Mapping histamine H4 receptor–ligand binding modes†
Sabine
Schultes‡
ab,
Saskia
Nijmeijer‡
a,
Harald
Engelhardt
ab,
Albert J.
Kooistra
a,
Henry F.
Vischer
a,
Iwan J. P.
de Esch
a,
Eric E. J.
Haaksma
ab,
Rob
Leurs
a and
Chris
de Graaf
*a
aLeiden/Amsterdam Center for Drug Research (LACDR), Division of Medicinal Chemistry, Department of Pharmacochemistry, Faculty of Exact Sciences, VU University Amsterdam, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands. E-mail: c.de.graaf@vu.nl
bBoehringer Ingelheim RCV GmbH & Co KG, Department of Medicinal Chemistry, Dr Boehringer-Gasse 5-11, Vienna, Austria
First published on 6th September 2012
Abstract
The increasing number of G protein-coupled receptor (GPCR) crystal structures offers new opportunities for histamine receptor homology modeling. However, computational prediction of ligand binding modes in GPCRs such as the histamine H4 receptor (H4R), a receptor that plays an important role in inflammation, remains a challenging task. In the current work we have combined complementary in silico receptor modeling approaches with in vitro ligand structure–activity relationship (SAR) and protein site-directed mutagenesis studies to elucidate the binding modes of different ligand classes in H4R. By systematically considering different H4R modelling templates, ligand binding poses, and ligand protonation states in combination with docking and MD simulations we are able to explain ligand-specific mutation effects and subtle differences in ligand SAR. Our studies confirm that a combined theoretical and experimental approach represents a powerful strategy to map ligand–protein interactions.
Introduction
Rational drug design requires detailed knowledge and understanding of the molecular interactions between ligands and proteins. Both ligand- and protein-based in silico modeling approaches have been successfully applied to rationalize structure–activity relationships (SAR) and receptor site-directed mutagenesis studies, for the histamine H4 receptor (H4R),1–7 a G protein-coupled receptor (GPCR) that plays an important role in inflammation.8–20 The increasing number of GPCR X-ray structures,21,22 including the recently solved histamine H1 receptor (H1R) crystal structure23 (Fig. 1A), offers new opportunities for histamine receptor homology modeling and the structure-based design of new histamine receptor ligands.24–27 However, computational H4R–ligand binding mode prediction still remains a challenging task. The symmetric distribution of the two acetic residues (aspartate D943.32 and glutamate E1825.46)2,3,7 in combination with different hydrophobic subpockets (I and II,28Fig. 1B and C) that are complementary to (two) basic and several hydrophobic groups in H4R ligands (Fig. 2)29–31 allows different plausible H4R–ligand binding modes. Site-directed mutagenesis studies have identified D943.32 (a conserved binding residue in bioaminergic GPCRs)32 as an essential residue in H4R to bind both the non-imidazole antagonist JNJ 7777120 1b3 and the small imidazole-containing agonist histamine 3.3,7 In addition, site-directed mutagenesis (SDM) studies showed that the anionic carboxylate group of E1825.46 plays an important role in binding of H4R ligands that contain two basic groups, including 3, VUF 8430 4, and to a smaller degree clobenpropit 5 and clobenpropit analogue VUF 5228 6, while 1b and clozapine 7 only require a H-bond acceptor at position 1825.46 (e.g. present in the E1825.46Q mutant).2,3 In the H1R co-crystal structure23 the inverse agonist doxepin forms an ionic H-bond interaction with the conserved D1073.32, but does not form a H-bond with N1985.46. The aromatic ring systems of doxepin bind to two connected subpockets in the hydrophobic cavity between transmembrane (TM) helices 3–6 (pocket II28), in the current study defined as subpocket IIa and subpocket IIb (Fig. 1A). Receptor mutagenesis, ligand SAR, and receptor–ligand interaction modeling studies have indicated that different ligands bind to these different subpockets.2,4,30,33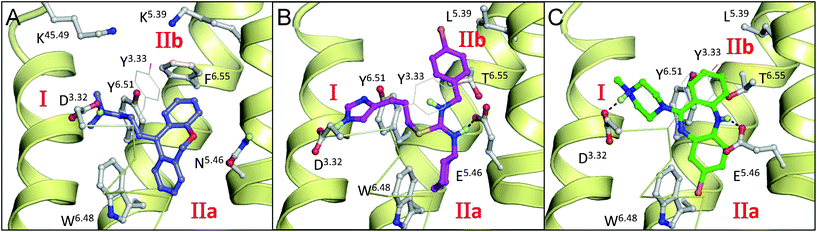 | ||
| Fig. 1 Binding modes of doxepin in the H1R co-crystal structure23 (A), and clozapine4 (B) and VUF 5228 (ref. 2) (C) in (ADRB2 crystal structure34 based) H4R homology models. Compounds and pocket residues are depicted as ball-and-sticks, whereas for clarity Y953.33 is shown as lines. H-bonds between the ligand and pocket residues are represented as black dotted lines. The backbone TM helices 5, 6, and 7 (right to left) are presented as yellow helices. Helix 3 is presented by yellow ribbons. Subpockets I, IIa and IIb are labeled in red. | ||
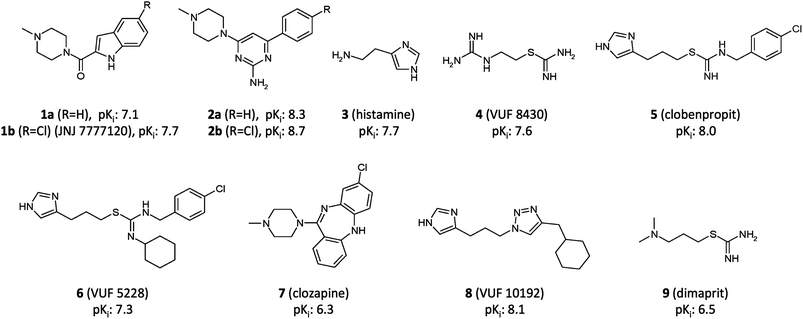 | ||
| Fig. 2 Structures of H4R ligands 1–9. Affinities (pKi) for H4R of 1 and 2 are from the current study, 3, 4 and 7 from ref. 4, 5 and 6 from ref. 2, 8 from ref. 33 and 9 from ref. 35. | ||
Ligands 6 and 7 occupy both subpockets IIa and IIb simultaneously (Fig. 1B and C).2,4 Triazole ligand 8 is proposed to bind only in subpocket IIa,33 while combined modeling and mutagenesis studies support two alternative binding modes of ligand 5 in which it accommodates its aromatic moiety in subpocket IIb, or, alternatively, in subpocket I.2
In this study we have focused on the elucidation of the binding interactions of two different selective H4R ligand classes: indolecarboxamides4,6,27,36–39 (1a,b) and 2-aminopyrimidines40–46 (2a,b) (Fig. 2). Recent ligand-binding mode predictions of dual H3R/H4R clobenpropit ligands 5 and 6 demonstrated that the integration of complementary in vitro and in silico modeling approaches is an efficient way to map protein–ligand interactions.2 In the current study we used in silico guided mutation studies to compare the binding modes of substituted and unsubstituted indolecarboxamides and 2-aminopyrimidines. To obtain more detailed insights into ligand-specific molecular recognition features, molecular dynamics (MD) simulations were employed, a technique that has been successfully applied to describe subtle differences in ligand binding (e.g. ligand regio-, stereo-, and protein-selectivity47,48). Furthermore, different ligand protonation states (i.e. 2-aminopyrimidines 2a,b with one versus two positively ionized basic groups) and different H4R modeling templates (i.e. the bioaminergic GPCR β2-adrenergic receptor (ADRB2)34versus the more closely related H1R)23 were considered to investigate the influence of modeling parameters. The combination of protein mutagenesis data with different binding mode hypotheses, molecular modeling and MD-simulation revealed similarities as well as differences in the molecular H4R binding determinants for both ligand classes. These insights are useful for rational optimization of indolecarboxamides and 2-aminopyrimidines, and the design of new H4R ligands.
