Structure–activity relationship studies of miniproteins targeting the androgen receptor–coactivator interaction†
Marta Dominguez
Seoane
b,
Katja
Petkau-Milroy
ab,
Belen
Vaz
b,
Sabine
Möcklinghoff
ab,
Simon
Folkertsma
c,
Lech-Gustav
Milroy
a and
Luc
Brunsveld
*ab
aLaboratory of Chemical Biology, Department of Biomedical Engineering, Eindhoven University of Technology, Den Dolech 2, 5612 AZ, Eindhoven, The Netherlands. E-mail: l.brunsveld@tue.nl; Fax: +31 40-247-8367
bChemical Genomics Centre of the Max Planck Society, Otto-Hahn Straße 15, 44227 Dortmund, Germany
cComputational Drug Discovery, Centre for Molecular and Biomolecular Informatics, Radboud University, Nijmegen, The Netherlands
First published on 13th August 2012
Abstract
A classical approach to treating prostate cancer uses antagonist ligands – so-called anti-androgens, such as bicalutamide – which block gene transcription through binding to a lipophilic pocket at the ligand binding domain of the androgen receptor (AR). An alternative strategy has been developed using compounds which directly target the surface charge clamp by mimicking the coactivator's highly conserved α-helical motif. Thus, to gain additional knowledge about the AR–coactivator interaction, the use of natural miniproteins as a source of novel AR–coactivator inhibitors incorporating the FXXLF motif was explored. Their stable well-defined α-helical secondary structures make miniproteins ideal candidates for development into AR–coactivator inhibitors. Therefore, starting from two potent miniprotein scaffold structures, identified from previous work, systematic point mutations aimed at improving AR affinity were introduced using solid-phase peptide synthesis (SPPS). Structure–activity relationship studies were performed, from which a number of high affinity inhibitors, typically in the low micromolar-to-high nanomolar range, with a ten-fold gain in potency compared with the reference compounds, were identified, thus highlighting the high potential for these scaffolds.
Introduction
The androgen receptor (AR) is a member of the steroid receptor sub-group of the nuclear receptor (NR) super-family of transcription factors, which regulate distinct physiological responses including development and dynamic homeostasis.1,2 Members of this family share a common structural and functional framework, including an N-terminal domain (NTD) harbouring activation function 1 (AF-1), a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) containing the activation function 2 (AF-2).3 Under normal physiological conditions, binding of the cognate endogenous ligand at the NR LBD (e.g. dihydrotestosterone, DHT to AR) induces a conformational change, which facilitates the recruitment of coactivator proteins and the activation of gene transcription. Unregulated AR function – the primary cause of prostate cancer in men – is typically treated with small drug molecules, so-called anti-androgens such as bicalutamide. These molecules bind to the lipophilic binding pocket of the LBD, block coactivator recruitment, and antagonize gene transcription.4 However, classical anti-androgens have their limitations, such as insensitivity to hormone refractory AR behaviour. Therefore, alternative treatments for prostate cancer, which function via alternative modes-of-action, are highly sought after.The majority of NRs, including the estrogen receptor (ER) recruit coactivators through the binding of a short LXXLL peptide sequence (where L is Leu/leucine and X can be any amino acid). The recruitment of specific coactivator proteins is mediated by differences in the amino acid residues flanking the LXXLL motif.5–8 AR is different, however. Though still capable of recognizing LXXLL, AR prefers to bind to FXXLF motifs (where F is Phe/phenylalanine) due to a deeper lipophilic groove at the AF-2 (compared with ER, for example).9 In light of the importance of this protein–protein interaction the development of molecular mimics capable of blocking coactivator binding would have dramatic impact on gene transcription, and might therefore provide a useful alternative to classical approaches targeting the ligand binding pocket. In this direction, significant strides have already been made using modified peptides10–17 and small molecules,10,18–29 especially for ER11–16,18–23,26–28 and the thyroid receptor (TR).11,24,25 However, small molecule inhibitors of the AR–coactivator interaction are only now starting to emerge.30–37 A structural analysis of the AR–cofactor interaction has additionally revealed that the FXXLF motif binds as a short well-defined α-helix between a charge clamp located at a fixed position at the AR AF-2.31,36,38–40 The length and stability of the helix thus appear to be crucial factors in determining the optimal binding of coactivator peptides, and provide useful indications for the design of small molecule inhibitors. Control over helix length and stability is typically challenging for short linear peptides. Thus methods which stabilize short peptides in a well-defined stable fold are likely to be advantageous in the development of novel inhibitors.12–14,16,41,42
