Design and synthesis of a novel series of cyclohexyloxy-pyridyl derivatives as inhibitors of diacylglycerol acyl transferase 1†
Alleyn T.
Plowright
*a,
Peter
Barton
b,
Stuart
Bennett
b,
Alan M.
Birch
b,
Susan
Birtles
b,
Linda K.
Buckett
b,
Roger J.
Butlin
b,
Robert D. M.
Davies
b,
Anne
Ertan
c,
Pablo Morentin
Gutierrez
b,
Paul D.
Kemmitt
b,
Andrew G.
Leach
b,
Per H.
Svensson
cd,
Andrew V.
Turnbull
b and
Michael J.
Waring
b
aCardiovascular and Gastrointestinal Innovative Medicines Unit, AstraZeneca R&D, Pepparedsleden 1, Mölndal, 43183, Sweden. E-mail: alleyn.plowright@astrazeneca.com; Tel: +46 (0)31 7761572
bCardiovascular and Gastrointestinal Innovative Medicines Unit, AstraZeneca R&D, Mereside, Alderley Park, Cheshire, SK10 4TG, UK
cMedicines Evaluation, AstraZeneca R&D, 151 85 Södertälje, Sweden
dDepartment of Applied Physical Chemistry, Royal Institute of Technology, SE-10044, Stockholm, Sweden
First published on 17th August 2012
Abstract
A novel series of potent diacylglycerol acyl transferase 1 inhibitors was developed from the clinical candidate AZD3988. Replacement of the phenyl cyclohexyl-ethanoate side chain with substituted oxy-linked side chains to introduce changes in shape and polarity, reduce lipophilicity and mask the hydrogen bond donors with internal hydrogen bond acceptors led to improvements in solubility, unbound clearance and excellent selectivity over the related enzyme acyl-coenzyme A:cholesterol acyltransferase 1. A comparison of the small molecule crystal structures of compound 4 and compound 28 is described. Compounds in this series have good ADMET properties and provide an exposure-dependent decrease in circulating plasma triglyceride levels in a rat oral lipid tolerance test.
Introduction
The rate of developing obesity is alarming considering the connection between a body mass index above 30 and diseases such as diabetes, hypertension, arterial cardiovascular disease, stroke, and some forms of cancer. Hence, there is strong demand for drugs capable of treating both obesity and type 2 diabetes. Inhibition of diacylglycerol acyl transferase-1 (DGAT-1), a membrane bound enzyme found in the endoplasmic reticulum, is of increasing interest as a mechanism for therapeutic treatment of diabetes, obesity and other diseases which together constitute metabolic syndrome.1–3 DGAT-1 deficient (Dgat-1-) mice are viable and resistant to weight gain when fed a high-fat diet and show increased insulin and leptin sensitivity.4 A number of small molecule inhibitors of DGAT-1 have been reported and the progress of some compounds into clinical development supports the potential for therapeutic use. Recently we have reported different series of DGAT-1 inhibitors,5–7 including a series of oxadiazole amides leading to the clinical candidate AZD3988 (1) (Fig. 1).6 Compound 1 contains a phenyl cyclohexyl-ethanoate side chain, a fragment which appears to be a privileged structure as it is contained within several DGAT-1 inhibitors,8–10 including LCQ-908 (2) and PF-4620110 (3) which have both progressed to the clinic.8,11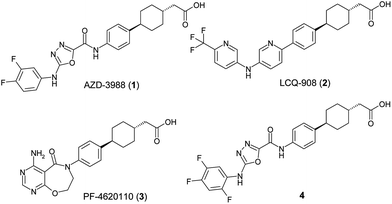 | ||
| Fig. 1 Some DGAT-1 inhibitors. | ||
As part of the ongoing research to develop the oxadiazole amide series, variations in this fragment were sought to provide structural variation and optimise the compounds further. Despite 1 displaying many positive criteria a potential area of improvement was in the physicochemical properties (thermodynamic solubility of crystalline 1 = 2 μM).12 Whilst 1 appeared to have good selectivity over its closest structural homologue, acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT-1),13 with an ∼40 fold in vitro enzyme selectivity, it was found to cause adrenal toxicities in dog toxicology studies which may impact clinical utility.14 Hence greater selectivity towards ACAT-1 was sought. Aqueous solubility is driven by a number of factors including lipophilicity and solid-state interactions in the crystal lattice. Strategies to explore both of these aspects were devised and herein our continued investigations to improve compound properties to provide an alternative series of acidic DGAT-1 inhibitors are reported.