Results
SAR (Table 1, Fig. 3) and mutagenesis (Table 2, Fig. 4) studies in combination with docking and MD-simulations (Fig. 5–7, Tables 3 and 4) were used to elucidate the binding modes of indolecarboxamides 1a,b and 2-aminopyrimidines 2a,b (Fig. 2) in the H4R binding site and to give insights into the molecular determinants of H4R–ligand interactions. For this procedure different starting poses of the ligands were generated (Fig. 5) in different H4R models (Fig. S1†). For the 2-aminopyrimidine ligands different protonation states were considered (Tables 3 and 4). The H4R–ligand binding mode models that could best explain the (ligand specific) mutation data were used to identify similarities and differences in the binding characteristics for the different ligand classes.
![Reagents and conditions: (i) dichloromethane, N-ethyl-diisopropylamine, 20 °C, 16 h; (ii) dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(ii) dichloromethane adduct, Cs2CO3, tetrahydrofuran, 1-methyl-2-pyrrolidinone, water, 100 °C, 16 h.](/image/article/2013/MD/c2md20212c/c2md20212c-f3.gif) | ||
| Fig. 3 Reagents and conditions: (i) dichloromethane, N-ethyl-diisopropylamine, 20 °C, 16 h; (ii) dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct, Cs2CO3, tetrahydrofuran, 1-methyl-2-pyrrolidinone, water, 100 °C, 16 h. | ||
![Radioligand displacement curves of ligands 1a (A), 1b (B), 2a (C), 2b (D) on H4R WT (), H4R-L5.39V (), H4R-E5.46Q (). E5.46Q binding studies were performed with the radioligand [3H]-JNJ 7777120, whereas [3H]-histamine was used for H4R and L5.39V. Data shown are representative specific binding curves of at least two experiments performed in triplicate. Error bars indicate SEM values. To enable a better visualization of the pKi shifts, each curve is corrected for the respective radioligand concentration and Kd values.](/image/article/2013/MD/c2md20212c/c2md20212c-f4.gif) | ||
Fig. 4 Radioligand displacement curves of ligands 1a (A), 1b (B), 2a (C), 2b (D) on H4R WT ( ), H4R-L5.39V ( ), H4R-L5.39V ( ), H4R-E5.46Q ( ), H4R-E5.46Q ( ). E5.46Q binding studies were performed with the radioligand [3H]-JNJ 7777120, whereas [3H]-histamine was used for H4R and L5.39V. Data shown are representative specific binding curves of at least two experiments performed in triplicate. Error bars indicate SEM values. To enable a better visualization of the pKi shifts, each curve is corrected for the respective radioligand concentration and Kd values. ). E5.46Q binding studies were performed with the radioligand [3H]-JNJ 7777120, whereas [3H]-histamine was used for H4R and L5.39V. Data shown are representative specific binding curves of at least two experiments performed in triplicate. Error bars indicate SEM values. To enable a better visualization of the pKi shifts, each curve is corrected for the respective radioligand concentration and Kd values. | ||
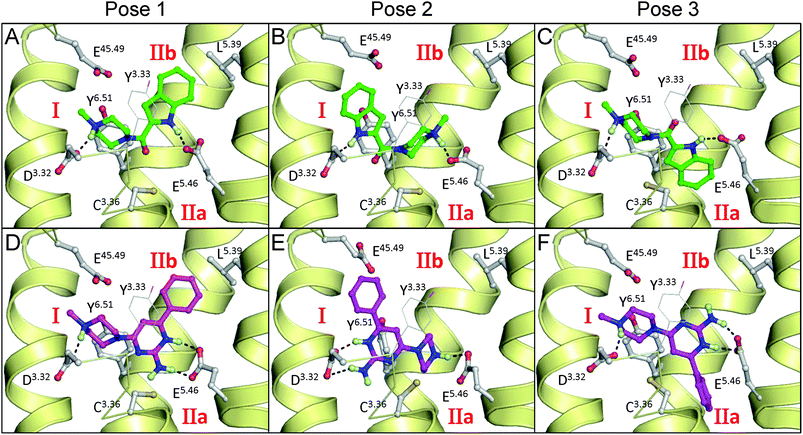 | ||
| Fig. 5 Initial binding poses of indolecarboxamide ligand 1a (A–C) and 2-aminopyrimidine ligand 2a (D–F) in H1R crystal structure based H4R models used as starting structures for MD simulations. Rendering and labels are the same as in Fig. 1. | ||
| Structure |
|
|
|
|||
|---|---|---|---|---|---|---|
| a A hydrophobic interaction is counted if the shortest distance between any heavy atom of the ligand and L1755.39 is ≤4 Å. b An H-bond is counted if the distance between the H-bond donor and acceptor heavy atom is below 3.5 Å and the angle between the H-bond donor heavy atom, hydrogen and H-bond acceptor heavy atom is between 135° and 225°. c See Fig. 5. | ||||||
| Protonation | 1+ | 1+ | 2+ | |||
| R | H | Cl | H | Cl | H | Cl |
| Name | 1a | 1b | 2a | 2b | 2a | 2b |
| H 1 R based H 4 R model | ||||||
| Pose 1c | 56.2 | 89.4 | 91.0 | 94.6 | 87.6 | 75.2 |
| Pose 2c | 0 | 0 | 15.4 | 14.4 | 22.6 | 24.0 |
| Pose 3c | 0 | 0 | 0 | 0 | 0 | 0 |
| ADRB2 based H 4 R model | ||||||
| Pose 1c | 21.0 | 70.6 | 90.2 | 80.0 | 51.2 | 88.8 |
| Pose 2c | 0 | 0 | 38.0 | 0 | 0.2 | 0 |
| Pose 3c | 0 | 0 | 0 | 0 | 0 | 0 |



Structure–activity relationships of substituted indolecarboxamides and 2-aminopyrimidines
To determine the binding mode of indolecarboxamides and 2-aminopyrimidines we synthesized H, Cl, NH2, NO2, and OCH3 substituted indolecarboxamide (1a–e) and 2-aminopyrimidine (2a–e) analogues and investigated the effect of different substituents for both ligand classes (Table 1). The synthesis of the indolecarboxamide ligands is described in the literature36,37,39 and Fig. 3 illustrates the synthesis route for the 2-pyrimidine-2-amine derivatives. Starting from 4,6-dichloro-pyrimidine-2-amine, methylpiperidine was introduced via a nucleophilic aromatic substitution reaction. In a second step the accordant phenyl moieties were installed using a Suzuki reaction.Radioligand displacement studies of [3H]-histamine binding to the human H4R of the indolecarboxamide (1a–e) and 2-aminopyrimidine (2a–e) analogues indicated that the SAR of the two ligand series is comparable for small substituents but deviates for the larger –OCH3 substituent (Table 1). Substitution of the indolecarboxamide with the methoxy group (1e) resulted in a 63-fold decrease compared to the unsubstituted ligand (1a), while comparison of the methoxy- (2e) and unsubstituted (2a) 2-aminopyrimidines gave only a 3-fold affinity change. The Cl-substituted ligands showed the highest H4R binding affinity for both the indolecarboxamide and 2-aminopyrimidine ligand classes.
