The effect of a thieno[2,3-b]pyridine PLC-γ inhibitor on the proliferation, morphology, migration and cell cycle of breast cancer cells†
Euphemia
Leung
a,
Joy M.
Hung
b,
David
Barker
b and
Jóhannes
Reynisson
*b
aAuckland Cancer Society Research Centre, The University of Auckland, New Zealand
bSchool of Chemical Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: j.reynisson@auckland.ac.nz; Fax: +64-9-373-7422; Tel: +64-9-373-7599 ext. 83746
First published on 22nd November 2013
Abstract
3-Amino-N-(3-chlorophenyl)-5-oxo-5,6,7,8-tetrahydrothieno[2,3-b]quinoline-2-carboxamide (compound 1) is a putative phosphoinositide specific-phospholipase C-γ (PLC-γ) enzyme inhibitor. This enzyme is a plausible anticancer target linked to cell motility, important for the invasion and dissemination of tumour cells. In this work it is shown that IC50 values of compound 1 are in the low nanomolar range against a host of breast cancer cell lines as a consequence of anti-proliferative activity. These results correlate well with previously published results (Feng et al., Eur. Med. Chem., 54, 2012, 463–469) on tumour cell viability confirming the efficacy of compound 1. Flow cytometry experiments revealed that compound 1 arrests the cell cycle in the G2/M phases. Furthermore, the morphology and cell migration for the MDA-MB-231 breast cancer cell line are severely affected by administration of compound 1, which fits the hypothesis of PLC-γ inhibition. Finally, a detailed docking study against PLC reveals that the side chains of the amino acids His356, Glu341, Arg549 and Lys438 are involved in hydrogen bonding with ligand 1 as well as a lipophilic pocket is occupied by the phenyl moiety. The results presented in this study are particularly interesting because compound 1 affects triple-negative breast cancer cells, which are difficult to treat in the clinic and are in a dire need for an effective targeted therapy. We believe that compound 1 and its thieno[2,3-b]pyridine derivatives demonstrate that such a therapy can be developed.
Introduction
Phosphoinositide specific-phospholipase C (pi-PLC) is a membrane bound protein that hydrolyses phosphatidylinositol 4,5-diphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3).1,2 DAG activates the phospholipid-dependent serine/threonine kinase, protein kinase C (PKC), and IP3 promotes the release of Ca2+ from intracellular stores, which mediates cell motility and proliferation.1–4 There are six subfamilies of the mammalian pi-PLC super-family, which are classified as PLC-β, PLC-γ, PLC-δ, PLC-ε, PLC-ζ and PLC-η.5–8 In particular, many cancer cellular functions have been discovered to be regulated by PLC-γ activation, including tumorigenesis,9 metastasis development,10 cancer cell invasion11 and tumour angiogenesis12 suggesting that it represents an important therapeutic target for development of anticancer drugs.13In 2009 Reynisson et al.14 reported that a family of thieno[2,3-b]pyridines were active inhibitors of the PLC-γ2 enzyme. The efficacy of these pyridines was tested against the National Cancer Institute's sixty human tumour cell lines (NCI60).15 It was found that six analogues were excellent growth inhibitors for many of the NCI60 cell lines.16 In particular compound 1 (3-amino-N-(3-chlorophenyl)-5-oxo-5,6,7,8-tetrahydrothieno[2,3-b]quinoline-2-carboxamide) showed nanomolar growth inhibition for the cell lines MDA-MB-435 (melanoma), MDA-MB-468 (breast), NCI-H522 (non-small cell lung cancer) and SF-295 (central nervous system).16 The structure of compound 1 is shown in Fig. 1.
 | ||
| Fig. 1 The structure of the PLC-γ2 inhibitor 1 (3-amino-N-(3-chlorophenyl)-5-oxo-5,6,7,8-tetrahydrothieno[2,3-b]quinoline-2-carboxamide). | ||
The discovery of tractable drug-like inhibitors of PLC is significant since it indicates the drugability of this enzyme. With only ∼325 known molecular targets for all classes of approved drugs a new target represents the possibility of a new therapeutic approach.17 Some inhibitors are known for the PLC enzymes18–22 but none of them are suitable for small molecule drug development highlighting the significance of the discovery of the thieno[2,3-b]pyridines as hypothetical PLC inhibitors.
The aim of this study is to establish the efficacy of 1 in breast cancer cells using DNA synthesis, cell cycle, morphology and migration assays. This is of particular interest since the breast tumour cell line MDA-MB-468 is triple negative, i.e., lacking the oestrogen, progesterone, and HER2/neu receptors,23 which are incentive to targeted drug treatments. Having a clinically approved drug to combat these difficult cases is highly desirable.