Results and discussion
Chemistry
The synthesis of the key oxy-linked phenyl and pyridyl derivatives is described in Schemes 1 and 2. Treatment of a mixture of cis- and trans-ethyl-4-hydroxycyclohexane carboxylate 5 with sodium hydride gave the alkoxide anion which underwent a nucleophilic aromatic substitution reaction with 1-fluoro-4-nitrobenzene 6. This provided a mixture of cis- and trans-intermediates, which were separated by flash column chromatography to provide the pure cis-cyclohexyl product, which was hydrogenated to give the substituted aniline 7. Acylation of 7 using methyl oxalylchloride gave the oxalate ester which was displaced with hydrazine monohydrate to give 8. The mono-hydrazide 8 was reacted with the required substituted aryl isothiocyanate and then cyclised using 1-ethyl-3-(3-di-methylaminopropyl)carbodiimide hydrochloride to give the 1,3,4-oxadiazole ring system which finally underwent ester hydrolysis to give the desired acid derivatives 9.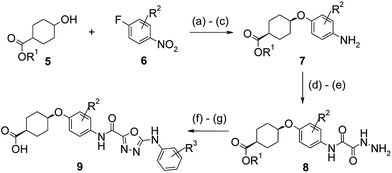 | ||
Scheme 1 Synthesis of oxy-linked phenyl derivatives. Reagents and conditions: (a) NaH (60% in oil), DMF, 0 °C to rt; (b) 6, DMF, 110 °C; (c) Pd (10% on C), H2, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1.5:1); (d) methyl oxalylchloride, pyridine, CH2Cl2; (e) N2H4·H2O, EtOH; (f) (i) R3C6H4NCS, DMA, (ii) EDC, 60 °C; and (g) LiOH, THF:H2O:MeOH (1:1:2), 35 °C. :THF (1.5:1); (d) methyl oxalylchloride, pyridine, CH2Cl2; (e) N2H4·H2O, EtOH; (f) (i) R3C6H4NCS, DMA, (ii) EDC, 60 °C; and (g) LiOH, THF:H2O:MeOH (1:1:2), 35 °C. | ||
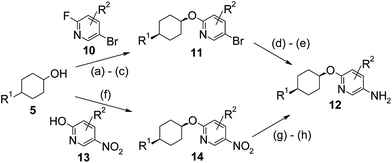 | ||
| Scheme 2 Synthesis of oxy-pyridyl derivatives. Reagents and conditions: (a) NaH (60% in oil), DMF, 0 °C to rt; (b) 10, DMF, 100 °C; (c) if R1 = COOH, then HCl, MeOH; (d) benzophenone imine, PdOAc2, Cs2CO3, (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, THF, 60 °C; (e) Pd (10% on C), H2, ethyl acetate; (f) 13, diisopropylazodicarboxylate, PPh3, THF, 180 °C, microwave; (g) for 28 where R2 = 6-Cl, Bu4NCN, DMSO, hexafluorobenzene, 0 °C to rt; and (h) Pd (10% on C), H2, ethyl acetate. | ||
The oxy-linked pyridyl derivatives were synthesized in one of the two ways (Scheme 2). Firstly, treating the anion of 5 with the substituted 2-fluoro-5-bromopyridine in an analogous fashion to the first step in Scheme 1 gave the 2-alkoxy-5-bromopyridine 11. Buchwald coupling of 11 with benzophenone imine under palladium catalysis followed by hydrogenation of the imine gave the required pyridylamine 12. Alternatively, performing a Mitsunobu reaction between the cyclohexanol 5 and the substituted 2-hydroxypyridine 13 using diisopropylazodicarboxylate and triphenylphosphine gave the intermediate 14 which was hydrogenated as before to give the required pyridylamine 12. In the case of compound 28, where a fluoro-substituent was required ortho to the pyridyl nitrogen, the Mitsunobu reaction was performed using 6-chloro-5-nitropyridin-2-ol (i.e. R2 = 6-Cl in 13) to give 14. The 6-chloro group was then converted to the 6-fluoro group using tetrabutylammonium cyanide and hexafluorobenzene in DMSO, which upon hydrogenation gave the pyridylamine intermediate 12 with the required substitution pattern. The pyridylamines 12 were transformed into the final oxadiazole acids 9 using essentially the same chemistry as described in steps (d)–(g) in Scheme 1.