Ligand binding affinities for H4R, L1755.39V and E1825.46Q
The binding affinities (pKi) of the unsubstituted and Cl-substituted indolecarboxamide and 2-aminopyrimidine ligands were analysed for H4R, L1755.39V and E1825.46Q (Table 2, Fig. 4) using heterologous [3H]histamine displacement binding experiments. Previous studies have shown that E1825.46Q affects the binding of H4R ligands that contain two basic groups (3–6),2,3,7 but does not affect binding of ligands that contain only one basic moiety (1b, 7).3 The L1755.39V mutant affects the affinity for larger ligands 1b and 7, but does not affect the binding of 3.4 The fact that these two mutants show ligand-dependent effects and that they have previously been used to investigate the binding mode of 1b makes them attractive tools for investigating similarities and differences in binding modes of 1a,b and 2a,b. Both ligand classes lost affinity for L1755.39V. The Cl-substituted ligands, 1b and 2b, were more affected (ΔpKi(1b): −1.3, ΔpKi(2b): −1.5) than the unsubstituted ligands, 1a and 2a, (ΔpKi(1a): −0.6, ΔpKi(2a): −0.6). In contrast, E1825.46Q had a different effect on the two ligand classes. While the affinities of the 2-aminopyrimidines for E1825.46Q significantly decreased (ΔpKi(2a): −1.1, ΔpKi(2b): −2) the mutations did not have a significant effect on the affinities of the indolecarboxamides (ΔpKi(1a): −0.4, ΔpKi(1b): −0.2). Again the Cl-substituted 2-aminopyrimidine ligand 2b was more affected than the unsubstituted ligand 2a.Molecular docking studies
We used molecular docking simulations49,50 to generate different plausible poses for the two indolecarboxamides (1a and 1b) and 2-aminopyrimidines (2a and 2b) in a H4R homology model that is based on the recently resolved H1R crystal structure (Fig. 5).23 For comparison we also investigated docking poses in a H4R model that is based on the ADRB2 crystal structure34 (Fig. S1†) as this model was as good as the H1R-based H4R model in retrospective virtual screening studies.51 While the TM helices of ADRB2 and H1R crystal structures share the same overall fold,23,34 the conformations of in particular the second extracellular loops (EL2) are different (Fig. S1†).As a result, the H4R models based on ADRB2 and H1R crystal structures have very similar TM domains and show overall the same receptor–ligand interactions, except in the EL2 loop region (Fig. S1†). Only docking poses where the ligands interact with both, aspartate D943.32 and glutamate E1825.46 (essential residues for binding of H4R ligands)3,7 were considered. Both residues were modelled in the deprotonated (negatively charged) state. Three binding poses were generated for both ligand classes in the two different H4R models as starting structures for MD simulations. The binding pose of indolecarboxamides in Fig. 5A (pose 1) is similar to the binding mode proposed by Lim et al.,4 in which the piperazine and indole nitrogen atoms of the ligand donate H-bonds to D943.32 and E1825.46, respectively, while the aromatic moiety of the ligand is accommodated in the upper hydrophobic subpocket IIb. The binding pose in Fig. 5B (pose 2) is similar to the pose proposed by Kiss et al.38 in which the piperazine and indole nitrogen atoms of the ligand donate H-bonds to E1825.46 and D943.32, respectively, while the aromatic moiety of the ligand is directed towards TM7 (subpocket I28). The binding pose in Fig. 5C (pose 3) is similar to the binding mode reported by Schneider et al.,6 in which the piperazine and indole nitrogen atoms of the ligand donate H-bonds to D943.32 and E1825.46, respectively (similar to pose 1), while the aromatic ring of the ligand binds to subpocket IIa. To obtain this latter pose the χ1 torsional angle of C983.36 was rotated from its t-conformation to the g+-conformation.
Three docking poses were selected for the 2-aminopyrimidines that correspond to the respective poses of the indolecarboxamides (Fig. 5D–F). The amino group of the pyrimidine interacts in a similar manner as the NH of the indole group in these poses. A binding pose for 2-aminopyrimidines that is comparable to pose 1 was proposed by Werner et al.,40–46 with the exception that the 2-aminopyrimidine moiety in the Werner H4R model is differently oriented in the H4R binding pocket and H-bonds to E1825.46 are formed via water molecules.40–46 The 2-aminopyrimidines can form an additional hydrogen bond with either E1825.46, in pose 1 or 3, or D943.32 in pose 2 when the pyrimidine ring is protonated. pKa measurements for ligand 2a (see Experimental section for procedure) revealed a pKa value of 7.5 for the piperazine nitrogen atom and a pKa value of 6.0 for the pyrimidine, indicating that in solution approximately 2.3% of ligand 2a will be double-protonated at physiological pH 7.4.