Results
Chemical synthesis
Thieno[2,3-b]pyridine 1 was prepared from 1,3-cyclohexanedione 2 using a modified literature method shown in Scheme 1.24 First, dione 2 was condensed with dimethylformamide dimethyl acetal (DMFDMA) to give enamine 3 (ref. 25) which was reacted immediately with cyanothioacetamide to give thiohexahydroquinoline 4 in 82% yield. Separately, bromoacetamide 6 was prepared in quantitative yield from the acylation, at 0 °C, of 3-chloroaniline 5 with bromoacetyl bromide. It was found that reaction at higher temperatures led to further amination at the α-bromide.26 Whilst thieno[2,3-b]pyridine-2-carboxamides such as 1 are often prepared through initial formation of 2-(substituted-thio)pyridines,24,27 such as 7 (Scheme 1), it was found that reaction of thiohexahydroquinoline 4 with bromoacetamide 6, using sodium carbonate, in refluxing ethanol, gave the desired thieno[2,3-b]pyridine 1 in 77% yield in a single step. | ||
| Scheme 1 General synthetic methods. Reagents and conditions: (i) DMFDMA, DMF, 24 h; (ii) cyanothioacetamide, NaH, DMF, 24 h, 82% (2 steps); (iii) bromoacetyl bromide, Et3N, CH2Cl2, 1 h, quant.; (iv) Na2CO3, EtOH, reflux, 18 h, 77%. | ||
Biological assays
T47D, KLP1, MCF7 and its sub-lines (TamC3, TamR3, TamC6, TamR6 and TamaR7)28 are classified as oestrogen receptor positive breast cancer cell lines (ER). The SKBr3 and MDA-MB-453 cell lines are with Human Epidermal Growth Factor Receptor 2 (HER2) amplification, however, MDA-MB-453 did not have HER2 over expression. Finally, BT20, HBL100 and MDA-MB-231/435/436/453/468 are triple-negative (TN) breast cancer cell lines except MDA-MB-453, which was previously described as a breast cancer cell line but later reclassified as melanoma.Cell proliferation
The effect of compound 1 on the proliferation of cancer cell lines was measured with the thymidine uptake assay. Fifteen breast cancer and one melanoma cell lines were tested and the IC50 results are shown in Fig. 2. A drastic inhibition of DNA synthesis is seen for all the cell lines with IC50 values ranging from 77 to 570 nM. The proliferation of oestrogen receptor positive (ER) cell line MCF7 and its sub-lines (TamR7, TamC3, TamR3, TamC6 and TamR6) is severely affected with inhibition in the 100–150 nM range. The NCI60 results for compound 1 are based on the sulphorhodamine B (SRB) colorimetric assay15,29 and MCF7 gives GI50 224 nM (n = 2)16 in good agreement with the values reported here (IC50 125 nM). The ER T47D cell line is the least affected with inhibition of ∼570 nM, which is also in good agreement with the SRB test at 422 nM (n = 1).16 The triple-negative breast cancer cell lines MDA-MB-231/468 are inhibited at 357 and 163 nM, respectively, with the thymidine uptake assay. The MDA-MB-468 cell line responds similarly to the SRB assay (GI50 108 nM) but to a much less extent for MDA-MB-231 (GI50 1.99 μM).16 Finally, the melanoma tumour cell line MDA-MB-453, which was found to be the most responsive of the NCI60 panel, has GI50 58 nM (n = 2)16 in good agreement with the DNA synthesis assay at 77 nM. The calculated Pearson's correlation (R2) between the two assays is 0.839 when the values for MDA-MB-231 are excluded (see the numerical values in Table S1 in the ESI†). This gives strong evidence that the same biological effect is observed with both methods suggesting that compound 1 inhibits growth and DNA synthesis efficiently. | ||
| Fig. 2 The effect of compound 1 on the proliferation of a panel of breast cancer cell lines and one melanoma cell line. ER, oestrogen receptor positive; HER2, Human Epidermal Growth Factor Receptor 2 positive; TN, triple negative. | ||
The TN breast cancer cells are inhibited in the 157–357 nM range, indicating that compound 1 has high efficacy. This is of great interest since TN breast cancer cells, defined as the absence of staining for the oestrogen receptor, progesterone receptor, and HER2/neu, are insensitive to some of the most effective therapies available for breast cancer treatment in the clinic.
The results shown in Fig. 2 demonstrate that 1 is highly anti-proliferative based on the thymidine incorporation assay, which is also reflected in the SRB measurements for the NCI60 cell lines.