Compound profiling
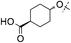
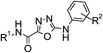
During the discovery of 1 it had been found that the carboxylic acid functionality was important for combining potent DGAT-1 inhibition in vitro with good oral activity in vivo and hence, for this study the SAR focused on acidic derivatives. The lipophilicity of 1 is not high (log D = 3, Table 1),12 but as the permeability of 1, as measured in MDCK cells (Papp A-B 12 × 10−6 cm s−1), is high there is a scope for lipophilicity to be reduced to improve compound characteristics including physicochemical properties.15 In parallel, crystals of the closely related derivative 4 (Fig. 1), itself being a potent DGAT-1 inhibitor (IC50 = 3 nM), suitable for X-ray structural determination were grown to understand the interactions existing in the solid state within this series (Fig. 2).16 A view of the X-ray structure in Fig. 2a shows the plane of molecules that are arranged adjacent to one another and reveals two interesting conformational features and a number of intermolecular interactions. The first conformational feature is that the amide carbonyl is oriented away from the oxadiazole nitrogens. Boström et al. have shown that the 1,3,4-oxadiazole has a large associated dipole moment.17 The amide carbonyl also has a large associated dipole moment and the overall conformational energy is minimized when these two dipole moments oppose one another as is seen in the observed conformation. The second conformational feature is that the N–H bond of the group linking the oxadiazole to the phenyl ring has the ortho-fluoro substituent towards it rather than the unsubstituted ortho position. Again, the dipole moment of the N–H bond is opposed to that of the C–F bond in this conformation whereas the alternative conformation would present a C–H bond with a reinforcing dipole which would be higher in energy. These conformational preferences are confirmed by quantum mechanical calculations (RHF/6-31G*) on a fragment of 4 with the cyclohexyl-ethanoic acid group removed, which shows that the lowest energy conformation is the one found in the crystal structure.18,19 Rotation by 180° about the oxadiazole–amide bond costs 5.0 kcal mol−1. Rotation about the oxadiazole–N bond by the same angle costs 2.1 kcal mol−1. No minimum energy structure could be found for rotation about the N-phenyl bond without also rotating about the oxadiazole–N bond.|
|
|||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | hDGAT-1 IC50a (μM) | hACAT-1 IC50a (μM) | Log D | Sol.b (μM) | LLE | |
| a All IC50 values reported are mean values from ≥2 experiments. b Measured by incubation of crystalline materials for 24 h at pH 7.4.12 | |||||||
| 1 | — | — | 0.002 | 0.08 | 3.0 | 2 | 5.6 |
| 15 |
|
3,4-diF | 0.14 | — | 2.3 | 6 | 4.6 |
| 16 |
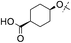
|
3,4-diF | 0.004 | 1.1 | — | 3 | — |
| 17 |
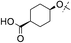
|
4-F | 0.005 | 1.8 | 1.2 | 5 | 7.1 |
| 18 |
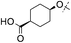
|
4-CN | 0.018 | 14.3 | 1.4 | — | 6.3 |
| 19 |
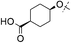
|
2,4,5-triF | 0.007 | 0.45 | 1.9 | 43 | 6.2 |
| 20 |
|
2,4,5-triF | 0.008 | 1.3 | 1.9 | 27 | 6.2 |
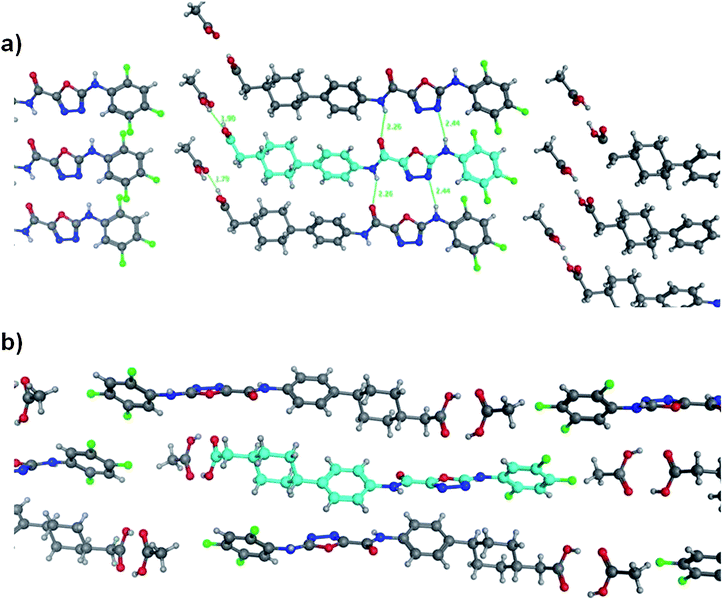 | ||
| Fig. 2 Two views of the crystal structure of the acetic acid co-complex of compound 4. Panel (a) shows a view from above the plane of adjacent molecules and panel (b) shows how these planes stack. | ||
The intermolecular interactions revealed by the crystal structure include (i) the amide NH interacting with the carbonyl of an adjacent amide; (ii) the aniline NH interacting with one of the oxadiazole nitrogens and (iii) the carboxylate residue in 4 forming a tight dimer with the acetic acid also present in the structure. The quantum mechanical calculations can also be used to investigate the strengths of intermolecular interactions. In particular, Kenny has shown that electrostatic potential calculations correlate closely with observed values of log Kα and log Kβ which are experimental measures of a molecule's hydrogen bonding capacity.20,21 These calculations reveal that the same features that drive the conformational preference for the molecule also influence its ability to interact with other molecules. The conformation of the same model compound described above that has fluorine oriented towards the NH has a computed log Kα for the NH of 2.02 while the same compound lacking the ortho fluoro has a log Kα of 2.24. Hence, any through-bond polarizing effect of fluorine is counteracted by a through-space masking of the dipole of the N–H bond by that of the C–F bond. The alternative view of the crystal structure in Fig. 2b shows how the molecules stack on top of one another and reveals that the various layers alternate the orientation of the molecule and that the molecules are slightly twisted with respect to one another indicating that there are no significant stacking interactions between the layers.
