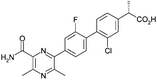
Optimisation of biphenyl acetic acid inhibitors of diacylglycerol acetyl transferase 1 – the discovery of AZD2353†
Michael J.
Waring
*,
Alan M.
Birch
,
Susan
Birtles
,
Linda K.
Buckett
,
Roger J.
Butlin
,
Leonie
Campbell
,
Pablo Morentin
Gutierrez
,
Paul D.
Kemmitt
,
Andrew G.
Leach
,
Philip A.
MacFaul
,
Charles
O'Donnell
and
Andrew V.
Turnbull
Cardiovascular and Gastrointestinal Innovative Medicines Unit, AstraZeneca R&D, Mereside, Alderley Park, Macclesfield, Cheshire, SK10 4TG, UK. E-mail: mike.j.waring@astrazeneca.com; Tel: +44 (0)1625 230942
First published on 25th October 2012
Abstract
Inhibition of diacylglycerol acetyl transferase 1 is of great interest in the treatment of diabetes, obesity and other diseases that constitute the metabolic syndrome. Small molecule inhibitors of the enzyme are often dogged with physicochemical property-related problems such as poor solubility. Herein, the optimisation of a series of biphenyl acetic acid inhibitors is reported. Focus on ligand efficiency and ligand lipophilicity efficiency and a strategy based on conformational restriction led to compounds with excellent potency and ADMET properties, culminating in the discovery of AZD2353.
Introduction
Diacylglycerol acyl transferase-1 (DGAT1) is a membrane bound enzyme found in the endoplasmic reticulum, notably in enterocytes and adipocytes. It catalyses the acylation of diacylglycerols with fatty acid-coenzyme A active esters to form triglycerides. Clearly, this is of critical importance in lipid biosynthesis and hence is a target of great interest in the search for new treatments for diabetes, obesity and other diseases that constitute the metabolic syndrome.1 Small molecule inhibitors of DGAT1 have progressed into the clinic from Novartis (LCQ-908, phase II), Pfizer (PF-4620110, phase I – discontinued) and AstraZeneca (AZD-7687, phase I – discontinued, structure undisclosed) and preclinical compounds have been disclosed by Roche (RO-6036), Abbott (A-922500) and AstraZeneca (AZD-3988), amongst others (Fig. 1).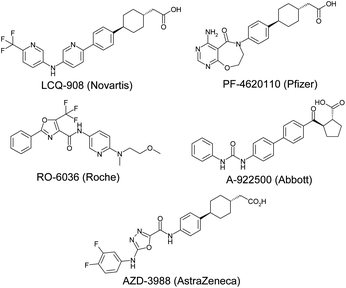 | ||
| Fig. 1 Known DGAT1 inhibitors. | ||
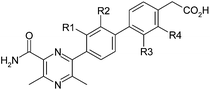
As part of an ongoing drug discovery programme, the biphenyl acetic acid 1 (Table 1) was identified as an inhibitor of DGAT1, which was promising as an attractive start point due to its low lipophilicity/high lipophilic ligand efficiency (LLE) and small size/high ligand efficiency (LE).2 These features were of critical importance for the optimisation of an acidic series in particular due to the need to maintain low lipophilicity and hence desirable physical properties and a small size to achieve acceptable permeability for such polar molecules.3 A second attractive feature was the very high solubility of the compounds given the precedented solubility issues with a number of DGAT1 inhibitor series previously disclosed.4 Herein, the programme of work to improve potency to the level required for a candidate drug, whilst maintaining these attractive features is described.
Results and discussion
The initial strategy to improve potency without significant increases in molecular size or lipophilicity was based on conformational analysis. The three aryl rings of 1 were expected to have little preference for any particular conformation and constraining the unbound molecule in its, albeit unknown, active conformation would likely improve potency by reducing the entropic penalty of binding associated with loss of rotational freedom. Accordingly, initial structural modifications were concerned with the introduction of a chloro-substituent in the R1-4 positions (Table 1) to perturb the torsional energy profile of the molecule.Since it had been observed previously within the project that DGAT1 inhibitors often carried ACAT1 activity,5 despite no ACAT1 activity being observed for 1, all compounds were assayed for both activities simultaneously.
The R1 and R4 chloro derivatives 2 and 5 lost DGAT1 potency relative to 1. The R2 chloro 3 gained almost an order of magnitude but the most marked effect was observed with R3 chloro 4, which exhibited a two log unit increase in potency relative to 1, achieving the level of activity required for a drug candidate. It is notable within this set that changing only the position of chloro substitution can alter the observed activity by three orders of magnitude overall with little change in log D and constant molecular weight (LLE and LE ranges of 2.4 and 0.15 units respectively).