Molecular dynamics simulations
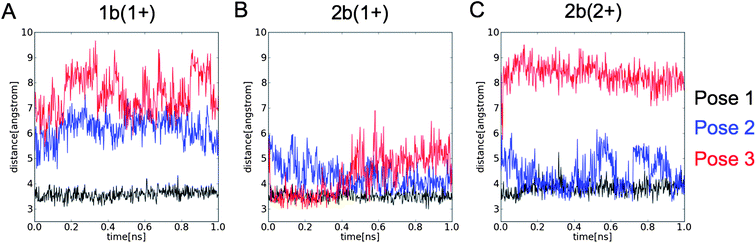 | ||
| Fig. 6 Shortest distance between any heavy atom of the L1755.39 and (A) 1b, and (B) mono-protonated (1+) and (C) double-protonated (2+) 2b in MD-simulations starting from pose 1 (black), pose 2 (blue), and pose 3 (red) (see Fig. 5). | ||
| Structure |
|
|
|
||||
|---|---|---|---|---|---|---|---|
| Protonation | 1+ | 1+ | 2+ | ||||
| R | H | Cl | H | Cl | H | Cl | |
| Name | 1a | 1b | 2a | 2b | 2a | 2b | |
| H 1 R based H 4 R model | |||||||
| WT | E5.46 | 1.2 | 1.1 | 1.1 | 1.3 | 2.5 | 2.2 |
| D3.32 | 0.9 | 0.9 | 1.0 | 1.0 | 1.0 | 1.0 | |
| E5.46Q | Q5.46 | 1.0 | 1.0 | 1.0 | 0.7 | 0.6 | 0.3 |
| D3.32 | 1.0 | 0.9 | 0.1 | 0.6 | 1.0 | 0.9 | |
| ADRB2 based H 4 R model | |||||||
| WT | E5.46 | 1.0 | 1.2 | 1.1 | 1.3 | 2.3 | 2.4 |
| D3.32 | 1.0 | 1.0 | 1.1 | 1.2 | 1.0 | 1.0 | |
| E5.46Q | Q5.46 | 0.9 | 1.2 | 1.3 | 1.4 | 1.1 | 0.7 |
| D3.32 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | |



Rationalization of differences in ligand structure–activity relationships
The SAR for different substitutions in both ligand series (Table 1) indicated that the larger substituent –OCH3 is less compatible with the H4R binding site than smaller apolar (–Cl) or polar (–NH2, –NO2) groups. This affinity decrease was much more pronounced for the indolecarboxamides than for 2-aminopyrimidines (Table 1). To explain this effect we monitored the pocket volume around 3.5 Å distance of the chlorine atom for 1b and 2b (Fig. 7). We observed that in the H1R-based H4R model the chlorine atom of 2b (independent of the modeled protonation state) occupies more frequently larger pockets than the chlorine atom of 1b. Average pocket volumes around the chlorine atom in the H1R-based H4R model were significantly smaller for ligand 1b (0.6 Å3) than for ligand 2b (8.4 Å3 (mono-protonated) and 3.2 Å3 (double-protonated)) while in the ADRB2-based H4R model the volume for 1b (11.0 Å3) was significantly larger than for 2b (6.4 Å3 (mono-protonated) and 3.9 Å3 (double-protonated)). The H1R-based H4R models therefore explain the fact that 2-aminopyrimidines can accommodate larger substituents than indolecarboxamides. A comparison of representative snapshots at the end of the MD-simulation showed that although the overall orientation of the ligands in the binding pocket is comparable, the chlorine atoms of both ligands are oriented in different pockets (Fig. 7A and B). The chlorine atom of 1b is directed towards the extracellular loop (EL2) and TM6 (surrounded by F16845.54, F16945.55, E16345.49, L1755.39, T3236.55) in a relatively tight binding pocket (Fig. 7A). The chlorine atom of 2b occupies a pocket, which is located more towards TM5 (surrounded by I1745.38, L1755.39 and T1785.42 in TM5, as well as F16845.54 and backbone atoms of EL2) and has a larger volume (Fig. 7B) that allows larger substituents (Table 1).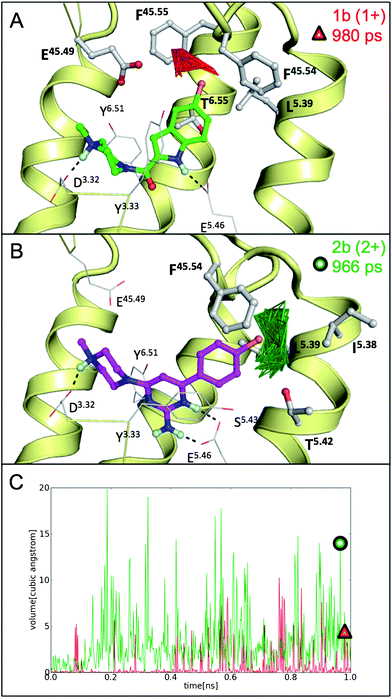 | ||
| Fig. 7 Representative snapshots of MD simulations starting from pose 1 in the H1R-based H4R model showing the pocket volume within 3.5 Å of the chlorine atom of ligand 1b (red, A) and ligand 2b (green, B). The snapshots presented in panels A (red triangle) and B (green circle) are indicated in panel C, showing pocket volumes within 3.5 Å of the chlorine atom of ligand 1b (red) and double-protonated ligand 2b (green) along the full 1 ns MD trajectories. | ||
Discussion
In the current study, ligand SAR and H4R SDM studies were combined with molecular docking and MD simulations to elucidate the binding modes of indolecarboxamides and 2-aminopyrimidines in the H4R binding pocket. Insights were obtained into the similarities and differences of molecular interactions between H4R and these two ligand classes.Overlap in indolecarboxamides and 2-aminopyrimidines binding modes
Our combined SDM and in silico studies indicate that the binding modes of indolecarboxamides and 2-aminopyrimidines overlap, but also show that there are subtle differences in the importance of different H4R interaction features within these two ligand series. Ligands of both ligand classes accommodate their aromatic ring moieties in subpocket IIb that is located in the upper half of the extracellular regions between TM helices 3–6 (pose 1), (Fig. 5A and D and Fig. S1†). Only in this pose all ligands (1a,b and 2a,b) form stable H-bond interactions with D943.32 and E1825.46 in combination with hydrophobic interactions with L1755.39. Current (Table 2 and Fig. 4) and previous SDM studies show that these interactions are important binding determinants for ligands 1a,b and 2a,b as well as other H4R ligands.2,3,7 The overall binding orientation was stable and not influenced by interactions with water molecules.It should be noted that effects of H4R mutations are often ligand specific. While the E1825.46Q mutant has a decreased affinity for ligands 2a, 2b, 3, and 4, this mutation does not affect binding of ligands 1a, 1b and 7 (Table 2 and Fig. 4).2,3 The L1755.39V mutant that mimics the monkey H4R has a decreased affinity for 1 and 2 (Table 2 and Fig. 4) but an increased affinity for ligand 7 (Fig. 1B), while binding of ligand 3 is not affected by this mutation.4 This reflects the diversity in H4R ligand binding modes (Fig. 1 and 7) that can match different parts of the H4R ligand pharmacophore.30 This pharmacophore contains: (i) 1 to 2 H-bond donors (e.g. piperazine and indole/aminopyrimidine nitrogen atoms of ligands 1 and 2) that are complementary to D943.32 (ref. 3 and 7) and E1825.46, (ii) a central aromatic moiety (e.g., indole (1) and 2-aminopyrimidine (2) moieties) that stacks between Y3.33 and Y6.51, and (iii) 1 to 2 hydrophobic moieties that can accommodate subpockets IIa (e.g., cyclohexyl groups of ligands 6 (Fig. 1C) and 8, the chlorinated benzene ring of ligand 7 (Fig. 1B)),2,4,33 IIb (benzene rings of ligands 1 and 2 (Fig. 7) and ligands 5 and -7 (Fig. 1B and C)2,4 between TM helices 3–6, or alternatively, subpocket I between TM helices 3 and 5–7 (benzene ring of ligand 5).2
Our MD-simulation showed that although the overall orientation of the ligands (1a,b and 2a,b) in the H4R binding pocket is comparable, the chlorine atoms of indolecarboxamide and 2-aminopyrimidine ligands are located in subpockets of different size (Fig. 7). This size difference explains why larger substituents have a more negative effect on the affinity for indolecarboxamides than for 2-aminopyrimidines (Table 1). Interestingly, SDM studies have shown ligand-dependent effects towards mutations in the same EL2 region that can explain H4R species selectivity differences.5
The role of ligand protonation states in H4R binding
In silico evaluation of different protonation states of 2-aminopyrimidines showed that the H4R binding models in which both the piperazine and 2-aminopyrimidine moieties are positively ionized best explain the affinity decrease of ligands 2a and 2b (but not of 1a and 1b) for the E1825.46Q mutant (Tables 2–4). The pKa value of the pyrimidine ring of 6.0, however, suggests that under physiological pH 7.4 only a small portion (∼2.3%) of ligands 2a and 2b contains a positively ionized pyrimidine ring. On the other hand, it has been reported that pKa values of functional groups in ligands can change upon binding, as demonstrated for the pteridine ring of methotrexate that becomes protonated upon binding to dihydrofolate reductase.54,55 Thus, it cannot be excluded that the pKa value of the 2-aminopyrimidine ring is shifted towards higher values upon binding to H4R.It should furthermore be noted that also other ligands have been proposed to bind in a double-protonated state to H4R. The two basic groups of ligand 9 have pKa values of 8.3 and 9.4 (ref. 56) indicating that most parts of this ligand will be double-protonated at pH 7.4. Agonist 4 (ref. 57), derived from ligand 9, has two chemically similar basic moieties, while the pKa values of the amine and imidazole groups of 3 (9.4–9.9 and 5.9–6.5, respectively)58–60 suggest that also 3 might bind in a double-protonated form. This is supported by the fact that the E1825.46Q H4R mutant has a significantly decreased affinity for small agonists 3 and 4 (ref. 3) that depend on strong H-bond interactions. The effects of the E1825.46Q mutation on H4R binding affinity are relatively smaller (5 and 6) or not significant (1a,b and 7) for ligands with larger hydrophobic moieties (Table 2, Fig. 4),2,3 as the binding of these molecules is more determined by hydrophobic interactions. Interestingly, in this current study we show that also the 2-aminopyrimidines (containing a hydrophobic benzene ring) are to a large extend dependent on ionic interactions with E1825.46.