Hence, the initial design strategies to improve physicochemical properties were focused on reducing the overall lipophilicity and disrupting the intricate framework of intra- and intermolecular hydrogen bonding observed in the crystal lattice of 4 by (i) introducing changes in shape and polarity into the side chain to reduce the efficiency of crystal packing; (ii) reducing the overall lipophilicity of the phenyl cyclohexyl-ethanoate side chain, whilst maintaining the high ligand lipophilicity efficiency (LLE) of 1; and (iii) masking the hydrogen bond donors which are required for DGAT-1 inhibition in this series with internal hydrogen bond acceptors to break up the hydrogen bonding network in the crystal lattice to increase solubility. Compounds were tested in vitro for inhibition towards recombinant human DGAT-1 (hDGAT-1).5 To ensure selectivity, inhibition of human ACAT-1 (hACAT-1) and human DGAT-2, which although serving the same function is only distantly related to DGAT-1 structurally,22 was monitored to find opportunities for enhancing selectivity. Solubility was measured on crystalline materials.12
Initial attempts to introduce changes in the shape and increase the polarity in the side chain were made through introduction of an oxygen atom. The phenyl-trans-cyclohexyl unit in 1 is linear and relatively flat and therefore packs well in the lattice. The introduction of an oxygen linker completely changes the shape to the one that packs less efficiently. This can be exaggerated further by changing from a trans-configuration in which both substituents can be placed in equatorial positions to a cis-configuration in which one of the substituents must become axial. Axially substituted cyclohexanes have a shape that tessellates less well and so might reduce the efficiency of packing in the lattice. Initially this was attempted on the bis-phenyl side chain substituted with a carboxylic acid in the ortho-, meta- or para-position. Whilst good hDGAT-1 inhibition was observed with both the meta- (15) and para-derivatives (data not shown) the solubility of both remained poor. In addition, 15 showed high rat plasma protein binding (Fu < 0.01%) and high intrinsic metabolic clearance in rat hepatocytes (Clint = 22 μL min−1 mg−1) so was not pursued. Introduction of an oxy-link between the phenyl and cyclohexyl rings of 1 to provide the cis-cyclohexyl derivative 16 proved more beneficial and excellent inhibition of hDGAT-1 was achieved. Compound 16 also showed excellent selectivity against hACAT-1 (280 fold).
Combining the cis-cyclohexyloxy side chain with alternative aniline substitution on the oxadiazole ring was explored extensively and a large variety of substitution was tolerated with a similar SAR to what was observed in the initial series.6 As increasing molecular weight and lipophilicity would be detrimental to physicochemical properties the focus was primarily on small variations of aniline (Table 1). Replacement of the meta-fluoro substituent of 16 with a hydrogen atom to give the 4-fluoro derivative 17 gave excellent inhibition of hDGAT-1 with a corresponding reduction in lipophilicity and hence improvement in LLE. However, the solubility was still poor. Substitution on aniline with a 4-CN group (18) led to reduced potency versus hDGAT-1 so was not pursued further.
Alternatively, in an attempt to disrupt the intermolecular hydrogen bond between the hydrogen bond donor of the aniline and an oxadiazole nitrogen acceptor in a similar fashion to that shown with 4 (Fig. 2), a fluoro substituent was added to the aniline ring ortho to the anilino-NH to give the 2,4,5-trifluoro derivative 19. Excellent inhibition of hDGAT-1 was achieved, the fraction unbound in rat plasma protein binding showed a 10-fold increase (0.4%) compared with 1 and solubility was increased to 43 μM. The corresponding trans-derivative, 20, had the same potency against hDGAT-1 but interestingly was 3-fold less potent than the cis-derivative against hACAT-1.