The conformational profiles computed with B3LYP/6-31G* for rotation about a phenyl-pyrazine bond (with fully substituted pyrazine) for model compounds with and without chlorine to mimic 2 were computed (Fig. 2 and 3). These show that if the change in LLE between 1 and 2 is due to a conformational effect then the bioactive conformation is likely to involve a twist about this bond of no more than ±30°. In this range of dihedral angles, the model for 1 has a low energy while that of the model for 2 is significantly higher. The effect of either R2 or R3 being Cl on the torsion linking the two phenyl rings is likely to be very similar; both favour a twist of about ±60° and flat conformations (twist less than about 45°) are excluded. That 4 has a higher LLE than 3 suggests that the Cl in the R3 position is able to form a constructive interaction with the DGAT enzyme. The drop in LLE caused by adding a Cl to R4 is consistent with a bioactive conformation that has the C–CO2H bond at an angle of ±60° twist, the region in which the R4 = H has an energy that is substantially lower than that of R4 = Cl.
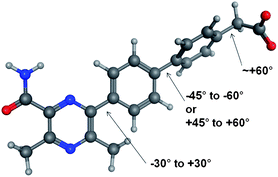 | ||
| Fig. 2 Optimal torsional angles for 2 about the critical rotatable bonds. | ||
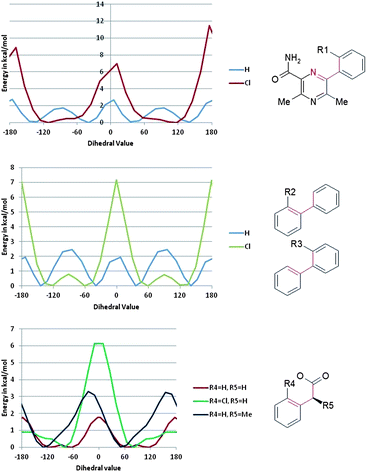 | ||
| Fig. 3 Torsional profiles computed with B3LYP/6-31G* for rotation of the indicated dihedral. | ||
Some ACAT1 activity was observed with the more potent DGAT1 inhibitors 3 and 4. Whilst this activity was only weak, the potential consequences of ACAT1 inhibition were believed to be severe enough to require a significant margin between the two activities for a compound to be safe enough to progress.6 This margin was set minimally at 100-fold, with an ideal compound having a margin significantly in excess of this figure.
To follow up these encouraging potency improvements, the effect of varying the R2 and R3 substituents was investigated. In the R2 position (Table 2), the methyl derivative 6 was broadly equivalent to 3 and the fluoro derivative 7 showed equipotency with a slightly improved ratio to ACAT1 and increased LLE. More polar substituents methoxy-(8) and cyano-(9) led to reduced potency but the magnitude of decrease was not consistent with the drop in log D. The cyano derivative actually showed increased LLE and a loss of measurable ACAT1 activity. These LLE changes are possibly suggestive of specific interactions being made by these substituents with the protein in addition to their effects on lipophilicity and conformation. However, both of these more polar substituents decreased the overall log D of the molecule to such an extent that compromised permeability was observed (for example, 8 and 9 had A to B Papp values in MDCK cells of 59 and 12 nm s−1 respectively) and hence were not followed up.
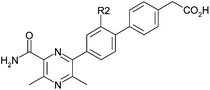
With this information in hand, modifications of the R3 position were selected so as not to lower the log D significantly (Table 3). In this case, chloro substitution proved to be optimal with methyl (10) and fluoro (11) both showing a loss of potency, LLE and ACAT1 selectivity.
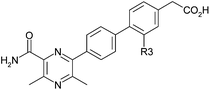
A further position of substitution likely to have an effect on conformation and hence potency was the methylene substituent α-to the carboxyl group. Substitution in this position would also obviously introduce chirality to the molecule and possibly affect the ACAT1 selectivity. In addition, phenylacetic acids are known to sometimes have a propensity to form reactive acyl glucuronide metabolites7 and it was postulated that α-substitution would decrease the risk associated with this potential issue.
Accordingly, the effect of methyl substitution in this position was investigated (Table 4). In this case, gratifyingly, the DGAT1 and ACAT1 potencies do indeed show orthogonal SAR with the S-enantiomer 12 showing increased DGAT1 potency (0.5 log increase, constant LLE) relative to 1, and the R-enantiomer 13 showing similar DGAT1 potency to 1 and introducing measurable ACAT1 activity.