Morphology
The morphology of the TN MDA-MB-231 cell line was investigated. The clinically approved MEK kinase inhibitor trametinib was used as a negative control due to its inhibition of TN breast cancer growth along with the knowledge that it does not affect cell morphology.28,30 Trametinib's IC50 against MEK1/2 is reported to be 3.4/1.6 nM (ref. 31) and the GI50 for MDA-MB-231 is 25 nM.30 The cells treated with compound 1 are rounded in comparison with the untreated control as well as for the trametinib treated cells as shown in Fig. 3. This shows that the cytoskeletons of the breast cancer cells are heavily affected by 1 and a clear dose response is seen. It has been reported that the knock down of PLC-γ1 via an inducible short hairpin RNA system promotes the same rounded-up morphology as the administration of compound 1 reported here.32 This observation suggests that compound 1 is targeting PLC-γ1. However, it is also possible that this morphological effect is caused by the initiation of apoptotic cell death or alternatively the rounded shape of adherent cells might be a mark of mitosis. | ||
| Fig. 3 Morphological changes in the red fluorescent labelled MDA-MB-231 cell line induced by compound 1. Trametinib is used as a reference and does not instigate morphological change. Cells were incubated for 16 h. Photographs were taken by Floid imaging station (460× magnification). | ||
Migration
As migration of cancer cells is a key step of tumour metastasis, the in vitro wound healing assay33 was used to investigate the effect of compound 1 for the TN MDA-MB-231 cell line. To enable easy visualization, red fluorescent MDA-MB-231 was used in parallel. Again trametinib was used as a control and the experiments were conducted with a concentration of 1 μM for both molecules. It can be inferred from the results shown in Fig. 4 that compound 1 suppresses the migration of the breast cancer cells by ∼50% compared to the controls but also causes a substantial reduction in cell number (see Fig. 4B and C), which can be induced by cytostatic, or even cytotoxic, activity at the concentrations used. Nevertheless, it is known that PLC-γ plays an important role in metastasis10 supporting the argument that 1 is its inhibitor. | ||
| Fig. 4 Effect of compound 1 on migration. (A) Relative migration of MDA-MB-231 in 24 h. (B) The images were acquired at 0 and 24 h in an in vitro scratch assay. The lines define the areas lacking cells. (C) Red fluorescent MDA-MB-231 mKATE2 cells were used to demonstrate wound healing. 1 μM of 1 and trametinib were used. *Significantly different from untreated control (p < 0.05). | ||
Cell cycle
The inhibition of DNA synthesis shown in Fig. 2 implicates compound 1 in cell cycle arrest. In order to verify this possibility flow cytometry experiments were conducted on four breast cancer cell lines, i.e., MCF-7, T47D, MDA-MB-231 and MDA-MB-468 and the results are shown in Fig. 5.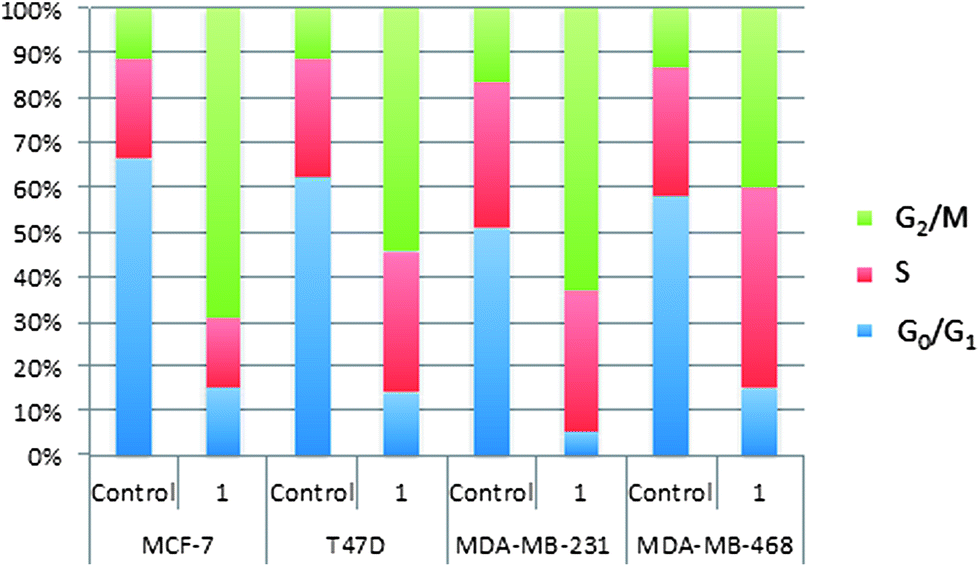 | ||
| Fig. 5 The cell cycle proportions of MCF-7, T47D, MDA-MB-231 and MDA-MB-468 breast cancer cells. Control: no inhibitors administered. 1.0 μM of 1 for 16 h. The results are the average of two independent experiments in duplicates. | ||
It is clear from the results shown in Fig. 5 that exposure to compound 1 shifts the breast cancer cells from the dominant G0/G1 phases to the G2/M phases. The DNA synthesis phase (S) is relatively unaffected. This means that the cell division is arrested during mitosis (M) or in the gap between the synthesis (S) and mitosis (M) phases, i.e., G2. It is known that the M-phase of the MDA-MB-231 cell cycle is regulated with the PLC-γ–Akt interaction triggered by fibroblast growth factor (FGF) receptors.34 Also, the nuclear PLC-β1 isoform is implicated in the G2/M cell cycle progression.35 This means that PLC inhibition can theoretically produce the results that are observed in Fig. 5. Interestingly, G2/M arrest is typical for cytotoxic microtubule inhibitors.36 Further experimental work is required to elucidate the specific mechanism of the cell cycle arrest caused by compound 1.