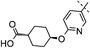
Attempts were made to reduce the lipophilicity further (Table 2). These included reducing the size of the cyclohexyl ring to the cyclobutyl, which led to a 10-fold reduction in potency against hDGAT-1 (data not shown), and introducing an additional heteroatom into the side chain by replacing the phenyl ring with a pyridyl ring as exemplified with 21. Compound 21 maintained good potency and achieved excellent selectivity versus hACAT-1 as well as improved solubility. The final strategy to improve solubility by disrupting the hydrogen bonding framework was to replicate the effect of the fluorine atom adjacent to the aniline NH and also mask the amide hydrogen bond donor with an ortho-fluoro substituent. This hypothesis was tested by synthesising the ortho-fluoro derivative 22. Compound 22 gave excellent inhibition of hDGAT-1, high LLE and gratifyingly a further small, but consistent, increase in solubility. Interestingly, 22 also had good potency against hACAT-1. This was similar to the des-fluoro derivative 19 suggesting that the 2-fluoro substituent in the aniline ring was good for hACAT-1 activity. As observed with previous examples, the corresponding trans-derivative 23 had significantly reduced potency towards hACAT-1. Introduction of a fluoro group into the meta-position relative to the amide (24) gave no beneficial effect on either hDGAT-1 potency or solubility.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | hDGAT-1 IC50a (μM) | hACAT-1 IC50b (μM) | Log D | Sol.b (μM) | LLE | |
| a All IC50 values reported are mean values from ≥2 experiments. b Measured by incubation of crystalline materials for 24 h at pH 7.4.12 | |||||||
| 21 |
|
3,4-diF | 0.005 | 6.5 | 1.9 | 49 | 6.5 |
| 22 |
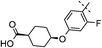
|
2,4,5-triF | 0.003 | 0.31 | 1.9 | 69 | 6.5 |
| 23 |
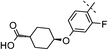
|
2,4,5-triF | 0.015 | 6.5 | 1.8 | 13 | 6.0 |
| 24 |
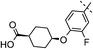
|
2,4,5-triF | 0.008 | 0.86 | 2.3 | 11 | 5.8 |
| 25 |
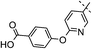
|
3,4-diF | 0.005 | 5.6 | 1.4 | 23 | 6.8 |
| 26 |
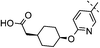
|
3,4-diF | 0.007 | 1.5 | 2.2 | 4 | 6.0 |
| 27 |
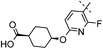
|
3,4-diF | 0.003 | 4.8 | 1.9 | 15 | 6.6 |
| 28 |
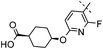
|
2,4,5-triF | 0.004 | 1.1 | 1.8 | 2200 | 6.6 |
| 29 |
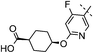
|
2,4,5-triF | 0.006 | 1.3 | 1.6 | 240 | 6.6 |
Further profiling revealed that as the lipophilicity was reduced over one log unit compared to the starting point 1 then the impact on both permeability and clearance would be of paramount importance (Table 3). Introduction of an oxy-link in combination with the 2,4,5-trifluoroanilino moiety, as in 19, led to a reduction of log D to 1.9 and still provided good permeability as measured in MDCK cells. Additional fluoro-substitution in the side chain, for example, 22, also gave high permeability.23 This was hypothesised as both hydrogen bond donors could now be masked through intramolecular hydrogen bonds. However, introduction of an additional heteroatom into the side chain such as the pyridyl derivative 21 gave a significant reduction in permeability, despite the compound having the same log D as 19 and 22. Further reduction of the lipophilicity of the side chain led to a significant reduction in permeability as exemplified with 25 (MDCK Papp A-B 1.1 × 10−6 cm s−1). The clearance of the compounds in human hepatocytes was low in most cases. However, differences emerged in in vivo DMPK experiments. The oxy-linked phenyl derivatives 19 and 22 had excellent DMPK parameters in rat including low clearance and high bioavailability. The clearance in the dog was significantly higher, particularly after normalizing for plasma protein binding to give the unbound clearance. This was in contrast to the pyridyl derivative 21, which had significantly higher clearance in the rat mainly due to increased renal clearance. In the dog the clearance, in particular the unbound clearance, was significantly lower. In summary, the cis-fluorophenyl derivatives 19 and 22 offered improvements in solubility and permeability and an excellent rat DMPK profile, whereas the pyridyl derivative 21 offered advantages in selectivity against hACAT-1 and lower unbound clearance in the dog.
| MDCK Papp A-B 10−6 (cm s−1) | Hu Hep Clint (μL min−1 mg−1) | Rat Cl (Clu) (mL min−1 kg−1) | Rat F% | Dog Cl (Clu) (mL min−1 kg−1) | Dog F% | |
|---|---|---|---|---|---|---|
| a Measured using Caco-2 cells. | ||||||
| 19 | 11 | <2 | 1.1 (470) | 115 | 11 (920) | 23 |
| 21 | 2.0 | <2 | 7.4 (740) | 33 | 4.7 (130) | 60 |
| 22 | 33a | <2 | 0.4 (200) | 85 | 6.5 (1700) | 76 |
| 26 | 5.1 | <2 | 0.7 (230) | 82 | 2.3 (100) | 35 |
| 27 | 16 | <2 | 3.6 (600) | 78 | 5.1 (300) | 84 |
| 28 | 15 | <2 | 1.8 (300) | 100 | 4.6 (200) | 61 |
To achieve a combination of these properties two strategies were undertaken. Firstly, slightly increasing the lipophilicity of the pyridyl derivative 21 to increase permeability and reduce renal clearance gave 26 (Table 2). Both excellent potency towards hDGAT-1 and selectivity over hACAT-1 were achieved as well as significantly reducing in vivo clearance in the rat. However, despite a moderate increase in permeability as measured in MDCK cells, solubility was also decreased so the optimal profile was not achieved. The second strategy involved replacing the benzene ring with pyridine in the side chain of 22 to bring the benefits of the pyridyl but with minimal impact on overall lipophilicity. This led to derivatives 27, 28 and 29. All three compounds had excellent potency against hDGAT-1, improved selectivity against hACAT-1 compared with 22 and maintained very low metabolic clearance in human hepatocytes. Moreover, good improvements in both rat DMPK parameters as well as a reduction in unbound clearance in the dog were achieved for 28. In addition, 28 brought together all strategies for improving solubility in this series by introducing shape changes and reducing lipophilicity and attempting to break the interactions in the crystal lattice through introducing heteroatoms and masking the hydrogen bond donors. Gratifyingly 28 achieved excellent solubility balanced with good permeability, which both contribute to the high bioavailability observed in rat and dog DMPK experiments.
