH 8.0) containing 5 mM Tris–HCl, 1 mM EDTA, and 12 mM MgCl2via heating to 85 °C for 5 min, and cooling to 4 °C for 2.5 h, followed by incubation at 4 °C for 12 h, the solution was diluted with folding buffer to 500 μL, and excess staple strands were removed by filtration at 10 °C, using an Amicon Ultra 0.5 mL centrifugal filter with a molecular weight cut-off of 100000 Da. The filter was washed three times with 500 μL of folding buffer. The origami was recovered (30 μL), the solution was diluted with folding buffer to 80 μL, and transferred to a UV/Vis microcuvette (10 mm path length). To prevent evaporation during the melting experiment, the sample was covered with 100 μL of mineral oil. The first derivatives were calculated using the spectrometer software (UV Winlab 3.0, Perkin Elmer).Acknowledgements
The authors are grateful to Dominik Kauert and Dr Ralf Seidel (TU Dresden) for joint exploratory experiments on the folding of the M1.3 sheet, Dr Ingo Stein and Prof. Philip Tinnefeld (L.M.U. Munich) for access to a single molecule laser set-up, and Dr M. Schweikert (U. Stuttgart) for assistance with TEM imaging. This work was supported by DFG (grant no. RI 1063/9-1 to C.R.; LI 1743/2-1 to T.L.), and the University of Stuttgart.Notes and references
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef.
- U. Feldkamp and C. M. Niemeyer, Angew. Chem., Int. Ed., 2006, 45, 1856–1876 CrossRef CAS.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- B. Sacca and C. M. Niemeyer, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef CAS.
- R. Chhabra, J. Sharma, Y. Ke, Y. Liu, S. Rinker, S. Lindsay and H. Yan, J. Am. Chem. Soc., 2007, 129, 10304–10305 CrossRef CAS.
- S. Pal, Z. Deng, B. Ding, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 2700–2704 CrossRef CAS.
- B. Ding, Z. Deng, H. Yan, S. Cabrini, R. N. Zuckermann and J. Bokor, J. Am. Chem. Soc., 2010, 132, 3248–3249 CrossRef CAS.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS.
- S. M. Douglas, J. J. Chou and W. M. Shih, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6644–6648 CrossRef CAS.
- M. J. Berardi, W. M. Shih, S. C. Harrison and J. J. Chou, Nature, 2011, 476, 109–113 CrossRef CAS.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS.
- V. J. Schüller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 12, 9696–9702 CrossRef.
- For a recent review, see: A. Rajendran, M. Endo and H. Sugiyama, Angew. Chem., Int. Ed., 2012, 51, 874–890 CrossRef CAS.
- M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2010, 132, 1592–1597 CrossRef CAS.
- N. V. Voigt, T. Torring, A. Rotaru, M. F. Jacobsen, J. B. Ravnsbaek, R. Subramani, W. Mamdouh, J. Kjems, A. Mokhir, F. Besenbacher and K. V. Gothelf, Nat. Nanotechnol., 2010, 5, 200–203 CrossRef CAS.
- I. H. Stein, C. Steinhauer and P. Tinnefeld, J. Am. Chem. Soc., 2011, 133, 4193–4195 CrossRef CAS.
- I. H. Stein, V. Schüller, P. Böhm, P. Tinnefeld and T. Liedl, ChemPhysChem, 2011, 12, 689–695 CrossRef CAS.
- H. Gu, J. Chao, S.-J. Xiao and N. C. Seeman, Nature, 2010, 465, 202–205 CrossRef CAS.
- C. Yanisch-Perron, J. Vieira and J. Messing, Gene, 1985, 33, 103–119 CrossRef CAS.
- M. Endo, T. Sugita, Y. Katsuda, K. Hidaka and H. Sugiyama, Chem.–Eur. J., 2010, 16, 5362–5368 CrossRef CAS.
- A. Rajendran, M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, ACS Nano, 2011, 5, 665–671 CrossRef CAS.
- M. Endo, T. Sugita, A. Rajendran, Y. Katsuda, T. Emura, K. Hidaka and H. Sugiyama, Chem. Commun., 2011, 47, 3213–3215 RSC.
- A. Rajendran, M. Endo and H. Sugiyama, Curr. Protoc. Nucleic Acid Chem., 2012, 48, 12.9.1–12.9.18 Search PubMed.
- Z. Zhao, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 1414–1417 CrossRef CAS.
- Z. Zhao, Y. Liu and H. Yan, Nano Lett., 2011, 11, 2997–3002 CrossRef CAS.
- W. Liu, H. Zhong, R. Wang and N. C. Seeman, Angew. Chem., Int. Ed., 2011, 50, 264–267 CrossRef CAS.
- H. Zhang, J. Chao, D. Pun, H. Liu, Q. Huang and C. Fan, Chem. Commun., 2012, 48, 6405–6407 RSC.
- B. Wei, M. Dai and P. Yin, Nature, 2012, 485, 623–626 CrossRef CAS.
- P. Rothemund and E. Sloth-Andersen, Nature, 2012, 485, 584–585 CrossRef CAS.
- E. Pound, J. R. Ashton, H. A. Becerril and A. T. Woolley, Nano Lett., 2009, 9, 4302–4305 CrossRef CAS.
- B. Högberg, T. Liedl and W. M. Shih, J. Am. Chem. Soc., 2009, 131, 9154–9155 CrossRef.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS.
- A. N. Kapanidis, N. K. Lee, T. A. Laurence, S. Doose, E. Margeat and S. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8936–8941 CrossRef CAS.
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS.
- Y. Ke, S. M. Douglas, M. Liu, J. Sharma, A. Cheng, A. Leung, Y. Liu, W. M. Shih and H. Yan, J. Am. Chem. Soc., 2009, 131, 15903–15908 CrossRef CAS.
- (a) C. F. Bleczinski and C. Richert, J. Am. Chem. Soc., 1999, 121, 10889–10894 CrossRef CAS; (b) Z. Dogan, R. Paulini, J. A. Rojas Stütz, S. Narayanan and C. Richert, J. Am. Chem. Soc., 2004, 126, 4762–4763 CrossRef CAS; (c) J. Tuma, W. H. Connors, D. H. Stitelman and C. Richert, J. Am. Chem. Soc., 2002, 124, 4236–4246 CrossRef CAS; (d) S. Egetenmeyer and C. Richert, Chem.–Eur. J., 2011, 17, 11813–11827 CrossRef CAS.
- (a) K. M. Guckian, B. A. Schweitzer, R. X.-F. Ren, C. J. Sheils, P. L. Paris, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 8182–8183 CrossRef CAS; (b) S. Hainke, S. Arndt and O. Seitz, Org. Biomol. Chem., 2005, 3, 4233–4238 RSC.
- S. Narayanan, J. Gall and C. Richert, Nucleic Acids Res., 2004, 32, 2901–2911 CrossRef CAS.
- S. Smirnov, T. J. Matray, E. T. Kool and C. de los Santos, Nucleic Acids Res., 2002, 30, 5561–5569 CrossRef CAS.
- M. E. Østergaard and P. J. Hrdlicka, Chem. Soc. Rev., 2011, 40, 5771–5788 RSC.
- Catalog no. 10-1987, Glen Research Inc., Sterling, VA 20164, USA Search PubMed.
- For earlier reports on UV-melting curves of origami, see: (a) A. Rajendran, M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2011, 133, 14488–14491 CrossRef CAS; (b) J. Song, J.-M. Arbona, Z. Zhang, L. Liu, E. Xie, J. Elezgaray, J.-P. Aime, K. V. Gothelf, F. Besenbacher and M. Dong, J. Am. Chem. Soc., 2012, 134, 9844–9847 CrossRef CAS.
- (a) G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230–3232 CrossRef CAS; (b) A. J. Boersma, B. de Bruin, B. L. Feringa and G. Roelfes, Chem. Commun., 2012, 48, 2394–2396 RSC.
- See e.g.: C. Kröner, M. Röthlingshoefer and C. Richert, J. Org. Chem., 2011, 76, 2933–2936 CrossRef.
- See e.g.: (a) C. Holzhauser and H. A. Wagenknecht, ChemBioChem, 2012, 13, 1136–1138 CrossRef CAS; (b) S. M. Biner and R. Häner, ChemBioChem, 2011, 12, 2733–2736 CrossRef CAS.
- C. Prestinari and C. Richert, Chem. Commun., 2011, 47, 10824–10826 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full sequence of M1.3, alternative restriction reactions, sequences and origami designs, ALEX data, estimated cost of producing M13, additional melting data for origami, and MALDI spectra of individual capped oligonucleotides. See DOI: 10.1039/c2nr32393a |
| This journal is © The Royal Society of Chemistry 2013 |