Ligand-binding mode predictions in H4R homology models were modelling template independent
To elucidate H4R-ligand binding modes we considered two H4R models, one based on the ADRB2 crystal structure34 and another based on the more recent H1R crystal structure.23 The ADRB2 and H1R crystal structures share a similar fold23 and consequently both models are constructed using the same modeling approach.2,4 It is therefore not surprising that the overall structure of the homology models is relatively similar, and gives comparable results in ligand-binding mode prediction2 and virtual screening studies.51 Also in the current study, the conformational ensembles derived from MD simulations of ADRB2- and H1R-based H4R models show the same overall binding mode (pose 1) for indolecarboxamides and 2-aminopyrimidines that matches our SDM studies (Table 2 and Fig. 4).In contrast, only the H1R-based H4R model is able to explain that larger substituents have a larger negative effect on the affinity for indolecarboxamides than for 2-aminopyrimidines (Table 1) because the chlorine atoms of indolecarboxamide and 2-aminopyrimidine ligands are located in different subpockets close to EL2 (as discussed above). Accurate GPCR loop modelling is still very challenging, as demonstrated by the recent GPCR DOCK competitions to predict the coordinates of GPCR–ligand co-crystal structures,61,62 and emphasized by the different extracellular loop structures displayed in the currently available GPCR crystal structures.21,63 Despite some recent reports of successful automated retrospective prediction of individual loops of GPCR crystal structures,64 GPCR loop modeling should be approached with much caution and should be reserved for cases where loop building can be guided and validated by experimental data.65 Moreover, structure-based virtual screening studies have shown that loopless TM models of GPCRs can be suitable targets for virtual screening as well.63 Nevertheless, our H4R modeling studies indicate that the crystal structure of the more closely related H1R provides a better template for the EL2 region (despite the fact that some parts of this loop were not resolved in the H1R crystal structure),23 and to construct H4R models that can describe subtle differences between ligand binding modes.
Docking in combination with MD simulations explains H4R mutation studies
In particular the symmetric distributions of two acetic residues (aspartate D943.32 and glutamate E1825.46) and two hydrophobic subpockets IIa and IIb that are complementary to (two) basic and several hydrophobic groups in H4R ligands allow different plausible binding H4R–ligand modes (Fig. 5). Previous66 and current studies suggest that molecular docking alone is not suitable for accurate determination of relative probabilities of different ligand binding modes. To explain the in vitro SDM data (Table 2, Fig. 4), which are an average of different ligand and receptor orientations with time, a dynamic treatment of both ligand and receptor is required (Tables 3 and 4). MD simulations can account for distributions of protein–ligand interactions and thus can give a more comprehensive explanation of ligand-dependent mutation effects (Tables 2–4) and subtle differences in ligand structure–activity relationships (Table 1, Fig. 7).67,68 By systematically considering different ligand binding mode hypotheses and protein models, and combining ligand SAR and protein mutagenesis experiments with extensive MD-simulation studies (48 independent MD runs in total) we could identify similarities and differences in the molecular determinants of H4R binding for both indolecarboxamide and 2-aminopyrimidine ligand classes.Conclusion
We have successfully combined complementary in silico H4R structural modeling approaches and ligand SAR with in vitro mutagenesis data to investigate the binding mode of indolecarboxamides and 2-aminopyrimidines in H4R. By systematically considering different H4R modelling templates, ligand binding poses, and ligand protonation states in combination with molecular docking and MD simulations we are able to explain ligand-specific mutation effects and subtle differences in ligand SAR. Our results improve the knowledge of H4R–ligand binding and provide valuable information for optimizing and developing other H4R-specific ligands. The combined in silico modelling and in vitro SAR and mutagenesis approach presents a promising approach to explore protein–ligand interactions and can be considered as a general method to elucidate protein ligand-binding modes.Experimental section
Synthetic methods
High resolution mass spectroscopy data were obtained on a LTQ Orbitrap XL (Thermo Scientific) equipped with a NSI Source (Advion Nanomate) in ESI positive mode.
Analytical HPLC-MS analyses were conducted using an Agilent 1100 series LC/MSD system. The analytic method A1 is defined in Table S3.† Ligand purities were calculated as the percentage peak area of the analyzed ligand by UV detection at 254 nm. If purity data are not explicitly mentioned the ligand displays a purity >95%. Flash column chromatography was carried out using hand packed silica gel 60 (230–400 mesh) or pre-packed silica gel columns from Biotage and the product was eluted under medium pressure liquid chromatography. Preparative high performance chromatography was carried out on a Gilson system (pump system: 333 and 334 prep-scale HPLC pump; fraction collector: 215 liquid handler; detector: Gilson UV/VIS 155) using pre-packed reversed phase silica gel columns from waters. The method for preparative high performance chromatography P1 is defined in Table S3.†
6-Chloro-4-(4-methylpiperazin-1-yl)pyrimidin-2-amine (2h). 4,6-Dichloro-pyrimidine-2-amine 2f (1.00 g, 5.79 mmol), N-ethyl-diisopropylamine (1.33 g, 11.58 mmol) and 1-methylpiperazine 2g (0.64 g, 6.37 mmol) were suspended in 30 ml dichloromethane and stirred for 16 h at 20 °C. The solvent was removed under reduced pressure and the crude material was purified using silica gel flash column chromatography with a solvent mixture of dichloromethane, methanol and 25% aqueous ammonia of 90
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9:1 for elution. The solvent of the corresponding fractions was evaporated under reduced pressure, yielding 0.9 g (70%) of the title ligand. Purity by method A1: >95%; RT = 1.01 min; MS (ESI+) m/z 228/230 [M + H]+, Cl distribution; HRMS (ESI+) m/z found 228.1015 [M + H]+, C9H15ClN5 requires M+ 228.1016; 1H NMR (DMSO) δ (ppm) 6.45 (s, 2H), 6.08 (s, 1H), 3.56–3.47 (m, 4H), 2.30 (t, J = 5.1 Hz, 4H), 2.19 (s, 3H).