Molecular modelling
The PLC-δ1 crystal structure was used as a model for the PLC-γ isoform. This is justified by the conservation of the binding domain between these isoforms.37,38 Also, using the PLC-δ1 crystal structure as a docking scaffold led to the successful identification of a number of PLC-γ2 isoforms.14 The ChemPLP scoring function showed the best correlation when the co-crystallized IP3 was re-docked into the binding pocket. This was shown by the root-mean-square deviation (RMSD) between the heavy atoms of the co-crystallised ligand and its docked counterparts, i.e., ChemPLP – 0.62 Å, ASP – 1.18 Å, CS – 1.66 Å and GS – 4.61 Å. When the highest scoring ChemPLP pose for 1 was used as a reference for the poses generated for the other scoring functions, the RMSD were: ASP – 1.55 Å, CS – 1.78 Å and GS – 1.79 Å. The core tricyclic system is predicted to have a similar conformation for all of the scoring functions. However, the phenyl moiety shows more discrepancy with the meta substituted chlorine pointing into the lipophilic pocket for all the scoring functions except for ChemPLP where it occupies a niche on the surface of the protein as shown in Fig. 6A explaining the RMSD difference. The modelling showed that compound 1 has hydrogen bonding with the side chains of possibly five amino acids: histidines (His311 and His356), glutamate (Glu341), lysine (Lys438) and arginine (Arg594). The histidines interact with the oxygen in the amide moiety and Glu341 with the amine group. Lys438 and Arg549 bond with the carboxyl moiety in the cyclohexanone ring. The highest scoring ChemPLP prediction is shown in Fig. 6B. | ||
| Fig. 6 The docked configuration of 1 in the binding site of PLC-δ1 using ChemPLP. (A) The protein surface is rendered and ligand 1 is overlain with the co-crystallised IP3 shown in green. The phenyl group of 1 occupies a lipophilic cavity to the left hand side and the chlorine substitute fills a niche pocket. Red depicts a positive partial charge on the surface, blue depicts negative partial charge and grey shows neutral/lipophilic areas. (B) Hydrogen bonds are depicted as green lines between ligand 1 and the amino acids His356, Glu341, Arg549 and Lys438. | ||
In general, very good binding between compound 1 and the PLC binding domain was observed indicating the specificity of the small molecule for these enzymes.
Discussion
Overall, the PLC isoforms have a low level of amino acid sequence conservation.39,40 However, the isoforms contain two highly related structural domains with conserved amino acid sequences, the X- and Y-domains, that form the catalytic site with the exception of PLC-ζ.37,38 This means that 1, and other potential PLC-γ inhibitors, can interact strongly with many PLC isoforms. E.g. a host of PLC-γ2 inhibitors were discovered based on a virtual high throughput screening using the crystal structure of the PLC-δ1 isoform.14 Compound 1 was identified in this way and shows a good fit to the PLC-δ1 binding pocket (see Fig. 6) and inhibited PLC-γ2 at 50 μM using the 3H-PIP2 flashplate biochemical assay. The cell based results presented by Feng et al.16 and in this work can be interpreted as PLC-γ1 is the main isoform being blocked. The PLC-γ2 minor isoform is solely expressed in hematopoietic cells and can therefore be dismissed from consideration whereas PLC-γ1 is present in most cell types.7,41 This leaves PLC-β, PLC-δ, PLC-ε, and PLC-η and their subtypes. Expression of all of the isoforms and consequent testing with biochemical assays and/or X-ray crystallography studies can answer the question regarding specificity. Furthermore, synergistic, cytotoxic and off-target effects on some completely different receptors in the breast cancer cells investigated are always a possibility, e.g., blocking of G protein-coupled receptors (GPCRs). Further experimental work is required to establish the exact biological mechanism of compound 1 and its analogues.For future work for the development of PLC-γ small molecule inhibitors phosphocholine has been suggested as a potential pharmacodynamics biomarker.32 Also, an interesting biological effect is that the PLC enzymes enhance superoxide-generating activity, which can be imitated by the small molecule 2,4,6-trimethyl-N-(meta-3-trifluoromethyl-phenyl)-benzenesulfonamide.42 This biological phenomenon has also the potential to be used for assay development. Recently, Huang et al.43 published a paper on a successful high throughput screening against the PLC-δ1 isoform using a fluorescent assay, which has the potential to be modified for all of the PLC isoforms.
