
Near-infrared Ag2Se quantum dots with distinct absorption features and high fluorescence quantum yields†
Abstract
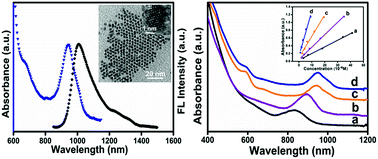
Near-infrared Ag2Se quantum dots (QDs) with distinct absorption features ranging between 830–954 nm and fluorescence quantum yields up to 23.4% were controllably synthesized using a phosphine-free approach. Based on the distinct absorption features, the molar extinction coefficients of the Ag2Se QDs have been determined.
 Please wait while we load your content...
Please wait while we load your content...