To assess the true impact on the interactions existing in the solid state, crystals of 28 were grown which were suitable for X-ray structural determination (Fig. 3).24 The X-ray structure reveals that compared to the structure of 4, the void density has increased from 10.1% of the unit cell to 12.7% of the unit cell,25 commensurate with a decreased packing efficiency. As before, the ortho-fluoro substituent masks the aniline NH but interestingly the amide NH is not masked. Instead, fluorine is twisted away from the NH which ensures that while the NH is a better donor, the pyridyl ring is twisted with respect to the amide and so adopts a shape that packs less well in the solid state. The consequence of all these changes is a much more complex structure in which the plane of adjacent molecules (Fig. 3a) is held together by van der Waals interactions, including some π–π stacking. These planes are in turn held together in a cross-hatched fashion because of the L shape of the molecule which arises because of the axial positioning of the oxy-pyridyl group. Each molecule is involved in a dimeric pairing (Fig. 3b, the light blue and pink molecules) and these dimers then present hydrogen bonding groups that interact with the acids of adjacent molecules. These are difficult to compare to the acid dimers in 4 but the data suggest that this network of interactions contributes less to the lattice energy.
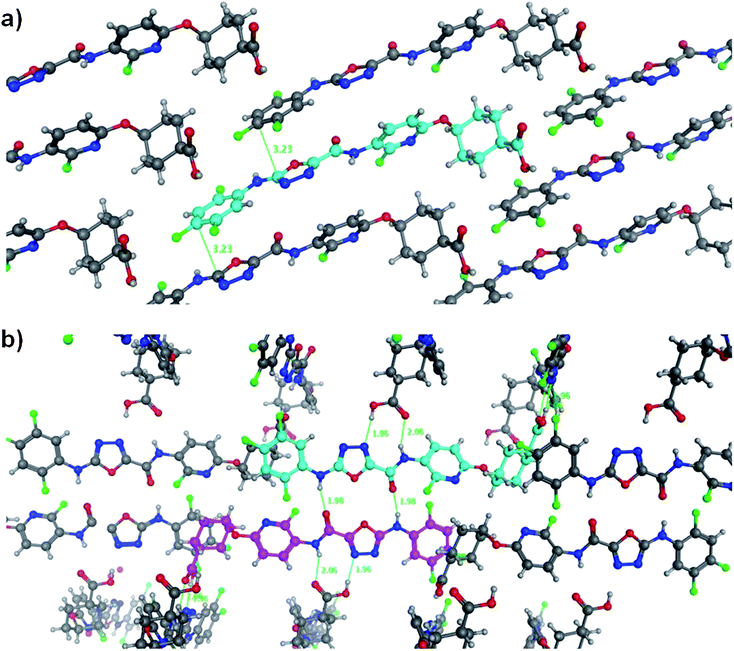 | ||
| Fig. 3 Two views of the crystal structure of compound 28. Panel (a) shows a view from above the plane of adjacent molecules and panel (b) shows how these planes stack. | ||
In general, compounds in this series showed no significant activity against human DGAT-2 (e.g.26; IC50 = 28 μM), the hERG encoded potassium channel (e.g.26; IC50 > 30 μM), or the cytochrome P450 enzyme isoforms CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 (e.g.26; all IC50s > 10 μM). Compounds in this series were also tested to confirm in vivo inhibition of DGAT-1 using a rat oral lipid tolerance test.5 Treatment with 26 resulted in an exposure-dependent decrease in circulating plasma triglyceride levels. Fig. 4 shows the relationship between plasma triglycerides and free compound levels in plasma for 26 in this model. PK-PD analysis indicates that the in vivo generated free IC50 (0.2 nM) is in good agreement with the in vitro IC50 in rat microsomes (0.9 nM) and a functional cell assay measuring triglyceride synthesis in the HuTu80 human cell line (4 nM).