It is noteworthy that compound 1 is a hit compound from a virtual high throughput screening (vHTS) with a very potent anticancer activity. This clearly demonstrates that vHTS is a powerful technique and can yield very bioactive small molecules. In addition, the thieno[2,3-b]pyridines are chemically tractable, i.e., further synthetic work can be easily conducted to improve the potency against TN breast cancer cells as well as pharmacokinetic concerns can be addressed.
Conclusion
It is clear that compound 1 impedes the DNA synthesis of the breast cancer lines investigated in the nanomolar range as a consequence of anti-proliferative activity. It is shown that cell cycle arrest is instigated in the G2/M phases. Furthermore, the morphology and motility are severely affected by compound 1 for the MDA-MB-231 breast tumour cell line. Based on the results presented here as well as previous molecular modelling, biochemical and cell based work, the most plausible biological target of 1 is PLC-γ with the strong possibility of other isoforms of PLC being affected leading to a synergistic effect on tumour cells.14,16 It is clear that the PLC enzymes serve as a hypothetical target for compound 1 and until experimental evidence suggests otherwise it would appear to be the most likely target. Further experimental work is clearly needed to elucidate the question of the mechanism of 1 and its derivatives, which we are currently engaged in. The next step is to screen this class of compounds against non-cancer cell lines such as MCF-10A breast epithelial cell line, which is defined as ‘normal’. In general, it is inherently challenging to introduce small organic molecules into complex biological systems and reveal their mechanisms of action (see e.g., Michieli and Di Nicolantonio36 and references therein). Finally, the chemical space around compound 1 is now being explored by the synthesis of its analogues in order to find even more potent ligands against triple-negative breast cancer cells, which are currently difficult to treat in the clinic.Experimental procedures
Synthesis
All reactions were carried out under a nitrogen atmosphere in dry, freshly distilled solvents unless otherwise noted. Chemical shifts are expressed in parts per million (ppm) relative to TMS in 1H NMR and to the deuterated solvent in 13C NMR. 1H NMR data are reported as position (δ), relative integral, multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet; br, broad peak), coupling constant (J, Hz), and the assignment of the atom. 13C NMR data are reported as position (δ) and assignment of the atom. NMR assignments were performed using heteronuclear single quantum coherence (HSQC) and heteronuclear multiple-bond correlation (HMBC) experiments. High-resolution mass spectroscopy (HRMS) was carried out by electrospray ionization (ESI) using a MicroTOF-Q mass spectrometer. Chemical reagents were used as purchased.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
The 1H-NMR data were in agreement with the literature values.25
M.p. 80–83 °C [lit.44 m.p. 80–81 °C]; 1H NMR (400 MHz; CDCl3; Me4Si) 4.02 (2H, s, H-2), 7.15 (1H, d, J = 8.0 Hz, H-4′), 7.26–7.30 (1H, m, H-5′), 7.39 (1H, d, J = 8.0 Hz, H-6′), 7.65 (1H, s, H-2′), 8.12 (1H, br s, NH); 13C NMR (100 MHz; CDCl3) 29.3 (C-2), 118.0 (C-6′), 120.1 (C-2′), 125.3 (C-4′), 130.1 (C-5′), 134.8 (C-3′), 138.0 (C-1′), 163.4 (CO). The 1H and 13C-NMR data were in agreement with the literature values.44
O), 164.2 (C-8a), 197.0 (C-5). IR: υmax (film)/cm−1: 3427, 3353, 3311, 3074, 2955, 2876, 1675, 1631, 1581, 1525, 1475, 1402, 1301, 1231, 1094. m/z (ESI+): 396 (37ClMNa+, 7%), 394 (35ClMNa+, 16), 360 (MNa+–Cl, 100). High resolution (ESI+): Found 37ClMNa+ 396.0350, C18H14N3O2S37ClNa requires 396.0359. Found 35ClMNa+ 394.0374, C18H14N3O2S35ClNa requires 394.0387.