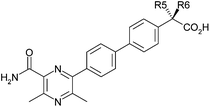
The addition of this methyl group shifts the conformational minimum into one of two regions that were hypothesised to be the bioactive conformation based on the effect of adding Cl at R4. By desymmetrising the rotational profile, the methyl group favours two of the four potential positions of the acid group and so provides an entropic advantage for the correct enantiomer and an enthalpic disadvantage for the other enantiomer.
These modifications gave confidence that a candidate suitable for development could be found within this series and that a combination of the exemplified substituents was likely to deliver the required profile. However, key considerations for selection of a development candidate were ACAT1 selectivity, predicted dose to man and acyl glucuronide risk. All of these factors were impossible to predict from existing SAR and hence an empirical approach to finding the best compound was adopted. The entire matrix of suitable R2, R3 and R5 substituents was enumerated computationally and predictive potency and log D data generated using Free–Wilson substructure based models (see ESI†).8 Candidates of appropriate predicted potency and log D were selected for synthesis and profiled using in vitro potency and ADMET, in vivo pharmacokinetic and in vitro metabolite identification studies.
Some of the best compounds emerging from this exercise illustrate the effects of combining the beneficial R2, R3 and R5 substituents (Table 5). The R2, R3 dichloro derivative 14 shows similar potency to 4, illustrating that the potency benefits of the individual chloro substituents do not necessarily add up. This could be a conformational effect; one chloro substituent adjacent to the biphenyl bond induces a certain amount of twist, having two chloros causes the twist to be enhanced, presumably beyond that which is optimal.
|
4
|
14
|
15
|
16/AZD2353
|
|
|---|---|---|---|---|
| DGAT1 pIC50 | 8.2 | 8.1 | 7.9 | 8.1 |
| ACAT pIC50 | 5.9 | 6.0 | 5.2 | 5.3 |
| ACAT1/DGAT1 ratio | 190 | 110 | 550 | 580 |
| Log D7.4 | 0.5 | 1.0 | 1.1 | 1.0 |
| LLE | 7.7 | 7.1 | 6.8 | 7.1 |
| LE | 0.4 | 0.38 | 0.37 | 0.37 |
| Solubility/μM | 510 | 520 | 570 | 270 |
| MDCK Papp/nm s−1 | 89 | 170 | 190 | 140 |
| %Free (rat, dog, human) | 0.5, 2.2, 1.5 | 0.2, 0.9, 0.3 | 0.6, 1.0, 0.8 | 0.3, 1.4, 0.7 |
| Rat Cl (Clu)/mL min−1 kg−1 | 1.0 (210) | 1.2 (560) | 1.8 (300) | 2.4 (700) |
| Rat Vss/L kg−1 | 0.6 | 0.5 | 0.6 | 1.3 |
| Rat F% | 88 | 100 | 98 | 100 |
| Dog Cl (Clu)/mL min−1 kg−1 | 5.2 (240) | 9.4 (1000) | 3.0 (290) | 1.9 (140) |
| Dog Vss/L kg−1 | 0.5 | 0.4 | 1.0 | 0.4 |
| Dog F% | 14 | 24 | 73 | 37 |


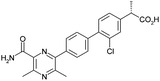
Combining R3 chloro with R5 methyl (15) retains potency with reduced ACAT1 activity relative to 4. This change, whilst clearly beneficial overall, is not consistent with that observed with the pair 1 and 12 previously where an increase in DGAT1 potency was observed with the R5 methyl substituent. A combination of all three substituents (16, R2 = fluoro, R3 = chloro, R5 = methyl) also showed retained DGAT1 potency and reduced ACAT1 activity.
All four of these compounds showed excellent ADME profiles with very high solubility and permeability characteristics. This translated into desirable pharmacokinetic profiles in both the rat and the dog. However, for acidic series such as these, human dose predictions are very sensitive to small changes in pharmacokinetic parameters (and associated errors) and hence not all apparently good profiles translate into desirable predictions. After rigorous scaling using a number of methods, 16 was deemed the compound most suitable for human dosing at an acceptable level (0.4 mg kg−1, twice daily) and was selected for development, being given the identifier AZD2353.
The acyl glucuronide formed from 16 was shown to be more stable than those derived from ibuprofen or diclofenac.9 Coupled with the low dose, this minimised the risk of this generating reactive metabolite issues.