:9:1 for elution. The solvent of the corresponding fractions was evaporated under reduced pressure, yielding 0.9 g (70%) of the title ligand. Purity by method A1: >95%; RT = 1.01 min; MS (ESI+) m/z 228/230 [M + H]+, Cl distribution; HRMS (ESI+) m/z found 228.1015 [M + H]+, C9H15ClN5 requires M+ 228.1016; 1H NMR (DMSO) δ (ppm) 6.45 (s, 2H), 6.08 (s, 1H), 3.56–3.47 (m, 4H), 2.30 (t, J = 5.1 Hz, 4H), 2.19 (s, 3H).
4-phenyl-6-(4-methylpiperazin-1-yl)pyrimidin-2-amine (2a). 4-Chloro-6-(4-methylpiperazin-1-yl)pyrimidin-2-amine 2h (100 mg, 0.44 mmol), 2-phenylboronic acid (66 mg, 0.53 mmol), caesium carbonate (286 mg, 0.88 mmol) and dichloro[1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (36 mg, 0.04 mmol) were suspended in 1 ml of a 3
:1:1 mixture of tetrahydrofuran, 1-methyl-2-pyrrolidinone and water. The reaction mixture was flushed with argon and stirred for 16 h at 100 °C. The crude product was purified using method P1, yielding 76 mg (64%, 0.28 mmol) of the title ligand. Purity by method A1: >95%; RT = 1.35 min; MS (ESI) m/z 270 [M + H]+; HRMS (ESI+) m/z found 270.1715 [M + H]+, C15H20N5 requires M+ 270.1718; 1H NMR (500 MHz, DMSO) δ (ppm) 8.05–8.01 (m, 2H), 7.45–7.41 (m, 3H), 6.57 (s, 1H), 6.06 (s, 2H), 3.64–3.59 (m, 4H), 2.37–2.34 (m, 4H), 2.21 (s, 3H).
Using the same method 2b–e were synthesized. Analytic data for these ligands are given in the ESI.†
Pharmacological assays
Computational methods
The entire system was subjected to a constant pressure (1 bar) MD-simulation. All bonds involving hydrogen atoms were frozen with the SHAKE algorithm. In the first 100 ps the temperature was increased in a stepwise manner to 300 K. During this procedure positional harmonic restraints on helical Cα atoms of 10 kcal mol−1 were applied which were reduced to 0.5 kcal mol−1 for the subsequent 1 ns production run (at 300 K). The temperature was controlled using the weak coupling approach74 with a coupling constant of 0.2 ps. For calculating the long range electrostatic interactions the Particle Mesh Ewald (PMW) method was applied. van der Waals interactions were calculated within a cut-off of 8 Å.
For the protein, the POPC and the water molecules the AMBER03 force field75 was used. For the ligand atoms the GAFF76 force field was used. Ligand force field parameters were derived using ANTECHAMBER.77 These parameters were adjusted and extended for 2-aminopyrimidine ligands to retain the amino group (and in the case of double protonation also the additional proton on the pyrimidine ring) in the plane of the pyrimidine ring. Partial charges for the ligand were derived using the AM1-BCC procedure in ANTECHAMBER. The tLEaP module of AMBER10 was used to generate the topology and coordinate files of the protein–ligand complex.
During the MD-simulation trajectories of every 1000th MD-step were saved. This results in 500 snapshots for a 1 ns MD-simulation. Using the PTRAJ module of AMBER10 distances and angles could be extracted which were used to analyse H-bond interactions between the protein and the ligand and interactions of the ligand with L1755.39. An H-bond was counted if the distance between the H-bond donor and acceptor was below 3.5 Å and the angle between the H-bond donor, hydrogen and H-bond acceptor was between 180 ± 45°.78 Hydrophobic interactions of the ligand with L1755.39 were counted if the distance between any heavy atom of the ligand and L1755.39 was ≤4 Å.
Acknowledgements
This research was financially supported by The Netherlands Organization for Scientific Research [NWO VENI Grant 700.59.408 to C. de G.], and COST Action BM0806. Obbe Zuiderveld is acknowledged for technical assistance.References
- P. Igel, R. Geyer, A. Strasser, S. Dove, R. Seifert and A. Buschauer, J. Med. Chem., 2009, 52, 6297–6313 CrossRef CAS.
- E. P. Istyastono, S. Nijmeijer, H. D. Lim, A. van de Stolpe, L. Roumen, A. J. Kooistra, H. F. Vischer, I. J. de Esch, R. Leurs and C. de Graaf, J. Med. Chem., 2011, 54, 8136–8147 CrossRef CAS.
- A. Jongejan, H. D. Lim, R. A. Smits, I. J. de Esch, E. Haaksma and R. Leurs, J. Chem. Inf. Model., 2008, 48, 1455–1463 CrossRef CAS.
- H. D. Lim, C. de Graaf, W. Jiang, P. Sadek, P. M. McGovern, E. P. Istyastono, R. A. Bakker, I. J. de Esch, R. L. Thurmond and R. Leurs, Mol. Pharmacol., 2010, 77, 734–743 CrossRef CAS.
- H. D. Lim, A. Jongejan, R. A. Bakker, E. Haaksma, I. J. de Esch and R. Leurs, J. Pharmacol. Exp. Ther., 2008, 327, 88–96 CrossRef CAS.
- E. H. Schneider, A. Strasser, R. L. Thurmond and R. Seifert, J. Pharmacol. Exp. Ther., 2010, 334, 513–521 CrossRef CAS.
- N. Shin, E. Coates, N. J. Murgolo, K. L. Morse, M. Bayne, C. D. Strader and F. J. Monsma, Jr, Mol. Pharmacol., 2002, 62, 38–47 CrossRef CAS.
- G. Coruzzi, M. Adami, E. Guaita, I. J. de Esch and R. Leurs, Eur. J. Pharmacol., 2007, 563, 240–244 CrossRef CAS.
- J. M. Cowden, J. P. Riley, J. Y. Ma, R. L. Thurmond and P. J. Dunford, Respir. Res., 2010, 11, 86 CrossRef.
- P. J. Dunford, N. O'Donnell, J. P. Riley, K. N. Williams, L. Karlsson and R. L. Thurmond, J. Immunol., 2006, 176, 7062–7070 CAS.
- P. J. Dunford, K. N. Williams, P. J. Desai, L. Karlsson, D. McQueen and R. L. Thurmond, J. Allergy Clin. Immunol., 2007, 119, 176–183 CrossRef CAS.
- F. Gantner, K. Sakai, M. W. Tusche, W. W. Cruikshank, D. M. Center and K. B. Bacon, J. Pharmacol. Exp. Ther., 2002, 303, 300–307 CrossRef CAS.
- C. L. Hofstra, P. J. Desai, R. L. Thurmond and W. P. Fung-Leung, J. Pharmacol. Exp. Ther., 2003, 305, 1212–1221 CrossRef CAS.