We recently reengineered miniproteins to specifically inhibit the AR40 and ER–coactivator interactions.16 In the case of AR, four natural miniproteins bearing α-helical secondary structures stabilized by two or three disulfide bridges – apamin,44 κ-hefutoxin,45 a scyllatoxin analogue CD4M3,46 and Om-toxin47 – were selected from the protein database (PDB)40 and subjected to mutation studies. Target mutants were chosen by overlaying with the parent miniprotein and a natural cofactor sequence (PDB: 1t7r)43 using YASARA48 and then synthesized by solid-phase peptide synthesis (SPPS). In this way, the AR selective FXXLF motif was inserted into the α-helical region, and specific point mutations made to tune the helix length and avert steric clash and potentially unfavourable charge–charge interactions at the protein surface.40 The result of this work was the development of novel sub-micromolar affinity AR–coactivator inhibitors. These findings not only confirm the importance of the FXXLF consensus motif for AR binding. They also demonstrate that the appropriate helical structure is by itself not sufficient to achieve effective inhibition, rather the length of the helical segment is highly critical for obtaining high binding affinities. The introduction of the FXXLF motif in the helix together with several additional mutations geared towards maintaining charge–charge interactions afforded inhibitor molecules with remarkable binding properties. Among them, analogue Omt-1, based on the structure of natural miniprotein Om-toxin, and Het-2, based on κ-hefutoxin, were ranked among the most potent AR–coactivator inhibitors tested (Fig. 1). To explore these promising initial findings further and to enable studies into the relationship between the miniprotein structure and binding affinity, an expanded library of miniproteins based on Omt-1 and Het-2 was targeted. In this case, modifications are focused in particular on the flanking regions of the FXXLF motif, while maintaining the same helical segment and length: both believed to be essential features for potent binding.
 | ||
| Fig. 1 Comparison of secondary structures of the FXXLF co-factor binding motif (centre, PDB: 1t7r)43 with miniprotein inhibitors of the AR–coactivator interaction, Omt-1 (left) and Het-2 (right).40 The FXXLF side-chain residues are highlighted in light blue and bridging disulfide bonds in yellow. | ||
Results and discussion
Library design and synthesis
To highlight AR's preference for the FXXLF consensus motif, FXXLF- and LXXLL-derived analogues of both miniprotein scaffold structures were synthesized thereby permitting a direct comparison (Omt-1e and Het-2d, Tables 1 and 2, respectively). Specific point mutations were introduced to study recognition specificity and to identify coactivator motif sequence variants preferential for AR.39,49 These included mutating R to L (Omt-1f) and N to S (Omt-1g) for the Omt-1 scaffold (Table 1) and Y to L or R (Het-2b and Het-2c, respectively) for Het-2 (Table 2). The effect of non-natural amino acids was additionally studied. For this, the phenylalanine residues present in the FXXLF motif were systematically substituted with either o-chlorophenylalanine, p-chlorophenylalanine or β-cyclohexylalanine (Omt-1k to Omt-1p and Het-2j to Het-2n); mutations inspired by the work of Guy and co-workers, in which highly potent NR coactivator inhibitors of the estrogen receptor (ER) and the thyroid receptor (TR) were identified through the systematic mutation of the Leu residues within the LXXLL motif.10 Wijngaart and co-workers previously established that FXXFF and FXXMF motifs present in different coactivators also bind with high affinity to AR, giving rise to similar or more specific interactions.50 This finding therefore motivated the synthesis of FXXMF mutants (Omt-1i, Table 1). Finally, in silico design of the scaffold miniproteins Omt-1 and Het-2 in complex with the AR-LBD surface (ESI†)39 resulted in a number of other target miniproteins bearing single point mutations designed to favour charge–charge interactions. Thus, the linear peptides were prepared by SPPS according to standard protocols and then folded in phosphate buffer (pH 8) under air oxidation conditions followed by RP-HPLC purification to yield the miniproteins with purity >90% (ESI†).| Name | Sequence | K i (μM) |
|---|---|---|
| a FXXLF motif italicized in grey-bold, mutations underlined in bold. F(pCl) = p-chlorophenylalanine, F(oCl) = o-chlorophenylalanine, Cha = cyclohexylalanine. | ||
| Omt-1 |
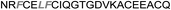
|
1.3 ± 0.2 |
| Omt-1a |
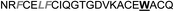
|
4.1 ± 0.3 |
| Omt-1b |

|
2.1 ± 0.4 |
| Omt-1c |

|
2.1 ± 0.5 |
| Omt-1d |

|
1.9 ± 0.4 |
| Omt-1e |
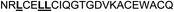
|
13.0 ± 2.0 |
| Omt-1f |
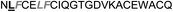
|
10.0 ± 2.0 |
| Omt-1g |

|
2.5 ± 0.3 |
| Omt-1h |
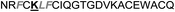
|
2.7 ± 0.5 |
| Omt-1i |

|
4.1 ± 0.3 |
| Omt-1j |
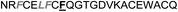
|
3.6 ± 0.6 |
| Omt-1k |

|
2.3 ± 0.4 |
| Omt-1l |
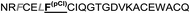
|
32.0 ± 15.0 |
| Omt-1m |

|
2.2 ± 0.4 |
| Omt-1n |
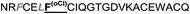
|
0.4 ± 0.1 |
| Omt-1o |

|
11.0 ± 2.0 |
| Omt-1p |

|
56.0 ± 5.0 |
| Omt-1q |

|
3.0 ± 1.0 |
| Name | Sequence | K i (μM) |
|---|---|---|
| a FXXLF motif italicized in grey-bold, point mutations underlined in bold. F(pCl) = p-chlorophenylalanine, F(oCl) = o-chlorophenylalanine, Cha = cyclohexylalanine. | ||
| Het-2 |

|
2.0 ± 0.5 |
| Het-2a |

|
1.7 ± 0.5 |
| Het-2b |

|
3.0 ± 0.8 |
| Het-2c |

|
12.0 ± 2.0 |
| Het-2d |

|
1.4 ± 0.5 |
| Het-2e |

|
0.2 ± 0.1 |
| Het-2f |
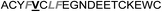
|
3.0 ± 1.0 |
| Het-2g |
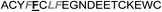
|
4.0 ± 1.0 |
| Het-2h |
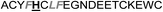
|
7.0 ± 2.0 |
| Het-2i |

|
n.a. |
| Het-2j |

|
3.0 ± 1.0 |
| Het-2k |

|
0.6 ± 0.3 |
| Het-2l |

|
3.0 ± 1.5 |
| Het-2m |

|
3.0 ± 1.0 |
| Het-2n |

|
2.3 ± 0.8 |
Biochemical evaluation
A competitive fluorescence-based polarization assay was used to evaluate the miniproteins' AR binding affinity. More specifically, a preformed complex of AR–LBD bound to a fluorescein-labelled FXXLF reference peptide (ESI†) was titrated with increasing amounts of the test miniprotein, leading to a displacement of the fluorescently labelled peptide and a corresponding decrease in polarization. In serial dilution, S-curves were measured (ESI†), which enabled IC50 determination, and the Ki was calculated (Tables 1 and 2) by first measuring the Kd of the fluorescently labelled reference peptide (Kd = 0.89 μM).40Structure–activity relationship studies
In the case of the κ-hefutoxin-derived miniprotein library, the introduction of a tryptophan (W) residue at the C-terminus, in a region not binding to the AR, did not change the AR binding affinity (compare Het-2 with Het-2a). The same mutation at the C-terminus of Omt-1 (Ki = 1.3 μM) did result in a three-fold decrease in binding affinity (Omt-1a, Ki = 4.1 μM). Even so, these data would suggest that for both protein scaffolds, mutating E to W did not extensively interfere with the AR binding and was therefore made to facilitate the spectroscopic determination of the final concentration of miniprotein stock. The similar activity observed for both Het-2a (Ki = 1.7 μM, Table 2) and mutant Het-2b (Y → L, Ki = 3.0 μM), while not reinforcing, at least does not contradict results from previous phage display screening performed by Chang et al., where L was found to be highly conserved at the −1 position relative to the AR FXXLF motif.49 A similar two-fold decrease in binding affinity compared to the parental structure was also measured for the equivalent Om-toxin analogue Omt-1f (Ki = 10.0 μM). Conversely, mutant Het-2c (Y → R, Ki = 12.0 μM), which was expected to favour charge interactions at the AR surface given its presence in all potent analogues of Omt-1, nevertheless measured a seven-fold lower binding affinity than Het-2a. In the case of Om-toxin miniproteins, derivatives Omt-1b (G → D), Omt-1c (Q → E) and Omt-1d (N → K) – each synthesized with the aim to probe specific charge interactions with K720, K717, and E709 residues at the AR surface based on previous modeling studies – were two-fold more active than the reference compound, Omt-1a. Point mutations probing charge interactions at different positions in and around the FXXLF motif of the Om-toxin scaffold yielded similarly active miniproteins (Omt-1g to Omt-1j). Taken together, these data for the Om-toxin series would suggest that the miniprotein's stable helical structure favours AR binding through a more pre-organized state, which lowers the entropic penalty for binding, and out-weighs potentially negative enthalpic contributions through disfavoured charge–charge interactions between the miniprotein side-chains and the AR-surface. Results from similar mutation studies on the hefutoxin scaffold (Het-2e to Het-2i), however, were more variable. Mutant Het-2e (N → E), for example, was approximately ten-fold more potent (Ki = 0.2 μM) than Het-2a, due potentially to a more favourable surface charge–charge interaction through the addition of the glutamic acid residue (Fig. 2), and gave rise to the most potent analogue from either of the miniprotein scaffolds. The glutamic acid mutant Omt-1q (Ki = 3.0 μM) was also more potent than the reference compound, Omt-1a, though less active than Het-2e. Yet mutant Het-2i (N → E) was completely impotent. Thus, the data discussed so far suggest that the activity of the miniprotein scaffold can be fine-tuned through rationally motivated point mutations in the flanking regions of the FXXLF motif, in some cases leading to dramatic improvements in activity. Replacing FXXLF by LXXLL (Omt-1e) diminished the binding affinity compared to Omt-1a. This result is in line with precedent literature, which pinpoints AR's preferential binding to FXXLF due to a deeper lipophilic groove at the protein surface. Surprisingly, therefore, the corresponding hefutoxin mutant, LXXLL-derived Het-2d was effectively equipotent with Het-2a. These interesting data highlight as well the fact that, aside from lipophilic surface interactions, other factors are at play, such as charge–charge interactions at the coactivator groove or the miniprotein's pre-organized α-helical secondary structure, which can strongly influence the miniprotein's binding affinity.51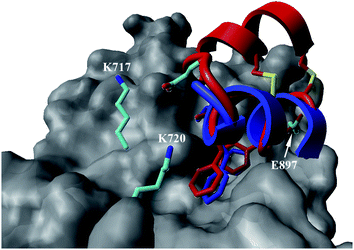 | ||
| Fig. 2 Proposed structural basis for the added affinity of Het-2e (red), where the flexible loop region linking the two helical segments may allow for the glutamic acid residue (E) to make a stabilizing charge interaction with surface lysine residues K717 and K720 of AR (grey), including at the charge clamp. Overlay with crystal structure of the linear FXXLF peptide (blue).43 | ||
The introduction of non-natural amino acids in the peptide sequence of both scaffold structures afforded interesting affinity data (Table 1). In the case of the Om-toxin series, the specific point mutation of either of the two phenylalanine residues of the FXXLF motif to more sterically hindered amino acids – such as β-cyclohexylalanine and ortho- or para-chlorophenylalanine – yielded mutants with varying binding affinities. For example, the p-chloro mutant Omt-1k (Ki = 2.3 μM) was effectively equipotent with the parental miniprotein, Omt-1a. Its constitutional isomer, Omt-1l, however, was at least one order of magnitude less active (Ki = 32.0 μM). Most intriguing of all for this short series, however, was the o-chloro analogue Omt-1n (Ki = 0.4 μM), which was ten-fold more potent than Omt-1a and nearly 100-fold higher in affinity than the corresponding p-chloro isomer Omt-1l, possibly due to a more perfect fit at the lipophilic surface cleft (Fig. 3).52 The p-chloro group in analogue Omt-1l appears to clash with the AR surface. In contrast the o-chloro group on the phenylalanine of Omt-1n shows a perfect fit, in both orientations, towards and away from the hydrophobic binding cleft on the AR surface. In stark contrast, the chlorophenylalanine isomers Omt-1k (Ki = 2.3 μM) and Omt-1m (Ki = 2.2 μM) were similarly active with one another and approximately two-fold more active than Omt-1a. A similar though less pronounced trend was observed for the hefutoxin series in the case of analogues Het-2j, Het-2k, Het-2l. Mutating either of the phenylalanine residues to a β-cyclohexylalanine did not in any case increase the binding affinity (Omt-1o, Omt-1p, Het-2m and Het-2n).
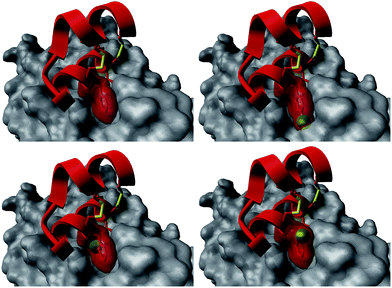 | ||
| Fig. 3 Structural basis for the differences in activity between the Omt-1a (top left) and Omt-1l (top right), featuring a steric clash of the p-chloro group (green) with the AR surface (grey), and Omt-1n (bottom, two orientations) showing nice space filling, especially with the o-chloro group pointing inward to the surface cleft. The protein surface (PDB: 1t7r)43 is represented as grey fill and the miniproteins as red ribbon. The phenylalanine residue is depicted as red cloud, with the chlorine atom in green. | ||
Previously, a series of novel inhibitors of the AR–coactivator interaction based on stable α-helical miniproteins was developed using a rational approach based on computational design, synthesis and in vitro biological evaluation. In this present study, synthetic point mutation studies were performed on two of the most potent miniproteins – Omt-1 and Het-2 – with a view to achieving more potent co-activator inhibition. For this both miniproteins are treated as conformationally stable α-helical scaffold structures and that modifications to side-chain residues are assumed not to change the secondary structure but instead favour increased binding through additional favourable interactions at the AR surface. Thus, the data presented in Tables 1 and 2 would suggest that in certain cases subtle structural modifications to the hydrophobic side-chain residues of the FXXLF motif, especially at the C-terminal phenylalanine amino acid, can lead to significant gains in potency.
Summary and conclusions
In conclusion, we have shown that miniproteins represent a viable approach to the discovery of inhibitors of the AR–coactivator interaction and provide understanding on the molecular characteristics and needs of peptide motifs binding the AF-2. Two miniprotein scaffold structures derived from toxins κ-hefutoxin and Om-toxin were used, around which a focused library of thirty-four analogues was synthesized (eighteen new analogues based on Om-toxin and sixteen derived from κ-hefutoxin). Structure–activity relationship studies were performed, and already with this focused analogue set, a number of high affinity (in the micromolar-to-nanomolar range) inhibitors could be identified. A ten-fold gain in potency, compared with the reference compounds, could be obtained by introducing only a single point mutation, thus highlighting the great potential for these scaffolds. In particular, rational point mutations in the regions flanking the FXXLF motif resulted in a fine-tuning of binding, typically in favour of slightly higher affinities. Interestingly for the hefutoxin series, mutating the FXXLF motif to LXXLL retained the high affinity of the parental compound. Finally, subtle mutation of the two phenylalanine residues of the FXXLF motif to sterically more bulky non-natural amino acids resulted in some dramatic changes in potency and some of the highest affinity compounds from both miniproteins series. The fact that such a dramatic increase in binding affinity can be achieved solely through manipulation of the lipophilic side chains within the FXXLF motif suggests that optimization of a suitable non-peptidic lipophilic small molecule might yield the same results. Results from this work have thus allowed for a better understanding of the effects of these amino acids on the AR–coactivator inhibition and might therefore prove useful in the future design of more potent and selective small molecule CBIs.Acknowledgements
We thank Dr Dorothee Wasserberg for help with protein expression and Dr Maarten Broeren for help with peptide synthesis. Funded by ERC grant 204554 – SupraChemBio (LB). We thank the Spanish Ministerio de Educación y Ciencia for a Postdoctoral Fellowship to M.D.S and Merck-Serono, MSD, and Bayer-Schering Pharma for financial support.Notes and references
- M. E. Baker, BioEssays, 2003, 25, 396 CrossRef CAS.
- H. Gronemeyer, J.-A. Gustafsson and V. Laudet, Nat. Rev. Drug Discovery, 2004, 3, 950 CrossRef CAS.
- P. Huang, V. Chandra and F. Rastinejad, Annu. Rev. Physiol., 2010, 72, 247 CrossRef CAS.
- Y. Chen, N. J. Clegg and H. I. Scher, Lancet Oncol., 2009, 10, 981 CrossRef CAS.
- D. M. Heery, E. Kalkhoven, S. Hoare and M. G. Parker, Nature, 1997, 387, 733 CrossRef CAS.
- B. D. Darimont, R. L. Wagner, J. W. Apriletti, M. R. Stallcup, P. J. Kushner, J. D. Baxter, R. J. Fletterick and K. R. Yamamoto, Genes Dev., 1998, 12, 3343 CrossRef CAS.
- E. M. McInerney, D. W. Rose, S. E. Flynn, S. Westin, T.-M. Mullen, A. Krones, J. Inostroza, J. Torchia, R. T. Nolte, N. Assa-Munt, M. V. Milburn, C. K. Glass and M. G. Rosenfeld, Genes Dev., 1998, 12, 3357 CrossRef CAS.
- M. Needham, S. Raines, J. McPheat, C. Stacey, J. Ellston, S. Hoare and M. Parker, J. Steroid Biochem. Mol. Biol., 2000, 72, 35 CrossRef CAS.
- B. He, J. A. Kemppainen and E. M. Wilson, J. Biol. Chem., 2000, 275, 22986 CrossRef CAS.
- T. W. Moore, C. G. Mayne and J. A. Katzenellenbogen, Mol. Endocrinol., 2010, 24, 683 CrossRef CAS.
- T. R. Geistlinger and R. K. Guy, J. Am. Chem. Soc., 2003, 125, 6852 CrossRef CAS.
- A.-M. Leduc, J. O. Trent, J. L. Wittliff, K. S. Bramlett, S. L. Briggs, N. Y. Chirgadze, Y. Wang, T. P. Burris and A. F. Spatola, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11273 CrossRef CAS.
- A. K. Galande, K. S. Bramlett, T. P. Burris, J. L. Wittliff and A. F. Spatola, J. Pept. Res., 2004, 63, 297 CrossRef CAS.
- A. K. Galande, K. S. Bramlett, J. O. Trent, T. P. Burris, J. L. Wittliff and A. F. Spatola, ChemBioChem, 2005, 6, 1991 CrossRef CAS.
- M. Carraz, W. Zwart, T. Phan, R. Michalides and L. Brunsveld, Chem. Biol., 2009, 16, 702 CrossRef CAS.
- T. Phan, H. D. Nguyen, H. Göksel, S. Möcklinghoff and L. Brunsveld, Chem. Commun., 2010, 46, 8207 RSC.
- C. Phillips, L. R. Roberts, M. Schade, R. Bazin, A. Bent, N. L. Davies, R. Moore, A. D. Pannifer, A. R. Pickford, S. H. Prior, C. M. Read, A. Scott, D. G. Brown, B. Xu and S. L. Irving, J. Am. Chem. Soc., 2011, 133, 9696 CrossRef CAS.
- A. L. Rodriguez, A. Tamrazi, M. L. Collins and J. A. Katzenellenbogen, J. Med. Chem., 2004, 47, 600 CrossRef CAS.
- J. Becerril and A. D. Hamilton, Angew. Chem., Int. Ed., 2007, 46, 4471 CrossRef CAS.
- H. B. Zhou, M. L. Collins, J. R. Gunther, J. S. Comninos and J. A. Katzenellenbogen, Bioorg. Med. Chem. Lett., 2007, 17, 4118 CrossRef CAS.
- A. L. LaFrate, J. R. Gunther, K. E. Carlson and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2008, 16, 10075 CrossRef CAS.
- J. R. Gunther, T. W. Moore, M. L. Collins and J. A. Katzenellenbogen, ACS Chem. Biol., 2008, 3, 282 CrossRef CAS.
- A. A. Parent, J. R. Gunther and J. A. Katzenellenbogen, J. Med. Chem., 2008, 51, 6512 CrossRef CAS.
- P. Sadana, J. Y. Hwang, R. R. Attia, L. A. Arnold, G. Neale and R. K. Guy, ACS Chem. Biol., 2011, 6, 1096 CrossRef CAS.
- J. Y. Hwang, R. R. Attia, F. Zhu, L. Yang, A. Lemoff, C. Jeffries, M. C. Connelly and R. K. Guy, J. Med. Chem., 2012, 55, 2301 CrossRef CAS.
- A. B. Williams, P. T. Weiser, R. N. Hanson, J. R. Gunther and J. A. Katzenellenbogen, Org. Lett., 2009, 11, 5370 CrossRef CAS.
- A. Sun, T. W. Moore, J. R. Gunther, M.-S. Kim, E. Rhoden, Y. Du, H. Fu, J. P. Snyder and J. A. Katzenellenbogen, ChemMedChem, 2011, 6, 654 CrossRef CAS.
- A. B. Williams and R. N. Hanson, Tetrahedron, 2012, 68, 5406 CrossRef CAS.
- Y. Mita, K. Dodo, T. Noguchi-Yachide, H. Miyachi, M. Makishima, Y. Hashimoto and M. Ishikawa, Bioorg. Med. Chem. Lett., 2010, 20, 1712 CrossRef CAS.
- M. D. Sadar, World J. Urol., 2012, 30, 311 CrossRef CAS.
- E. Estébanez-Perpiñá, L. A. Arnold, P. Nguyen, E. Delgado Rodrigues, E. Mar, R. Bateman, P. Pallai, K. M. Shokat, J. D. Baxter, R. K. Guy, P. Webb and R. J. Fletterick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16074 CrossRef.
- B. Vaz, S. Möcklinghoff and L. Brunsveld, in Methods in Principles in Medicinal Chemistry, Nuclear Receptors as Drug Targets, ed. E. Otto and H. Weinmann, Wiley-VCH, Weinheim, 2008, pp. 25–45 Search PubMed.
- C. Féau, L. A. Arnold, A. Kosinski, F. Zhu, M. Connelly and R. K. Guy, ACS Chem. Biol., 2009, 4, 834 CrossRef.
- H. Göksel, D. Wasserberg, S. Möcklinghoff, B. Vaz-Araujo and L. Brunsveld, Bioorg. Med. Chem., 2011, 19, 306 CrossRef.
- J. R. Gunther, A. A. Parent and J. A. Katzenellenbogen, ACS Chem. Biol., 2009, 4, 435 CrossRef CAS.
- P. Axerio-Cilies, N. A. Lack, M. R. Nayana, K. H. Chan, A. Yeung, E. Leblanc, E. S. Guns, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2011, 54, 6197 CrossRef CAS.
- L. Caboni, G. K. Kinsella, F. Blanco, D. Fayne, W. N. Jagoe, M. Carr, D. C. Williams, M. J. Meegan and D. G. Lloyd, J. Med. Chem., 2012, 55, 1635 CrossRef CAS.
- B. He, R. T. Gampe Jr, A. J. Kole, A. T. Hnat, T. B. Stanley, G. An, E. L. Stewart, R. I. Kalman, J. T. Minges and E. M. Wilson, Mol. Cell, 2004, 16, 425 CrossRef CAS.
- E. Hur, S. J. Pfaff, E. S. Payne, H. Grøn, B. M. Buehrer and R. J. Fletterick, PLoS Biol., 2004, 2, e274 CrossRef.
- B. Vaz, S. Mocklinghoff, S. Folkertsma, S. Lusher, J. de Vlieg and L. Brunsveld, Chem. Commun., 2009, 5377 RSC.
- (a) A. Patgiri, K. K. Yadav, P. S. Arora and D. Bar-Sagi, Nat. Chem. Biol., 2011, 7, 585 CrossRef CAS; (b) D. Wang, K. Chen, J. L. Kulp III and P. S. Arora, J. Am. Chem. Soc., 2006, 128, 9248 CrossRef CAS; (c) D. Wang, K. Chen, G. Dimartino and P. S. Arora, Org. Biomol. Chem., 2006, 4, 4074 RSC; (d) L. K. Henchey, S. Kushal, R. Dubey, R. N. Chapman, B. Z. Olenyuk and P. S. Arora, J. Am. Chem. Soc., 2010, 132, 941 CrossRef CAS.
- (a) L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. W. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466 CrossRef CAS; (b) F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456 CrossRef CAS.
- E. Hur, S. J. Pfaff, E. S. Payne, H. Grøn, B. M. Buehrer and R. J. Fletterick, PLoS Biol., 2004, 2, 1303 CrossRef.
- J. H. B. Pease and D. E. Wemmer, Biochemistry, 1988, 27, 8491 CrossRef CAS.
- K. N. Srinivasan, V. Sivaraja, I. Huys, T. Sasaki, B. Cheng, T. K. S. Kumar, K. Sato, J. Tytgat, C. Yu, B. C. C. San, S. Ranganathan, H. J. Bowie, R. M. Kini and P. Gopalakrishnakone, J. Biol. Chem., 2002, 277, 30040 CrossRef CAS.
- C. Vita, E. Drakopoulou, J. Vizzavona, S. Rochette, L. Martin, A. Ménez, C. Roumestand, Y.-S. Yang, L. Ylisastigui, A. Benjouad and J. C. Gluckman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13091 CrossRef CAS.
- B. Chagot, C. Pimentel, L. Dai, J. Pil, J. Tytgat, T. Nakajima, G. Corzo, H. Darbon and G. Ferrat, Biochem. J., 2005, 388, 263 CrossRef CAS.
- E. Krieger and G. Vriend, Bioinformatics, 2002, 18, 315 CrossRef CAS.
- C.-Y. Chang, J. Abdo, T. Hartney and D. P. McDonnell, Mol. Endocrinol., 2005, 19, 2478 CrossRef CAS.
- D. J. van de Wijngaart, M. E. van Royen, R. Hersmus, A. C. W. Pike, A. B. Houtsmuller, G. Jenster, J. Trapman and H. J. Dubbink, J. Biol. Chem., 2006, 281, 19407 CrossRef CAS.
- H. J. Dubbink, R. Hersmus, C. S. Verma, H. A. van der Korput, C. A. Berrevoets, J. van Tol, A. C. Ziel-van der Made, A. Brinkmann, A. C. Pike and J. Trapman, Mol. Endocrinol., 2004, 18, 2132 CrossRef CAS.
- Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide synthesis, fluorescence polarization measurements and Ki determination. See DOI: 10.1039/c2md20182h |
| This journal is © The Royal Society of Chemistry 2013 |