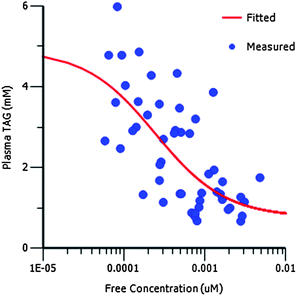 | ||
| Fig. 4 PK/PD analysis showing the relationship between plasma triglycerides (TAG) and free compound levels in plasma for 26 in a rat oral lipid tolerance test. A direct response (Emax) model was used to fit the PK/PD data (see ESI†). | ||
Conclusion
In summary, a novel series of hDGAT-1 inhibitors is described. Design hypotheses were devised to introduce changes in shape and polarity into the side chain, reduce lipophilicity and mask the hydrogen bond donors with internal hydrogen bond acceptors to improve the solubility and unbound clearance profile of the clinical candidate 1. These changes led to the replacement of the phenyl cyclohexyl-ethanoate side chain, a fragment present in a number of hDGAT-1 inhibitors, with substituted oxy-linked side chains. Combining the best features of two sub-series, including balancing lipophilicity and permeability, provided potent inhibitors of hDGAT-1 and displayed increased solubility and excellent ADMET parameters, whilst maintaining good properties such as LLE. Simultaneously, greater selectivity against the related enzyme hACAT-1 was achieved. The comparison of the small molecule X-ray structures of 4 and 28 shows marked differences in the solid state. Compound 28 adopts a different shape due to the axial positioning of the cis-oxy-pyridyl group. The planes of the molecules are held together by van der Waals interactions, including π–π stacking, and each molecule is involved in a dimeric pairing leading to an overall decreased packing efficiency of 28. Finally, inhibition of DGAT-1 in vitro translates to the in vivo setting as compound 26 is shown to decrease circulating plasma triglyceride levels in an exposure-dependent manner in a rat oral lipid tolerance test.Acknowledgements
The research was sponsored by AstraZeneca. All authors were employees of AstraZeneca at the time of the research. The authors thank Scott Boyd, Craig Donald, Kristin Goldberg and Adele Lloyd for the synthesis and purification of test compounds, Usha Chauhan for the testing of compounds in biological assays and colleagues in Physical Chemistry, DMPK and safety assessment for the provision of secondary ADMET data.Notes and references
- V. A. Zammit, L. K. Buckett, A. V. Turnbull, H. Wure and A. Proven, Pharmacol. Ther., 2008, 118, 295 CrossRef CAS.
- B. K. Hubbard, I. Enyedy, T. A. Gilmore and M. Serrano-Wu, Expert Opin. Ther. Pat., 2007, 17, 1331 Search PubMed.
- D. Matsuda and H. Tomoda, Curr. Opin. Invest. Drugs, 2007, 8, 836 Search PubMed.
- H. C. Chen and R. V. Farese, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 482 CrossRef CAS.
- A. M. Birch, S. Birtles, L. K. Buckett, P. D. Kemmitt, G. J. Smith, T. J. D. Smith, A. V. Turnbull and S. J. Y. Wang, J. Med. Chem., 2009, 52, 1558 CrossRef CAS.
- W. McCoull, M. S. Addie, A. M. Birch, S. Birtles, L. K. Buckett, R. J. Butlin, S. S. Bowker, S. Boyd, S. Chapman, R. D. M. Davies, C. S. Donald, C. P. Green, C. Jenner, P. D. Kemmitt, A. G. Leach, G. C. Moody, P. Morentin Gutierrez, N. J. Newcombe, T. Nowak, M. J. Packer, A. T. Plowright, J. Revill, P. Schofield, C. Sheldon, S. Stokes, A. V. Turnbull, S. J. Y. Wang, D. P. Whalley and J. M. Wood, Bioorg. Med. Chem. Lett., 2012, 22, 3873 CrossRef CAS.
- M. J. Waring, A. M. Birch, S. Birtles, L. K. Buckett, R. J. Butlin, L. Campbell, P. Morentin Gutierrez, P. D. Kemmitt, A. G. Leach, P. A. MacFaul, C. O'Donnell and A. V. Turnbull, Med. Chem. Commun., submitted for publication Search PubMed.
- R. L. Dow, J. Li, M. P. Pence, E. M. Gibbs, J. L. LaPerle, J. Litchfield, D. W. Piotrowski, M. J. Munchhof, T. B. Manion, W. J. Zavadoski, G. S. Walker, R. K. McPherson, S. Tapley, E. Sugarman, A. Guzman-Perez and P. DaSilva-Jardine, ACS Med. Chem. Lett., 2011, 2, 407 Search PubMed.
- R. L. Dow, M. Andrews, G. E. Aspnes, G. Balan, E. M. Gibbs, A. Guzman-Perez, K. Karki, J. L. LaPerle, J. Li, J. Litchfield, M. J. Munchhof, C. Perreault and L. Patel, Bioorg. Med. Chem. Lett., 2011, 21, 6122 CrossRef CAS.
- V. S. C. Yeh, D. W. A. Beno, S. Brodjian, M. E. Brune, S. C. Cullen, B. D. Dayton, M. K. Dhaon, H. D. Falls, J. Gao, N. Grihalde, P. Hajduk, T. M. Hansen, A. S. Judd, A. J. King, R. C. Klix, K. J. Larson, Y. Y. Lau, K. C. Marsh, S. W. Mittelstadt, D. Plata, M. J. Rozema, J. A. Segreti, E. J. Stoner, M. J. Voorbach, X. Wang, X. Xin, G. Zhao, C. A. Collins, B. F. Cox, R. M. Reilly, P. R. Kym and A. J. Souers, J. Med. Chem., 2012, 55, 1751 CrossRef CAS.
- http://clinicaltrials.gov/ct2/show/NCT01146522?term=LCQ-908&rank=1 .
- log D, plasma-protein binding and solubility measurements were made as described in: D. Buttar, N. Colclough, S. Gerhardt, P. A. MacFaul, S. D. Phillips, A. T. Plowright, P. Whittamore, K. Tam, K. Maskos, S. Steinbacher and H. Steuber, Bioorg. Med. Chem., 2010, 18, 7486 Search PubMed.
- S. Cases, S. J. Smith, Y. W. Zheng, H. M. Myers, T.-Y. Chang, B.-L. Li, C. C. Y. Chang, Y. Urano and Y. Am., J. Endocrinol. Metab., 2009, 297, E1–E9.
- E. Floetmann, L. K. Buckett, A. V. Turnbull, C. Hammond, C. Hallberg, A. Birch, D. Lees and H. B. Jones, manuscript in preparation.
- M. J. Waring, Bioorg. Med. Chem. Lett., 2009, 19, 2844 CrossRef CAS.
- X-ray data for compound 4: molecular formula = C23H21F3N4O4, C2H4O2, formula weight = 534.491, crystal system = triclinic, space group = P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 5.1885(5) Å, b = 11.4587(10) Å, c = 21.469(2) Å, α = 75.373(5), β = 86.058(5), γ = 83.124(4), V = 1225.2(2) Å3, T = 200 K, Z = 2, Dc = 1.449 Mg m−3, (Mo-Kα) λ = 0.71073 Å, μ = 0.12 mm−1, 5552 reflections measured, 3628 independent reflections, 1988 observed reflections [I> 2.0 σ (I)], R1_obs = 0.053, goodness of fit = 0.92.
, a = 5.1885(5) Å, b = 11.4587(10) Å, c = 21.469(2) Å, α = 75.373(5), β = 86.058(5), γ = 83.124(4), V = 1225.2(2) Å3, T = 200 K, Z = 2, Dc = 1.449 Mg m−3, (Mo-Kα) λ = 0.71073 Å, μ = 0.12 mm−1, 5552 reflections measured, 3628 independent reflections, 1988 observed reflections [I> 2.0 σ (I)], R1_obs = 0.053, goodness of fit = 0.92. - J. Boström, A. Hogner, A. Llinás, E. Wellner and A. T. Plowright, J. Med. Chem., 2012, 55, 1817 CrossRef.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino and G. Zheng, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. W. Kenny, J. Chem. Soc., Perkin Trans. 2, 1994, 199 RSC.
- P. W. Kenny, J. Chem. Inf. Model., 2009, 49, 1234 CrossRef CAS.
- K. D. Lardizabal, J. T. Mai, N. W. Wagner, A. Wyrick, T. Voelker and D. J. Hawkins, J. Biol. Chem., 2001, 276, 38862 CrossRef CAS.
- D. Dalvit and A. Vulpetti, ChemMedChem, 2012, 7, 262 CrossRef.
- X-ray data for compound 28: molecular formula = C21H17F4N5O5, formula weight = 495.40, crystal system = monoclinic, space group = C2/c, a = 29.7773(14) Å, b = 6.3200(3) Å, c = 23.2587(11) Å, β = 100.671(3), V = 4301.4(4) Å3, T = 200 K, Z = 8, Dc = 1.530 Mg m−3, (Mo-Kα) λ = 0.71073 Å, μ = 0.13 mm−1, 55003 reflections measured, 4007 independent reflections, 2557 observed reflections [I> 2.0 σ (I)], R1_obs = 0.043, goodness of fit = 1.02.
- Computed in mercury with a probe radius of 0.3 Å on a grid of 0.2 Å spacing on the solvent accessible surface.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for the preparation of compounds and crystallographic data. CCDC 886319 and 886320. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c2md20187a |
| This journal is © The Royal Society of Chemistry 2013 |