- G. C. Hsieh, P. Chandran, A. K. Salyers, M. Pai, C. Z. Zhu, E. J. Wensink, D. G. Witte, T. R. Miller, J. P. Mikusa, S. J. Baker, J. M. Wetter, K. C. Marsh, A. A. Hancock, M. D. Cowart, T. A. Esbenshade, J. D. Brioni and P. Honore, Pharmacol., Biochem. Behav., 2010, 95, 41–50 CrossRef CAS.
- H. D. Lim, R. A. Smits, R. Leurs and I. J. De Esch, Curr. Top. Med. Chem., 2006, 6, 1365–1373 CAS.
- P. Ling, K. Ngo, S. Nguyen, R. L. Thurmond, J. P. Edwards, L. Karlsson and W. P. Fung-Leung, Br. J. Pharmacol., 2004, 142, 161–171 CrossRef CAS.
- C. Liu, X. Ma, X. Jiang, S. J. Wilson, C. L. Hofstra, J. Blevitt, J. Pyati, X. Li, W. Chai, N. Carruthers and T. W. Lovenberg, Mol. Pharmacol., 2001, 59, 420–426 CAS.
- M. O'Reilly, R. Alpert, S. Jenkinson, R. P. Gladue, S. Foo, S. Trim, B. Peter, M. Trevethick and M. Fidock, J. Recept. Signal Transduction, 2002, 22, 431–448 Search PubMed.
- K. Rossbach, S. Wendorff, K. Sander, H. Stark, R. Gutzmer, T. Werfel, M. Kietzmann and W. Baumer, Exp. Dermatol., 2009, 18, 57–63 CrossRef CAS.
- R. L. Thurmond, P. J. Desai, P. J. Dunford, W. P. Fung-Leung, C. L. Hofstra, W. Jiang, S. Nguyen, J. P. Riley, S. Sun, K. N. Williams, J. P. Edwards and L. Karlsson, J. Pharmacol. Exp. Ther., 2004, 309, 404–413 CrossRef CAS.
- V. Katritch, V. Cherezov and R. C. Stevens, Trends Pharmacol. Sci., 2012, 33, 17–27 CrossRef CAS.
- A. J. Kooistra, L. Roumen, R. Leurs, I. J. P. de Esch and C. de Graaf, Methods Enzymol., 2012 Search PubMed , in press.
- T. Shimamura, M. Shiroishi, S. Weyand, H. Tsujimoto, G. Winter, V. Katritch, R. Abagyan, V. Cherezov, W. Liu, G. W. Han, T. Kobayashi, R. C. Stevens and S. Iwata, Nature, 2011, 475, 65–70 CrossRef.
- C. de Graaf, A. J. Kooistra, H. F. Vischer, V. Katritch, M. Kuijer, M. Shiroishi, S. Iwata, T. Shimamura, R. C. Stevens, I. J. de Esch and R. Leurs, J. Med. Chem., 2011, 54, 8195–8206 CrossRef CAS.
- R. Kiss, B. Kiss, A. Konczol, F. Szalai, I. Jelinek, V. Laszlo, B. Noszal, A. Falus and G. M. Keseru, J. Med. Chem., 2008, 51, 3145–3153 CrossRef CAS.
- F. Sirci, E. P. Istyastono, H. F. Vischer, A. J. Kooistra, S. Nijmeijer, M. Kuijer, M. Wijtmans, R. Mannhold, R. Leurs, I. J. P. de Esch and C. de Graaf, 2012, Poster presentation: 30th Noorwijkerhout–Camerino–Cyprus Symposium: Trends in Drug Research, http://f1000.com/posters/browse/summary/1090195 Search PubMed.
- Y. Tanrikulu, E. Proschak, T. Werner, T. Geppert, N. Todoroff, A. Klenner, T. Kottke, K. Sander, E. Schneider, R. Seifert, H. Stark, T. Clark and G. Schneider, ChemMedChem, 2009, 4, 820–827 CrossRef CAS.
- J. S. Surgand, J. Rodrigo, E. Kellenberger and D. Rognan, Proteins, 2006, 62, 509–538 Search PubMed.
- H. Engelhardt, R. A. Smits, R. Leurs, E. Haaksma and I. J. de Esch, Curr. Opin. Drug Discovery Dev., 2009, 12, 628–643 CAS.
- E. P. Istyastono, C. de Graaf, I. J. de Esch and R. Leurs, Curr. Top. Med. Chem., 2011, 11, 661–679 CAS.
- R. A. Smits, R. Leurs and I. J. de Esch, Drug Discovery Today, 2009, 14, 745–753 CrossRef CAS.
- L. Shi and J. A. Javitch, Annu. Rev. Pharmacol. Toxicol., 2002, 42, 437–467 CrossRef CAS.
- M. Wijtmans, C. de Graaf, G. de Kloe, E. P. Istyastono, J. Smit, H. Lim, R. Boonnak, S. Nijmeijer, R. A. Smits, A. Jongejan, O. Zuiderveld, I. J. de Esch and R. Leurs, J. Med. Chem., 2011, 54, 1693–1703 CrossRef CAS.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS.
- H. D. Lim, R. M. van Rijn, P. Ling, R. A. Bakker, R. L. Thurmond and R. Leurs, J. Pharmacol. Exp. Ther., 2005, 314, 1310–1321 CrossRef CAS.
- H. Engelhardt, I. J. P. de Esch, D. Kuhn, R. A. Smits, O. P. Zuiderveld, J. Dobler, M. Mayer, S. Lips, H. Arnhof, D. Scharn, E. E. J. Haaksma and R. Leurs, Eur. J. Med. Chem., 2012, 54, 660–668 CrossRef CAS.
- J. A. Jablonowski, C. A. Grice, W. Chai, C. A. Dvorak, J. D. Venable, A. K. Kwok, K. S. Ly, J. Wei, S. M. Baker, P. J. Desai, W. Jiang, S. J. Wilson, R. L. Thurmond, L. Karlsson, J. P. Edwards, T. W. Lovenberg and N. I. Carruthers, J. Med. Chem., 2003, 46, 3957–3960 CrossRef CAS.
- R. Kiss, B. Noszal, A. Racz, A. Falus, D. Eros and G. M. Keseru, Eur. J. Med. Chem., 2008, 43, 1059–1070 CrossRef CAS.
- J. D. Venable, H. Cai, W. Chai, C. A. Dvorak, C. A. Grice, J. A. Jablonowski, C. R. Shah, A. K. Kwok, K. S. Ly, B. Pio, J. Wei, P. J. Desai, W. Jiang, S. Nguyen, P. Ling, S. J. Wilson, P. J. Dunford, R. L. Thurmond, T. W. Lovenberg, L. Karlsson, N. I. Carruthers and J. P. Edwards, J. Med. Chem., 2005, 48, 8289–8298 CrossRef CAS.
- R. J. Altenbach, R. M. Adair, B. M. Bettencourt, L. A. Black, S. R. Fix-Stenzel, S. M. Gopalakrishnan, G. C. Hsieh, H. Liu, K. C. Marsh, M. J. McPherson, I. Milicic, T. R. Miller, T. A. Vortherms, U. Warrior, J. M. Wetter, N. Wishart, D. G. Witte, P. Honore, T. A. Esbenshade, A. A. Hancock, J. D. Brioni and M. D. Cowart, J. Med. Chem., 2008, 51, 6571–6580 CrossRef CAS.