Compound 16 showed dose proportional effects on triglyceride excursion in rat oral lipid tolerance (OLT) tests (Fig. 4)10,11 and on triglyceride synthesis (Fig. 5),10,12 achieving maximal efficacy at 1–3 mg kg−1 in both cases. PK-PD analysis indicates that the IC50 values generated for OLT (Fig. 6) and triglyceride synthesis (Fig. 7) are in very good agreement with the level of inhibition of recombinant human DGAT1, inhibition in rat liver microsomes and with a functional cell assay measuring triglyceride synthesis in the HuTu80 human gastrointestinal cell line (Table 6). This shows that 50% inhibition of DGAT1 in adipose tissue or in the GI tract can be achieved if the free plasma concentration of 16 is maintained at the in vitro IC50 value.
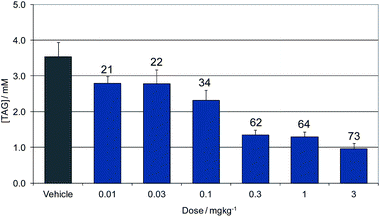 | ||
| Fig. 4 Dose response of 16 for inhibition of triglyceride excursion in a rat oral lipid tolerance test. Data show concentration of plasma triglycerides (TAG). Labels show the % reduction from vehicle dosed animals. | ||
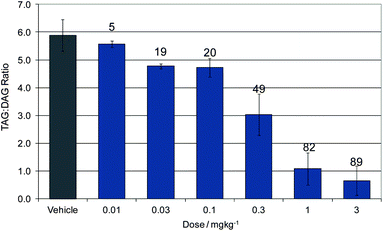 | ||
| Fig. 5 Dose response of 16 for inhibition of triglyceride synthesis. Data show the ratio of plasma triglycerides (TAG) to diglycerides (DAG). Labels show the % reduction from vehicle dosed animals. | ||
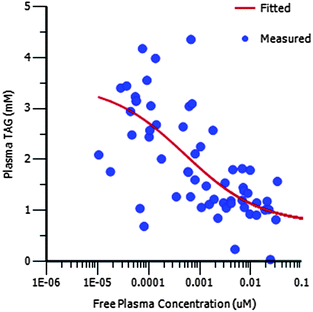 | ||
| Fig. 6 PK/PD showing the relationship between free plasma concentration and plasma TAG concentration for 16 in the OLT assay. | ||
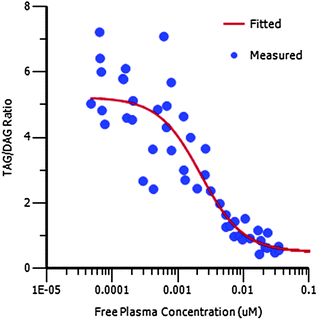 | ||
| Fig. 7 PK/PD showing the relationship between free plasma concentration and plasma TAG to DAG ratio for 16 in the TAG synthesis assay. | ||
| In vivo OLT pIC50 | 9.3 |
| In vivo TAG synthesis pIC50 | 8.7 |
| DGAT1 pIC50 | 8.1 |
| Rat microsomal pIC50 | 8.4 |
| HuTu80 TAG synthesis pIC50 | 8.4 |
Compound 16 showed a benign safety profile with no significant activity in a panel of more than 100 in vitro secondary pharmacology assays and no significant histopathological findings in in vivo safety studies.
Synthesis
The synthesis of 16 commenced with sequential palladium mediated couplings of 17, firstly to install the pyrazine moiety and then, after conversion of the phenol to the triflate, the phenyl acetic acid to afford 18 (Scheme 1). The α-methyl group was installed in an enantioselective manner by interception of the enolate formed from 18 with formaldehyde to afford the alcohol 19. The hydroxyl group of 19 was then eliminated to form the acrylate 20 which was saponified to acid 21. Finally, 21 was reduced by stereoselective hydrogenation in the presence of (S)-Ru(OAc)2(BINAP) as catalyst to afford homochiral 16. The absolute configuration of 16 was established by X-ray crystallography.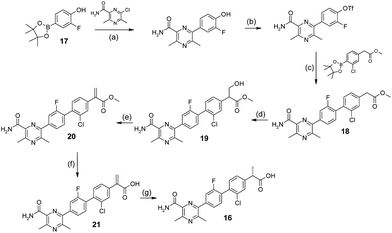 | ||
| Scheme 1 Synthesis of 16. Reagents and conditions: (a) K3PO4, DME/EtOH/H2O, 80 °C; 91%; (b) PhN(CF3SO2)2, K2CO3, THF, 120 °C; 75%; (c) Pd(dppf)Cl2·DCM, LiCl, K3PO4, DME/MeOH/H2O, 65 °C; 55%; (d) (CH2O)n, K2CO3, DMF, 0 °C to rt; 100%; (e) CH3SO2Cl, Et3N, THF, rt; 55%; (f) KOH, t-BuOH, rt; 67%; (g) (S)-Ru(OAc)2(BINAP), H2, MeOH, 5 bar, 35 °C; 35%. | ||
Conclusions
In summary, it has been shown that substituents which provide conformational restriction provided a highly effective means of optimising this series avoiding inflation of molecular size or lipophilicity. Significant improvements in potency and selectivity were achievable with this strategy resulting in dramatic increases in LE and LLE. Finally, Free–Wilson analysis provided an effective means of predicting the best compounds within the series resulting in the identification of 16 as a highly potent, selective inhibitor of DGAT1 with promising in vivo activity and a candidate for progression to the clinic.Acknowledgements
The research was sponsored by AstraZeneca. All authors were employees of AstraZeneca at the time of the conduct of the research. The authors thank Craig Donald, Neal Fazakerley, Mark D. Fenwick, Adele Lloyd, Adrian Pickup, Wendy Snelson and Rolf P. Walker for the synthesis and purification of test compounds, Usha Chauhan for the testing of compounds in biological assays and colleagues in physical chemistry, DMPK and safety assessment for the provision of secondary ADMET data.Notes and references
- A. M. Birch, L. K. Buckett and A. V. Turnbull, Curr. Opin. Drug Discovery Dev., 2010, 13, 489 Search PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Drug Discovery Today, 2004, 9, 430 CrossRef.
- M. J. Waring, Bioorg. Med. Chem. Lett., 2009, 19, 2844 CrossRef CAS.
- W. McCoull, M. S. Addie, A. M. Birch, S. Birtles, L. K. Buckett, R. J. Butlin, S. S. Bowker, S. Boyd, S. Chapman, R. D. M. Davies, C. S. Donald, C. P. Green, C. Jenner, P. D. Kemmitt, A. G. Leach, G. C. Moody, P. Morentin Gutierrez, N. J. Newcombe, T. Nowak, M. J. Packer, A. T. Plowright, J. Revill, P. Schofield, C. Sheldon, S. Stokes, A. V. Turnbull, S. J. Y. Wang, D. P. Whalley and J. M. Wood, Bioorg. Med. Chem. Lett., 2012, 22, 3873 CrossRef CAS.
- A. T. Plowright, P. Barton, S. Bennett, A. M. Birch, S. Birtles, L. K. Buckett, R. J. Butlin, R. Davies, A. Ertan, P. M. Gutierrez, P. D. Kemmitt, A. G. Leach, P. H. Svensson, A. V. Turnbull and M. J. Waring, Med. Chem. Commun. 10.1039/c2md20187a.
- E. Floetmann, L. K. Buckett, A. V. Turnbull, C. Hammond, C. Hallberg, A. Birch, D. Lees and H. B. Jones, in preparation.
- S. Bolze1, N. Bromet, C. Gay-Feutry, F. Massiere, R. Boulieu and T. Hulot, Drug Metab. Dispos., 2002, 30, 404 Search PubMed.
- S. M. J. Free and J. W. Wilson, J. Med. Chem., 1964, 7, 395 CrossRef CAS.
- L. Z. Benet, H. Spahn-Langguth, S. Iwakawa, C. Volland, T. Mizuma, S. Mayer, E. Mutschler and E. T. Lin, Life Sci., 1993, 53, PL141 Search PubMed.
- A. M. Birch, S. Birtles, L. K. Buckett, P. D. Kemmitt, G. J. Smith, T. J. D. Smith, A. V. Turnbull and S. J. Y. Wang, J. Med. Chem., 2009, 52, 1558 CrossRef CAS.
- K. K. Buhman, S. J. Smith, S. J. Stone, J. J. Repa, J. S. Wong, F. F. Knapp Jr, B. J. Burri, R. L. Hamilton, N. A. Abumrad and R. V. Farese Jr, J. Biol. Chem., 2002, 277, 25474 Search PubMed.
- C. Silversand and C. Haux, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 703, 7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols and references thereof are given in the ESI. See DOI: 10.1039/c2md20190a |
| This journal is © The Royal Society of Chemistry 2013 |