Cell culture
The culture conditions are described by Leung et al.28 The cell lines MCF7, T47D, MDA-MB-231, MDA-MB-468, MDA-MB-435, MDA-MB-436, SKBr3 and BT20 were purchased from the American Type Culture Collection (ATCC). KPL-1 and HBL-100 were kind gifts from Dr Chanel Smart at the University of Queensland, Australia. Cells were grown in α-MEM containing 5% foetal bovine serum (FBS). All growth media contained insulin/transferrin/selenium supplement, added according to the manufacturer's instructions (Roche), as well as penicillin (100 U mL−1) and streptomycin (100 μg mL−1). The previously published MCF7 sub-lines TamR7, TamC3, TamR3, TamC6 and TamR6 cell lines were isolated by growth of MCF7 cells in phenol-red-free RPMI containing 5% charcoal-stripped foetal bovine serum (Invitrogen, Auckland, NZ) over a period of three months to progressively increased concentrations of tamoxifen (10 nM to 1000 nM in ethanol) and then maintained for >15 months in 1000 nM tamoxifen.28 All experiments were carried out on cells grown in their respective growth media but without tamoxifen. For migration assays, MDA-MB-231 was transfected with pmKATE2-N vector (Evrogen, Russia) encoding the far-red fluorescent protein mKate2 and stable transfected cells were selected using G418.Cell proliferation assay
Cell proliferation was measured using a thymidine incorporation assay by seeding 3000 cells in each well using 96 well plates with varying concentrations of inhibitors for three days. Experiments were performed in triplicate with a minimum of two experimental repeats. Briefly, 0.04 μCi of 3H-thymidine was added to each well and incubated for five hours, after which the cells were gathered onto glass fibre filters using an automated TomTec harvester. The filters were incubated with Betaplate Scint and thymidine incorporation determined with a Trilux/Betaplate counter showing the percentage of cells incorporated with 3H-thymidine into the DNA helix.In vitro wound healing assay
Cells were cultured as confluent monolayers for 24 h followed by the performance of the wound healing assay.45 Briefly, wounds were created by removing a ∼1 mm strip of cells across the well with a standard p200 pipette tip. The wounded monolayers were washed twice to remove non-adherent cells. Light and fluorescent micrographs were taken immediately after cell removal and after 24 h. Image J (National Institute of Health freeware) was used for the analysis. The experiments were performed in triplicate wells twice. Wound healing was quantified as the relative displacement of the wound edges after 24 h.Flow cytometric analysis
Cells (1 × 106 cells) were grown in six-well plates and incubated with inhibitors for 24 h. Cells were harvested, washed with 1% FCS/PBS, resuspended in 200 μl of PBS, fixed in 2 mL of ice-cold 100% ethanol, and stored overnight at −20 °C. The cells were washed and resuspended in 1 mL of 3% FCS/PBS containing RNase (1 μg mL−1) and propidium iodide (PI) (10 μg mL−1) for 30 min at room temperature. DNA content was determined using forward scatter (FSC) intensity by PI staining based on a total 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 acquired events by FACScan cytometry.
000 acquired events by FACScan cytometry.
Statistical analysis
The data were analysed using a one-way ANOVA coupled with multiple comparisons versus treatment control applying the Holm–Sidak method correction, where p < 0.05 denotes a statistically significant difference.Modelling
Compound 1 was docked to the crystal structure of PLC-δ1 (PDB ID: 1DJX, resolution 2.3 Å),46 which was obtained from the Protein Data Bank (PDB).47,48 The Scigress Ultra version 7.7.0.47 program49 was used to prepare the crystal structure for docking, i.e., hydrogen atoms were added, the co-crystallised ligand (D-myo-inositol-1,4,5-triphosphate, IP3) as well as crystallographic water molecules were removed. The Scigress software suite was also used to build compound 1 and the MM2 (ref. 50) force field was used to optimise its structure. The center of the binding pocket was defined as the position of the Ca2+ ion (x = 126.257, y = 38.394, z = 22.370) with 10 Å radius. Hundred docking runs were allowed for each ligand with very flexible search efficiency (200%). The basic amino acids lysine and arginine were defined as protonated. Furthermore, aspartic and glutamic acids were assumed to be deprotonated. The GoldScore (GS),51 ChemScore (CS),52,53 ChemPLP54 and ASP55 scoring functions were implemented to validate the predicted binding modes and relative energies of the ligands using the GOLD v5.1 software suite.Acknowledgements
Funding for this work was obtained from the Maurice and Phyllis Paykel trust, the McClelland Trust and Auckland Medical Research Foundation. This work is also supported by the Auckland Cancer Society Research Centre.References
- S. G. Rhee, Annu. Rev. Biochem., 2001, 70, 281–312 CrossRef CAS PubMed
.
- A. Wells and J. R. Grandis, Clin. Exp. Metastasis, 2003, 20, 285–290 CrossRef CAS
- N. P. Jones, J. Peak, S. Brader, S. A. Eccles and M. Katan, J. Cell Sci., 2005, 118, 2695–2706 CrossRef CAS PubMed
- P. J. Cullen and J. Lockyer, Nat. Rev. Mol. Cell Biol., 2002, 3, 339–348 CrossRef CAS PubMed
- S. G. Rhee and K. D. Choi, J. Biol. Chem., 1992, 267, 12393–12396 CAS
- C. M. Saunders, M. G. Larman, J. Parrington, L. J. Cox, J. Royse, L. M. Blayney, K. Swann and F. A. Lai, Development, 2002, 129, 3533–3544 CAS
- F. Sekiya, B. Poulin, Y. J. Kim and S. G. Rhee, J. Biol. Chem., 2004, 279, 32181–32190 CrossRef CAS PubMed
- P. G. Suh, J. I. Park, L. Manzoli, L. Cocco, J. C. Peak, M. Katan, K. Fukami, T. Kataoka, S. Yun and S. H. Ryu, BMB Rep., 2008, 41, 415–434 CrossRef CAS
- A. Wells, Adv. Cancer Res., 2000, 78, 31–101 CAS
- G. Sala, F. Dituri, C. Raimondi, S. Previdi, T. Maffucci, M. Mazzoletti, C. Rossi, M. Iezzi, R. Lattanzio, M. Piantelli, S. Iacobelli, M. Broggini and M. Falasca, Cancer Res., 2008, 68, 10187–10196 CrossRef CAS PubMed
- C. Raimondi, A. Chikh, R. A. Wheeler, T. Maffucci and M. Falasca, J. Cell Sci., 2012, 125, 3153–3163 CrossRef CAS PubMed
- T. Maffucci, C. Raimondi, S. Abu-Hayyeh, V. Dominguez, G. Sala, I. Zachary and M. Falasca, PLoS One, 2009, 4, e8285 Search PubMed
- V. Kölsch, P. G. Charest and R. A. Firtel, J. Cell Sci., 2008, 121, 551–559 CrossRef PubMed
- J. Reynisson, W. Court, C. O'Neill, J. Day, L. Patterson, E. McDonald, P. Workman, M. Katan and S. A. Eccles, Bioorg. Med. Chem., 2009, 17, 3169–3176 CrossRef CAS PubMed
- R. H. Shoemaker, Nat. Rev. Drug Discovery, 2006, 6, 813–823 CrossRef CAS PubMed
- L. Feng, I. Reynisdóttir and J. Reynisson, Eur. J. Med. Chem., 2012, 54, 463–469 CrossRef CAS PubMed
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed
- W. Xie, H. Peng, L. H. Zalkow, Y. H. Li, C. Zhu, G. Powis and M. Kunkel, Bioorg. Med. Chem., 2000, 8, 699–706 CrossRef CAS
- W. Xie, H. R. Peng, D. I. Kim, M. Kunkel, G. Powis and L. H. Zalkow, Bioorg. Med. Chem., 2001, 9, 1073–1083 CrossRef CAS
- M. K. Homma, M. Yamasaki, S. Ohmi and Y. Homma, J. Biochem., 1997, 122, 738–742 CrossRef CAS
- J. S. Lee, M. Y. Yang, H. Yeo, J. B. Kim, H. S. Lee and J. S. Ahn, Bioorg. Med. Chem. Lett., 1999, 9, 1429–1432 CrossRef CAS
- M. Aoki, Y. Itezonzo, H. Shirai, N. Nakayama, A. Sakai, Y. Tanaka, A. Yamaguchi, N. Shimma, K. Yokose and H. Seto, Tetrahedron Lett., 1991, 32, 4737–4740 CrossRef CAS
- C. R. Tate, L. V. Rhodes, H. C. Segar, J. L. Driver, F. N. Pounder, M. E. Burow and B. M. Collins-Burow, Breast Cancer Res., 2012, 14, R79 CrossRef CAS PubMed
- V. V. Dotsenko, S. G. Krivokolysko, A. N. Chernega and V. P. Litvinov, Russ. Chem. Bull., 2002, 51, 1556–1561 CrossRef CAS
- F. A. Abu-Shanab, A. D. Redhouse, J. R. Thompson and B. J. Wakefield, Synthesis, 1995, 5, 557–560 CrossRef PubMed
- G. S. Khan, B. D. Dickson and D. Barker, Tetrahedron, 2012, 68, 1790–1801 CrossRef CAS PubMed
- F. K. Abdel-Wadood, M. I. Abdel-Monem, A. M. Fahmy and A. A. Geies, J. Chem. Res., 2008, 2, 89–94 Search PubMed
- E. Leung, N. Kannan, G. W. Krissansen, M. P. Findlay and B. C. Baguley, Cancer Biol. Ther., 2010, 9, 717–724 CrossRef CAS
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed
- J. Jing, J. Greshock, J. D. Holbrook, A. Gilmartin, X. Zhang, E. McNeil, T. Conway, C. Moy, S. Laquerre, K. Bachman, R. Wooster and Y. Degenhardt, Mol. Cancer Ther., 2012, 11, 720–729 CrossRef CAS PubMed
- T. Yamaguchi, R. Kakefuda, N. Tajima, Y. Sowa and T. Sakai, Int. J. Oncol., 2011, 39, 23–31 CAS
- M. Beloueche-Babari, J. C. Peak, L. E. Jackson, M.-Y. Tiet, M. O. Leach and S. A. Eccles, Mol. Cancer Ther., 2009, 8, 1305–1311 CrossRef CAS PubMed
- L. G. Rodriguez, X. Wu and J. L. Guan, Methods Mol. Biol., 2005, 294, 23–29 Search PubMed
- E. Broways-Poly, D. Perdereau, A. Lescuyer, A. F. Burnol and K. Cailliau, Anticancer Res., 2009, 29, 4965–4970 Search PubMed
- R. Fiume, G. Ramazzotti, G. Teti, F. Chiarini, I. Faenza, G. Mazzotti, A. M. Billi and L. Cocco, FASEB J., 2009, 23, 957–966 CrossRef CAS PubMed
- P. Michieli and F. Di Nicolantonio, Nat. Rev. Clin. Oncol., 2013, 10, 372–374 CrossRef CAS PubMed
- M. V. Ellis, S. R. James, O. Perisic, C. P. Downes, R. L. Williams and M. Katan, J. Biol. Chem., 1998, 273, 11650–11659 CrossRef CAS PubMed
- R. L. Williams and M. Katan, Structure, 1996, 4, 1387–1394 CrossRef CAS
- J. B. Park, C. S. Lee, J. H. Jang, J. Ghim, Y. J. Kim, S. You, D. Hwang, P. G. Suh and S. H. Ryu, Nat. Rev. Cancer, 2012, 12, 782–792 CrossRef CAS PubMed
- C. M. Vines, Adv. Exp. Med. Biol., 2012, 740, 235–254 CrossRef CAS
- J. I. Wilde and S. P. Watson, Cell. Signalling, 2001, 13, 691–701 CrossRef CAS
- Y.-S. Bae, T. G. Lee, J. C. Park, J. H. Hur, Y. Kim, K. Heo, J.-Y. Kwak, P.-G. Suh and S. H. Ryu, Mol. Pharmacol., 2003, 63, 1043–1050 CrossRef CAS
- W. Huang, M. Barrett, N. Hajicek, S. Hicks, T. Kendall Harden, J. Sondek and Q. Zhang, J. Biol. Chem., 2013, 288, 5840–5848 CrossRef CAS PubMed
- K. K. Adhikary, C. K. Kim, B. Lee and H. W. Lee, Bull. Korean Chem. Soc., 2008, 29, 191–195 CrossRef CAS
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS PubMed
- L. O. Essen, O. Perisic, M. Katan, Y. Wu, M. F. Roberts and R. L. Williams, Biochemistry, 1997, 36, 1704–1718 CrossRef CAS PubMed
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed
- H. Berman, K. Henrick and H. Nakamura, Nat. Struct. Biol., 2003, 10, 980 CrossRef CAS PubMed
- Scigress Explorer Ultra Version 7.7.0.47, Fijitsu Limited, 2000–2007 Search PubMed.
- N. L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127–8134 CrossRef CAS
- G. Jones, P. Willet, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed
- M. D. Eldridge, C. Murray, T. R. Auton, G. V. Paolini and P. M. Mee, J. Comput.-Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed
- O. Korb, T. Stützle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS PubMed
- W. T. M. Mooij and M. L. Verdonk, Proteins, 2005, 61, 272–287 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3md00290j |
| This journal is © The Royal Society of Chemistry 2014 |