- M. D. Cowart, R. J. Altenbach, H. Liu, G. C. Hsieh, I. Drizin, I. Milicic, T. R. Miller, D. G. Witte, N. Wishart, S. R. Fix-Stenzel, M. J. McPherson, R. M. Adair, J. M. Wetter, B. M. Bettencourt, K. C. Marsh, J. P. Sullivan, P. Honore, T. A. Esbenshade and J. D. Brioni, J. Med. Chem., 2008, 51, 6547–6557 CrossRef CAS.
- J. Liu, Y. Li, H. X. Zhang, S. W. Zhang and L. Yang, J. Mol. Model., 2012, 18, 991–1001 CrossRef CAS.
- S. P. Meduna, B. M. Savall, H. Cai, J. P. Edwards, R. L. Thurmond and P. M. McGovern, Bioorg. Med. Chem. Lett., 2011, 21, 3113–3116 CrossRef CAS.
- C. E. Mowbray, A. S. Bell, N. P. Clarke, M. Collins, R. M. Jones, C. A. Lane, W. L. Liu, S. D. Newman, M. Paradowski, E. J. Schenck, M. D. Selby, N. A. Swain and D. H. Williams, Bioorg. Med. Chem. Lett., 2011, 21, 6596–6602 CrossRef CAS.
- K. Sander, T. Kottke, Y. Tanrikulu, E. Proschak, L. Weizel, E. H. Schneider, R. Seifert, G. Schneider and H. Stark, Bioorg. Med. Chem., 2009, 17, 7186–7196 CrossRef CAS.
- T. Werner, K. Sander, Y. Tanrikulu, T. Kottke, E. Proschak, H. Stark and G. Schneider, Chembiochem, 2010, 11, 1850–1855 CrossRef CAS.
- P. H. Keizers, C. de Graaf, F. J. de Kanter, C. Oostenbrink, K. A. Feenstra, J. N. Commandeur and N. P. Vermeulen, J. Med. Chem., 2005, 48, 6117–6127 CrossRef CAS.
- A. C. Kruse, J. Hu, A. C. Pan, D. H. Arlow, D. M. Rosenbaum, E. Rosemond, H. F. Green, T. Liu, P. S. Chae, R. O. Dror, D. E. Shaw, W. I. Weis, J. Wess and B. K. Kobilka, Nature, 2012, 482, 552–556 CrossRef CAS.
- O. Korb, T. Stutzle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS.
- O. Korb, T. Stützle and T. E. Exner, Lect Notes Comput Sci, 2006, 4150, 247–258 Search PubMed.
- E. P. Istyastono. PhD Thesis, 2012.
- I. Gantz, J. DelValle, L. D. Wang, T. Tashiro, G. Munzert, Y. J. Guo, Y. Konda and T. Yamada, J. Biol. Chem., 1992, 267, 20840–20843 CAS.
- K. Ohta, H. Hayashi, H. Mizuguchi, H. Kagamiyama, K. Fujimoto and H. Fukui, Biochem. Biophys. Res. Commun., 1994, 203, 1096–1101 CrossRef CAS.
- L. Cocco, J. P. Groff, C. Temple, Jr, J. A. Montgomery, R. E. London, N. A. Matwiyoff and R. L. Blakley, Biochemistry, 1981, 20, 3972–3978 CrossRef CAS.
- P. Czodrowski, I. Dramburg, C. A. Sotriffer and G. Klebe, Proteins, 2006, 65, 424–437 Search PubMed.
- G. J. Sterk, H. van der Goot and H. Timmerman, Eur. J. Med. Chem., 1984, 19, 545 CAS.
- H. D. Lim, R. A. Smits, R. A. Bakker, C. M. van Dam, I. J. de Esch and R. Leurs, J. Med. Chem., 2006, 49, 6650–6651 CrossRef CAS.
- B. Lenarcik and M. Wisniewski, Polish. J. Chem, 1983, 57, 735–744 Search PubMed.
- P. R. Reddy, N. Raju, K. S. Rao and A. Shilpa, Indian J. Chem., 2009, 48, 761–768 Search PubMed.
- T. Vitali, M. Impicciatore, C. Ferrari and G. Morini, Farmaco, Ed. Sci., 1980, 35, 366–379 Search PubMed.
- I. Kufareva, M. Rueda, V. Katritch, Participants, G. D., R. C. Stevens and R. Abagyan, Structure, 2011, 19, 1108–1126 Search PubMed.
- M. Michino, E. Abola, Participants, G. D., C. L. Brooks, III, J. S. Dixon, J. Moult and R. C. Stevens, Nat. Rev. Drug Discovery, 2009, 8, 455–463 Search PubMed.
- C. de Graaf and D. Rognan, Curr. Pharm. Des., 2009, 15, 4026–4048 CrossRef CAS.
- D. A. Goldfeld, K. Zhu, T. Beuming and R. A. Friesner, Proc Natl Acad Sci USA, 2011, 108, 8275–8280 CrossRef CAS.
- C. de Graaf, N. Foata, O. Engkvist and D. Rognan, Proteins, 2008, 71, 599–620 Search PubMed.
- E. Stjernschantz and C. Oostenbrink, Biophys. J., 2010, 98, 2682–2691 CrossRef CAS.
- M. Bruysters, A. Jongejan, A. Akdemir, R. A. Bakker and R. Leurs, J. Biol. Chem., 2005, 280, 34741–34746 CrossRef CAS.
- A. Heifetz, G. B. Morris, P. C. Biggin, O. Barker, T. Fryatt, J. Bentley, D. Hallett, D. Manikowski, S. Pal, R. Reifegerste, M. Slack and R. Law, Biochemistry, 2012, 51, 3178–3197 CrossRef CAS.
- R. I. Allen, K. J. Box, J. E. Comer, C. Peake and K. Y. Tam, J. Pharm. Biomed. Anal., 1998, 17, 699–712 CrossRef CAS.
- G. Volgyi, R. Ruiz, K. Box, J. Comer, E. Bosch and K. Takacs-Novak, Anal. Chim. Acta, 2007, 583, 418–428 CrossRef.
- Chemical Computing Group (CCG) MOE (Molecular Operating Environment) v.2009.10, Montreal, 2009, http://www.chemcomp.com/software.htm.
- D. A. Case, T. A. Darden, T. E. Chatman III, C. W. J. Simmerling, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvary, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, AMBER10, 2008 Search PubMed.
- E. Urizar, S. Claeysen, X. Deupi, C. Govaerts, S. Costagliola, G. Vassart and L. Pardo, J. Biol. Chem., 2005, 280, 17135–17141 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS.
- G. Marcou and D. Rognan, J. Chem. Inf. Model., 2007, 47, 195–207 CrossRef CAS.
- J. D. Durrant, C. A. de Oliveira and J. A. McCammon, J. Mol. Graphics Modell., 2011, 29, 773–776 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional analyses of molecular dynamics simulations, purity of compounds by LC-MS and NMR, and comparison of ADRB2 vs. H1R crystal structure based H4R homology models. See DOI: 10.1039/c2md20212c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |