 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
BH3 helix-derived biophotonic nanoswitches regulate cytochrome c release in permeabilised cells†
Robert J. Martab,
Rachel J. Erringtonc,
Catherine L. Watkinscd,
Sally C. Chappellc,
Marie Wiltshirec,
Arwyn T. Jonesd,
Paul J. Smithc and
Rudolf K. Allemann*ab
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: allemannrk@cardiff.ac.uk; Fax: +44 (0)29 208 74030; Tel: +44 (0)29 208 79014
bCardiff Catalysis Institute, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
cInstitute of Cancer and Genetics, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK
dCardiff School of Pharmacy and Pharmaceutical Sciences, Cardiff University, Redwood Building, King Edward VII Avenue, Cardiff CF10 3NB, UK
First published on 13th August 2013
Abstract
Dynamic physical interactions between proteins underpin all key cellular processes and are a highly attractive area for the development of research tools and medicines. Protein–protein interactions frequently involve α-helical structures, but peptides matching the sequences of these structures usually do not fold correctly in isolation. Therefore, much research has focused on the creation of small peptides that adopt stable α-helical structures even in the absence of their intended protein targets. We show that short peptides alkylated with azobenzene crosslinkers can be used to photo-stimulate mitochondrial membrane depolarization and cytochrome c release in permeabilised cells, the initial events of the intrinsic apoptosis pathway.
Introduction
The specific recognition and binding of one protein to another is a fundamental biochemical mechanism for the regulation of complex cellular pathways. With over half a million potential targets, protein–protein interactions attract wide attention for the development of precision research tools and new medicines. Despite their importance, intracellular protein–protein interactions have proven inherently difficult to target because protein interfaces are often extensive, shallow and mainly hydrophobic with only limited opportunities for selective chemical intervention with small molecules. The development of peptide-based reagents for protein recognition provides a credible biomimetic strategy for selectively targeting functional regions of protein surfaces. Since many biomolecular interactions involve α-helical structures, much research has focused on the creation of small peptides that adopt stable α-helical structures even in the absence of their intended protein targets. Restricting the conformational freedom of an α-helical peptide minimizes the need for structural reorganization upon target binding and hence increases complex stability. Short peptides have been alkylated with a photoisomerisable azobenzene crosslinker to endow them with light-responsive α-helical structures.1–9 A considerable advantage of this design concept is the possibility to achieve predictable and reversible control over the target affinity of a constrained peptide, thereby overcoming both the limited protein recognition and the all-on or all-off activity modes of most small organic molecules.Programmed cell death is normally activated by different forms of cellular stress signals such as genotoxic or oxidative damage, but it is frequently dysregulated in cancerous cells, allowing aberrant cells to survive and multiply. Interactions between members of the B-cell leukaemia-2 (Bcl-2) family of proteins, which comprises both pro-apoptotic (Bak, Bim, Bid, Bad, PUMA, NOXA, Bik, Bmf) and anti-apoptotic (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1) members, are critical for the regulation of programmed cell death. Members of the Bcl-2 family are characterised by the presence of up to four conserved Bcl-2 homology (BH) domains that contain α-helical peptide segments. These regions mediate interactions between pro- and anti-apoptotic proteins as illustrated by the solution NMR structure of the complex of Bcl-xL with a peptide derived from the BH3 region of Bak showing that the Bak peptide forms an amphipathic α-helix that binds to a hydrophobic groove on the surface of Bcl-xL.10 Normally, the concentration of proteins in a cell favour the formation of such inactive heterodimeric complexes, but stress triggers can result in increased production of pro-apoptotic BH3-containing proteins11 or the unmasking of previously buried BH3 regions. These additional signals tip the balance towards apoptosis and either liberate the key pore-forming proteins Bak and Bax from inactive complexes with anti-apoptotic proteins or otherwise activate them.12,13 Bak and Bax then homo-oligomerize in the mitochondrial outer membrane and form pore complexes that initiate loss of mitochondrial outer membrane polarisation (MOMP) and a catastrophic release of cytochrome c into the cytosol.14–16
In contrast to their well-defined structure in protein complexes, BH3 derived peptides are generally unstructured in solution and hence bind to their targets with relatively low affinity. Their α-helicity can be increased in an irreversible fashion by covalently attaching ‘staples’ in a metathesis reaction.17,18 Initial reports suggested that such stapled peptides could effectively cross cell membranes and positive annexin V assays were taken to indicate the onset of apoptosis in Jurkat19,20 and mouse embryonic fibroblast (MEF)21 cells. However, recent findings indicate that neither MEF nor Jurkat cells treated with stapled peptides become permeable to propidium iodide,23 suggesting that a combination of markers are required to assess the progress towards full apoptosis induction by BH3 peptides. The reversible photo-control of α-helicity of BH3 peptides and their affinities for target proteins has been achieved with an azobenzene-derived crosslinker (Fig. 1), thereby providing the potential to activate apoptotic processes in cellular systems in a reversible fashion.24 Stabilisation of the α-helical conformation was observed when the cross-linker was in the cis-configuration for peptides linked through cysteine residues with i, i + 7 spacing, while the trans-configuration was α-helix-stabilizing for i, i + 11 spacings. For complexes of Bcl-xL and fluorescent derivatives (N-terminal fluoresceinamide; FAM) of the Bak-derived peptides BakI81Fi,i+11-XL and BakI81Fi,i+7-XL, small changes in α-helix stabilisation have been shown to be mirrored by large differences in the stability of the peptide–protein complexes.24 The NMR solution structure of BakI81Fi,i+11-XL in complex with Bcl-xL revealed the molecular mechanism, by which the increased affinity of such biophotonic nanoswitches is achieved, and showed why the I81F mutation does not increase the affinity of the peptides for i, i + 11 spacing.25
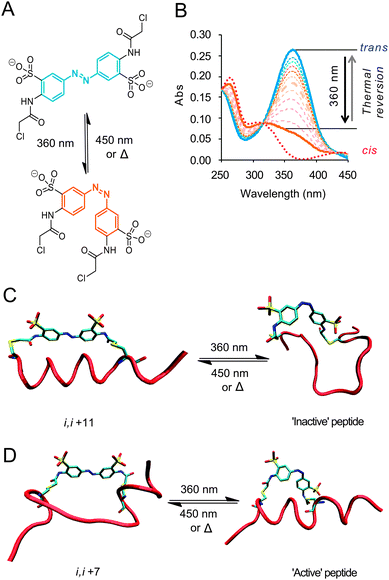 | ||
| Fig. 1 (A) Photo-isomerization of the water-soluble, cysteine-reactive azobenzene crosslinker with 360 nm light (or 720 nm two-photon irradiation)22 (B) UV-Vis spectra showing ∼75% switching to the cis-isomer followed by thermal reversion to the trans form. Idealised cartoons illustrating: (C) peptide crosslinked through i, i + 11 spaced cysteines; the trans-conformation encourages an α-helical in the dark state (FAM-Baki,i+11-XL, FAM-Bidi,i+11-XL) and a non-α-helical photoactivated state. (D) i, i + 7 Spaced cysteines (Ac-BakI81Fi,i+7-XL) reverse this with α-helical conformation is disfavoured in the dark state; whilst the light state stabilizes the α-helical conformation. | ||
Results and discussion
To investigate the ability of photoswitchable peptides to induce effects mimicking the earliest events of apoptosis, photoswitchable peptides derived from Bak and Bid, Ac-BakI81Fi,i+7-XL, FAM-Baki,i+11-XL and FAM-Bidi,i+11-XL (Table 1) were synthesised using Fmoc solid phase chemistry (see ESI†). The peptides were purified by reverse phase chromatography, alkylated with 3,3′-bis(sulfo)-4,4′-bis(chloroacetamido)azobenzene, repurified and their identity confirmed by mass spectrometry. In their dark states, FAM-Baki,i+11-XL and FAM-Bidi,i+11-XL bound to Bcl-xL with dissociation constants of 15 nM and 43 nM, respectively, as determined in fluorescence anisotropy titrations (Fig. S4, ESI†). The half-lives of the cis-forms at 37 °C were 18 minutes for FAM-Baki,i+11-XL and 15 minutes for FAM-Bidi,i+11-XL. The relative stabilities towards protease digestion of acetylated i, i + 7 and i, i + 11 peptides with and without crosslinker were assessed; addition of the crosslinker reduced the proteolytic stability of the i, i + 7 peptide and increased that of an equivalent i, i + 11 peptide (Fig. S5, ESI†). In agreement with previous reports, no evidence of susceptibility to reduction by physiological concentrations of glutathione was observed (Fig. S6, ESI†).22,26| Peptidea | Sequenceb |
|---|---|
| a Where Ac denotes N-terminal acetyl and FAM N-terminal fluoresceinamide.b Cysteines used for crosslinking are bold while other deviations from the wild type sequences are underlined (see ESI). | |
| Ac-BakI81Fi,i+7 | Ac-72G![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) VGRCLAI VGRCLAI![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) GDCINR87-NH2 GDCINR87-NH2 |
| FAM-Baki,i+11 | FAM-72GCVGRLAIGDCINR87-NH2 |
| FAM-Bidi,i+11 | FAM-81DIIRCIA![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) HLAVGDC HLAVGDC![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) DRSI101-NH2 DRSI101-NH2 |
Cancer cell lines can be divided into three general classes according to their apoptotic competence when challenged by BH3 mimics.27 Class A comprises cells with functional loss/dysfunction of sensing pathways or critical signal aggregators such as p53 resulting in a loss of the trigger signals which initiate apoptosis. Class B cells display loss of both Bak and Bax function and compromised pore formation. Class C cells show upregulation of anti-apoptotic Bcl-2 family members, which sequester elevated levels of pro-apoptotic molecules. In classes A and C, the ability of aberrant cells to commit to apoptosis remains intact but is latent. Here we have focused on class C cells, described as ‘primed for death’,28 since our peptides need only bind competitively to anti-apoptotic Bcl-2 proteins to release full-length activating pro-apoptotic to attempt to trigger the intrinsic apoptosis pathway. Human lymphoma SU-DHL-4 cells29,30 were made permeable through treatment with digitonin and the status of MOMP was monitored at the single cell level.31,32
Permeabilised cell systems may not fully reflect the behaviour of intact cells, but represent a compromise between complexity and accessibility. They provide quite well defined systems that can be analyzed without the complexities encountered in live cells.21,22 Apoptotic susceptibility can vary across a population of cells according to their position in the cell cycle and other as yet unknown factors. This heterogeneity is of crucial interest in the development of improved chemotherapeutics, making it highly desirable to see the effect of an identical apoptotic stimulus on an entire population of cells. However, azobenzene-modified peptides are typically delivered by lipidic transfection reagents22,33 or microinjection,26 producing an unavoidably heterogeneous peptide loading in target cells. Determining the differences in peptide loading and availability, e.g. whether the peptide resides in vesicles or free in the cytosol, at the same time as cell-to-cell variations in susceptibility are extremely challenging. Using permeablised cells ameliorates disparities brought about by variations in peptide delivery, compartmentalisation and potential active export of peptides from cells, whilst still representing a leap in complexity over in vitro testing with many more potential partners for BH3–protein interactions available.
Flow cytometry techniques allow correlation between the susceptibility of cells to our peptide agents and concentrations of other molecules or cellular properties via multi-wavelength fluorescence activated cell sorting, e.g. correlation between susceptibility to cytochrome c loss via immunofluorescence and cell cycle position as reported through the use of the spectrally compatible far-red DNA dye DRAQ5.34,35 Control experiments indicated that digitonin-permeablised cells in respiration buffer36 maintained their complement of anti-apoptotic Bcl-2 over at least a 30 minute period, with only a 1.5% fraction of cells exhibiting Bcl-2 levels below those found in intact cells (Fig. S7, ESI†) and hence provide a window of opportunity to tentatively assess activity in the presence of the cells complement of antiapoptotic proteins. MOMP was assayed using the mitochondria associating dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine iodide), which exhibits a green fluorescence in its positively-charged monomeric state, while association with healthy, membrane-polarised mitochondria neutralises the charge on the dye, promoting the formation of orange fluorescent J-aggregates.37,38 The level of cellular fluorescence at 580 nm and its distribution in the cell directly reports on mitochondrial health and morphology. SU-DHL-4 cells were pre-loaded with JC-1, then permeablised and maintained in respiration buffer before being treated with FAM-Baki,i+11-XL or FAM-Bidi,i+11-XL. In the dark state, FAM-Baki,i+11-XL and FAM-Bidi,i+11-XL adopt a mostly α-helical conformation (Fig. S3, ESI†) and hence can act as indicators of the potential efficiency for peptide-induced apoptosis. FAM-Baki,i+11-XL and FAM-Bidi,i+11-XL caused a sustained (>120 minute) decrease of membrane potential in a dose-dependent manner (Fig. 2A and B). FAM-Baki,i+11-XL was a more effective ‘initiator’ than FAM-Bidi,i+11-XL, despite both parent proteins being able to interact with the full spectrum of anti-apoptotic proteins. Confocal microscopy at 60 minutes showed a dramatic dual effect on the JC-1 aggregate signal (Fig. 2C); firstly, a severe loss of total fluorescence per cell and secondly the rearrangement of organelle morphology from a connected network to residual small punctuate structures. Clearly BH3-derived peptides alkylated with azobenzene cross-linkers have the ability to induce detectable physiological changes paralleling the early events in apoptotic cell death.
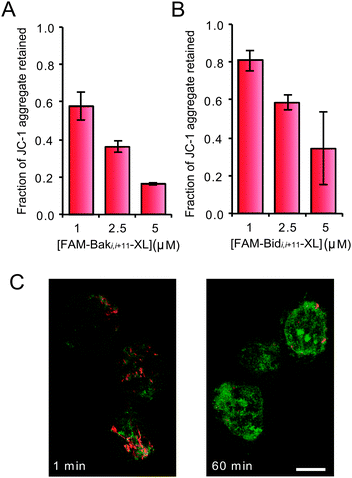 | ||
| Fig. 2 Retention of orange fluorescent JC-1 signal after a 120 min incubation with (A) FAM-Baki,i+11-XL and (B) FAM-Bidi,i+11-XL (±s.d.). (C) Confocal laser scanning microscope image of SU-DHL-4 cells labeled with JC-1 and treated with 7.5 μM FAM-Bidi,i+11-XL; the orange, aggregated form of the dye initially showed complex, morphological mitochondrial patterns typical of a healthy cell. After 1 min of exposure peptide rapidly accumulated in the cell in a non-specific pattern and after 60 min the aggregate signal was greatly reduced and punctate (bar = 10 μm). | ||
FAM-Baki,i+11-XL proved to be a potent inducer of apoptosis, but acted less well as a photoswitch (Fig. 3A) with a maximal conversion of 35% to the less active cis-form. FAM-BakI81Fi,i+7-XL on the other hand displays a much larger change in its affinity to Bcl-xL upon irradiation than FAM-BakI81Fi,i+11-XL24 or FAM-Baki,i+11-XL.25 Ac-BakI81Fi,i+7-XL was synthesized and displayed superior photo-switching properties (65–70% cis) (Fig. 3B) and an increased half-life of the cis-form of 28 min at 37 °C. Photo-activation of Ac-BakI81Fi,i+7-XL molecules triggered a measurable decay in the mitochondrial membrane potential (Fig. 3C) despite the fraction of the ‘active’ peptide diminishing rapidly even with an improved initial switching efficiency of 70%. The ‘switch-on’ nature of the i, i + 7 spacing photoswitch is also useful for observing cytochrome c release as this irreversible change is induced with light, rather than merely delayed by a ‘switch-off’ approach.
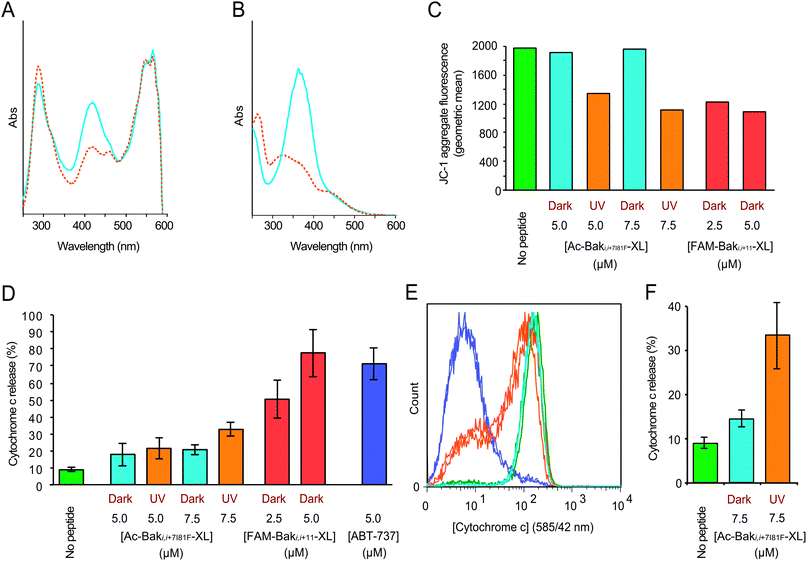 | ||
| Fig. 3 Typical absorbance spectra of cross-linked peptides in the dark (blue) and light-activated (red) state of (A) FAM-Baki,i+11-XL and (B) Ac-BakI81Fi,i+7-XL. (C) Typical cellular MOMP response after peptide activation. Geometric mean analysis of JC-1 aggregate fluorescence signal in permeabilized SU-DHL-4 cells 60 min after addition of Ac-BakI81Fi,i+7-XL in the dark and UV irradiated states and FAM-Baki,i+11-XL as a positive control. (D) Graph of the percentage of cells exhibiting cytochrome c release below a threshold in permeabilized SU-DHL-4 cell populations after 30 min exposure to 5.0 or 7.5 μM concentrations of Ac-BakI81Fi,i+7-XL with (red) and without (cyan) photoswitching. Controls include buffer only (green), FAM-Baki,i+11-XL (2.5 μM and 5.0 μM, red) and 5 μM ABT-737 (blue) (±s.d.). (E) Chart representing cytochrome c levels in permeabilized SU-DHL-4 cells exposed to ABT-737 (blue), irradiated (orange) and dark (cyan) 7.5 μM Ac-BakI81Fi,i+7-XL and no peptide (green) for 30 minutes depicting the sensitive sub-population that completes cytochrome c release before the bulk population. (F) Region analysis of cytochrome c release shows that delivery of a pulse of active peptide has a significant effect on apoptosis induction (±s.d.). | ||
Although MOMP necessarily precedes cytochrome c release we found, as has been observed by others,39 that the release of mitochondrial cytochrome c was more readily detected at early time points than MOMP changes. Using 30 minute incubation times to observe cytochrome c release, an early and committed step in apoptosis, allowed the matching of protein retention timescales with a single half-life of photoswitched Ac-BakI81Fi,i+7-XL. Our aim was to determine whether the test system showed evidence of a threshold for peptide-triggered cytochrome c release as a surrogate for apoptotic commitment or displayed any evidence of sub-population resistance. For the cytochrome c assay, SU-DHL-4 cells were permeablised and treated with the peptides for 30 minutes, then fixed, processed for cytochrome c detection and analysed by flow cytometry (see Materials and methods). The results suggest a finely balanced threshold for rapid commitment to apoptosis such that treatment with 5 μM of irradiated Ac-BakI81Fi,i+7-XL peptide was indistinguishable from its dark counterpart, while 7.5 μM of irradiated Ac-BakI81Fi,i+7-XL showed an increase in the fraction of cells releasing cytochrome c (Fig. 3D). Activation of components of the intrinsic apoptosis pathway resulted in the formation of a population of entirely cytochrome c depleted cells rather than an intermediate population (Fig. 3E); this result was similar to that obtained with the small molecule Bax BH3 mimic, ABT-737.40 Comparative analysis of a number of paired dark/irradiated responses showed that treatment with dark-adapted, non-α-helical Ac-BakI81Fi,i+7-XL produced only a minimal response (14.5 ± 2.0%) that was only slightly higher than that obtained with untreated cells (8.8 ± 1.3%). After irradiation, cytochrome c release increased significantly (33.2 ± 7.7%) (Fig. 3F). Nanoswitch effects were also evident by changes in the mitochondrial morphology.41 In control cells or cells exposed to unswitched Ac-BakI81Fi,i+7-XL, mitochondria formed a network of ring-like structures, whereas mitochondria exposed to photoswitched Ac-BakI81Fi,i+7-XL, FAM-Baki,i+11-XL or ABT-737 lost definition, forming dim punctuate and diffuse globular structures (Fig. S8, ESI†).
Conclusions
The cell population based approach adopted here lends itself to wider correlative studies with compatible fluorescent molecular probes and the potential for separation of cellular sub-populations of interest by fluorescence activated cell sorting. In this case, despite the entire population being capable of apoptosis when treated with ABT-737 (Fig. 3F), transient exposure to light-activated Ac-Baki,i+7-XL revealed a super-sensitive sub-population even within the larger primed population. To extend the potential of the platform, the permeabilised cell assay was paired with the DNA specific dye DRAQ5 to profile cell cycle related events. Plotting the proportion of cells in the G1, S and G2 phases of the cell cycle against the number of cells with depleted cytochrome c revealed differences in susceptibility to apoptosis. Both FAM-Baki,i+11-XL and photoactivated Ac-BakI81Fi,i+7-XL showed consistent cell cycle specific patterns for the induction of cytochrome c depletion (Fig. S10, ESI†) with implications for the origins of resistance to cell death triggering induced anticancer cytotoxic agents.Peptide-based nanoswitches have key advantages over small molecule agents for modulating cellular pathways controlled by protein–protein interactions. Firstly, nanoswitches exploit native sequences replicating the target selectivity of the parent protein. This is critical in cases where a protein of interest binds to many different partners as is the case with Bcl-2 family members whose patterns of affinity for each-other establish a pre-set for the apoptotic trigger. Secondly, the controlled delivery of a pulse of ‘activated’ peptide both matches the temporal scale for protein–protein interactions and provides an in situ perturbation that is discrete, predictable, programmable and not subject to a ‘wash-out’ procedure. The need for generic programmable tools has been highlighted by the appreciation that cell fate, for example determined by the stress signal integrator p53, can be related to the periodicity versus amplitude of the signaling pathway output.42 The design and synthesis of photoswitches with longer switching wavelengths and either rapid43 or extended44,45 relaxation times as required, coupled with improved methods for peptide delivery to live cells,46,47 may eventually lead to photo-controlled drugs and invaluable tools to probe critical protein–protein interactions in intact cells and whole organisms.48,49
Materials and methods
Synthesis of Bak and Bid peptides
BH3-derived peptides were synthesized by standard solid phase Fmoc chemistry, purified by HPLC and analyzed by MALDI mass spectroscopy.Cell lines and culture
The human follicular B-lymphoma cell line SU-DHL-4 used in this study was provided by P. F. E. Cotter50 and cultured in RPMI 1640 supplemented with 10% fetal calf serum. All growth media contained with 100 units mL−1 penicillin, 100 μg mL−1 streptomycin and 2 mM glutamine. The cells were passaged twice weekly at an initiating density of 5 × 104 cells mL−1 and cultured at 37 °C in a humidified atmosphere of 5% CO2/95% air. The p53 status of SU-DHL-4 (stabilized mutant) was confirmed by immunoblotting.Respiration buffer
Mitochondrial viability was maintained in MiR05 buffer (0.5 mM EGTA, 3 mM MgCl2, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KPO4, 20 mM HEPES, 110 mM sucrose and 1 g L−1 bovine serum albumin).51UV switching of peptides
The peptide was diluted from a stock solution in potassium phosphate (5 mM, pH 7) with respiration buffer. Peptide concentrations were determined by UV/Vis with a Nanodrop 2000 (LabTech) using an extinction coefficient of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 363 nm.
000 M−1 cm−1 at 363 nm.
Method 1
Peptide solution not exceeding 10 μL was pipetted into a glass-bottomed chamber (Nunc Lab Tek II), the chamber put onto a microscope stage and exposed to UV light filtered through a standard DAPI filter (350/50 nm) for 5 minutes.Method 2
10 μL of a peptide solution were placed in a clear PCR Eppendorf tube and irradiated with an array of ten FoxUV 360 nm LEDs for 5 minutes. A second UV/Vis absorbance spectrum was acquired after irradiation to determine the extent of switching.Permeabilised cell system for single cell mitochondrial membrane potential analysis using JC-1
SU-DHL-4 cells were seeded at a concentration of 2 × 105 mL−1 in complete RPMI 1640 media. JC-1 (Invitrogen Life Technologies) was added to the cells to a concentration of 1 μg mL−1 for 30 minutes at 37 °C in a 5% CO2 atmosphere. Cells were centrifuged (MSE Centaur 2, swinging bucket) at 900 rpm for 10 min, the supernatant was removed and replaced with 1 mL of fresh complete media. The cells were rested for a further 60 min at 37 °C in a 5% CO2 atmosphere then spun at 900 rpm for 10 min and the supernatant replaced with respiration buffer containing 10 μg mL−1 of digitonin (Sigma) and peptide. Control cells were run immediately on the flow cytometer to establish JC-1 signal ‘starting point’ then the samples were incubated for 60/120 min at 37 °C under a 5% CO2 atmosphere. Samples were analyzed with a FACS Calibur flow cytometer (Becton-Dickinson Immunocytometry Systems, San Jose, CA, USA) equipped with a 488 nm laser until 10000 total events had been collected using CellQuest software (Becton-Dickinson Immunocytometry Systems, San Jose, CA, USA). FSC, SSC (488/10 nm), FL-1 (530/30) channels and FL-2 (585/42) with compensation were acquired to quantify the J-monomer–FAM-peptide and J-aggregate, respectively.
Cytochrome c release assay
Cells were suspended at a concentration of 2 × 105 mL−1 in flow tubes and centrifuged at 900 rpm for 10 min. The supernatant was removed and replaced with 0.5 mL of respiration buffer with digitonin (10 μg mL−1) and peptides as required. The cells were mixed once to resuspend them and then incubated for 30 min. The samples were fixed by mixing with 0.5 mL of 8% paraformaldehyde (final concentration 4%) and left to stand for 30 min at room temperature. The flow tubes were filled to approximately 5 mL with phosphate buffered saline (PBS), then spun at 1300 rpm for 10 min and the supernatant removed. The cells were treated with 0.5 mL 0.1% Triton X-100 in PBS, the pellet resuspended and incubated at room temperature for 5 min. The flow tubes were again filled to approximately 5 mL with PBS and spun at 1300 rpm for 10 min, the supernatant was removed and 1 mL of PBS containing 5% BSA was added. The pellet was resuspended and incubated for 1 hour at room temperature. The tubes were spun at 1300 rpm for 10 min, the supernatant removed and 200 μL of primary antibody (BD mouse anti-cytochrome c) at a 1:200 dilution in PBS containing 0.6% bovine serum albumin added. The tubes were incubated at 4 °C overnight then filled to approximately 5 mL with PBS and spun at 1300 rpm for 10 min. The supernatant was removed and 500 μL of secondary antibody added (Life Tech goat anti-mouse R-PE) at a 1:500 dilution in PBS (0.6% BSA). After incubating for 1 hour in the dark, the tubes were filled with PBS and spun at 1300 rpm for 10 min, the supernatant aspirated and 500 μL of PBS added. DRAQ5 (Biostatus) dye was added to 20 μM and the samples were incubated for 10 min before being analyzed using a FACS Calibur flow cytometer (Becton-Dickinson Immunocytometry Systems, San Jose, CA, USA) equipped with a 488 nm laser until 25000 total events had been collected using CellQuest software (Becton-Dickinson Immunocytometry Systems, San Jose, CA, USA). FSC, SSC (488/10 nm), FL-1 (530/30 nm); FL-2 (585/42 nm) and FL-3 (>670 nm) with compensation to acquire the peptide, cytochrome c and DRAQ5 fluorescent signal, respectively.
Mitochondria morphology
Optical sections of doubly labeled cells were acquired using a confocal laser scanning microscope (CLSM) (BioRad Microsciences Ltd, Hemel Hempsted, UK), equipped with a Kr/Ar ion laser and attached to a Zeiss Axiovert 135. DRAQ5 (DNA) was visualized using 647 nm excitation and detected at 680/30 nm.Acknowledgements
We wish to thank Emeline Furon for her assistance during the early phase of this project. The financial support of the Engineering and Physical Sciences Research Council (EPSRC) through the Basic Technology Grant EP/F04095, the Biotechnology and Biological Sciences Research Council (BBSRC) through grant BB/I1021396/1 and Cardiff University is gratefully acknowledged.Notes and references
- J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS.
- E. C. Turner, C. H. Cureton, C. J. Weston, O. S. Smart and R. K. Allemann, Chem. Biol., 2004, 11, 69–77 CrossRef CAS.
- U. Kusebauch, L. Moroder and C. Renner, Biopolymers, 2005, 80, 575 Search PubMed.
- L. Guerrero, O. S. Smart, C. J. Weston, D. C. Burns, G. A. Woolley and R. K. Allemann, Angew. Chem., Int. Ed., 2005, 44, 7778–7782 CrossRef CAS.
- L. Guerrero, O. S. Smart, G. A. Woolley and R. K. Allemann, J. Am. Chem. Soc., 2005, 127, 15624–15629 CrossRef CAS.
- G. A. Woolley, A. S. I. Jaikaran, M. Berezovski, J. P. Calarco, S. N. Krylov, O. S. Smart and J. R. Kumita, Biochemistry, 2006, 45, 6075–6084 CrossRef CAS.
- U. Kusebauch, S. A. Cadamuro, H. J. Musiol, L. Moroder and C. Renner, J. Pept. Sci., 2006, 12, 199 CrossRef.
- C. Renner and L. Moroder, ChemBioChem, 2006, 7, 869–878 CrossRef.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- M. Sattler, H. Liang, D. Nettesheim, R. P. Meadows, J. E. Harlan, M. Eberstadt, H. S. Yoon, S. B. Shuker, B. S. Chang, A. J. Minn, C. B. Thompson and S. W. Fesik, Science, 1997, 275, 983–986 CrossRef CAS.
- A. Shamas-Din, H. Brahmbhatt, B. Leber and D. W. Andrews, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 508–520 CrossRef CAS.
- B. Leber, J. Lin and D. W. Andrews, Oncogene, 2010, 29, 5221–5230 CrossRef CAS.
- C. Bogner, B. Leber and D. W. Andrews, Curr. Opin. Cell Biol., 2010, 22, 845–851 CrossRef CAS.
- M. D. Esposti and C. Dive, Biochem. Biophys. Res. Commun., 2003, 304, 455–461 CrossRef.
- J. Plati, O. Bucur and R. Khosravi-Far, Integr. Biol., 2011, 3, 279–296 RSC.
- A. Strasser, S. Cory and J. M. Adams, EMBO J., 2011, 30, 3667–3683 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS.
- M. L. Stewart, E. Fire, A. E. Keating and L. D. Walensky, Nat. Chem. Biol., 2010, 6, 595–601 CrossRef CAS.
- L. D. Walensky, K. Pitter, J. Morash, K. J. Oh, S. Barbuto, J. Fisher, E. Smith, G. L. Verdine and S. J. Korsmeyer, Mol. Cell, 2006, 24, 199–210 CrossRef CAS.
- E. Gavathiotis, D. E. Reyna, M. L. Davis, G. H. Bird and L. D. Walensky, Mol. Cell, 2010, 40, 481–492 CrossRef CAS.
- E. Gavathiotis, M. Suzuki, M. L. Davis, K. Pitter, G. H. Bird, S. G. Katz, H. C. Tu, H. Kim, E. H. Y. Cheng, N. Tjandra and L. D. Walensky, Nature, 2008, 455, 1076 CrossRef CAS.
- Y. X. Zhang, F. Erdmann and G. Fischer, Nat. Chem. Biol., 2009, 5, 724–726 CrossRef CAS.
- T. Okamoto, K. Zobel, A. Fedorova, C. Quan, H. Yang, W. J. Fairbrother, D. C. S. Huang, B. J. Smith, K. Deshayes and O. E. Czabotar, ACS Chem. Biol., 2013, 8, 297–302 CrossRef CAS.
- S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9, 3046–3054 CrossRef CAS.
- P. Wysoczanski, R. J. Mart, E. J. Loveridge, C. Williams, S. B.-M. Whittaker, M. P. Crump and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 7644–7647 CrossRef CAS.
- A. A. Beharry, L. Wong, V. Tropepe and G. A. Woolley, Angew. Chem., Int. Ed., 2011, 50, 1325–1327 CrossRef CAS.
- J. Deng, N. Carlson, K. Takeyama, P. Dal Cin, M. Shipp and A. Letai, Cancer Cell, 2007, 12, 171–185 CrossRef CAS.
- M. Certo, V. D. Moore, M. Nishino, G. Wei, S. Korsmeyer, S. A. Armstrong and A. Letai, Cancer Cell, 2006, 9, 351–365 CrossRef CAS.
- J. I. J. Leu, P. Dumont, M. Hafey, M. E. Murphy and D. L. George, Nat. Cell Biol., 2004, 6, 443–450 CrossRef CAS.
- E. C. Pietsch, S. M. Sykes, S. B. McMahon and M. E. Murphy, Oncogene, 2008, 27, 6507–6521 CrossRef CAS.
- M. D. Boersma, H. S. Haase, K. J. Peterson-Kaufman, E. F. Lee, O. B. Clarke, P. M. Colman, B. J. Smith, W. S. Horne, W. D. Fairlie and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 315–323 CrossRef CAS.
- J. A. Ryan, J. K. Brunelle and A. Letai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12895–12900 CrossRef CAS.
- F. Z. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49, 3943–3946 CrossRef CAS.
- P. J. Smith, N. Blunt, M. Wiltshire, T. Hoy, P. Teesdale-Spittle, M. R. Craven, J. V. Watson, W. B. Amos, R. J. Errington and L. H. Patterson, Cytometry, 2000, 40, 280–291 CrossRef CAS.
- K. L. Njoh, L. H. Patterson, M. Zloh, M. Wiltshire, J. Fisher, S. Chappell, S. Ameer-Beg, Y. H. Bai, D. Matthews, R. J. Errington and P. J. Smith, Cytometry, Part A, 2006, 69A, 805–814 CrossRef CAS.
- A. V. Kuznetsov, V. Veksler, F. N. Gellerich, V. Saks, R. Margreiter and W. S. Kunz, Nat. Protocols, 2008, 3, 965–976 CAS.
- S. T. Smiley, M. Reers, C. Mottolahartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3671–3675 CrossRef CAS.
- A. Cossarizza, M. Baccaranicontri, G. Kalashnikova and C. Franceschi, Biochem. Biophys. Res. Commun., 1993, 197, 40–45 CrossRef CAS.
- K. Roberg, U. Johansson and K. Ollinger, Free Radical Biol. Med., 1999, 27, 1228–1237 CrossRef CAS.
- T. Oltersdorf, S. W. Elmore, A. R. Shoemaker, R. C. Armstrong, D. J. Augeri, B. A. Belli, M. Bruncko, T. L. Deckwerth, J. Dinges, P. J. Hajduk, M. K. Joseph, S. Kitada, S. J. Korsmeyer, A. R. Kunzer, A. Letai, C. Li, M. J. Mitten, D. G. Nettesheim, S. Ng, P. M. Nimmer, J. M. O'Connor, A. Oleksijew, A. M. Petros, J. C. Reed, W. Shen, S. K. Tahir, C. B. Thompson, K. J. Tomaselli, B. L. Wang, M. D. Wendt, H. C. Zhang, S. W. Fesik and S. H. Rosenberg, Nature, 2005, 435, 677–681 CrossRef CAS.
- E. H. Shroff, C. M. Snyder, G. R. S. Budinger, M. Jain, T. L. Chew, S. Khuon, H. Perlman and N. S. Chandel, PLoS One, 2009, 4, e5646 Search PubMed.
- J. E. Purvis, K. W. Karhohs, C. Mock, E. Batchelor, A. Loewer and G. Lahav, Science, 2012, 336, 1440–1444 CrossRef CAS.
- O. Sadovski, A. A. Beharry, F. Z. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484–1486 CrossRef CAS.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS.
- D. Bleger, J. Shwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef CAS.
- H. A. Rendall, R. F. Marchington, B. B. Praveen, G. Bergmann, Y. Arita, A. Heisterkamp, F. J. Gunn-Moore and K. Dholakia, Lab Chip, 2012, 12, 4816–4820 RSC.
- D. Wlodkowic, K. Khoshmanesh, J. C. Sharpe, Z. Darzynkiewicz and J. M. Cooper, Anal. Chem., 2011, 83, 6439–6446 CrossRef CAS.
- S. Y. A. Cheung, N. D. Evans, M. J. Chappell, K. R. Godfrey, P. J. Smith and R. J. Errington, Math. Biosci., 2008, 213, 119–134 CrossRef CAS.
- P. J. Smith, I. A. Khan and R. J. Errington, Drug Discovery Today, 2009, 14, 271–277 CrossRef CAS.
- A. L. Epstein, R. Levy, H. Kim, W. Henle, G. Henle and H. S. Kaplan, Cancer, 1978, 42, 2379–2391 CrossRef CAS.
- E. Gnaiger, A. V. Kuznetsov, S. Schneeberger, R. Seiler, G. Brandacher, W. Steurer and R. Margreiter, Life Cold, 2000, 431–442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of peptide synthesis, purification and identification, proteolytic and reductive stability, UV-Visible and circular dichroism spectra. Cell culture, and assay conditions. See DOI: 10.1039/c3mb70246d |
| This journal is © The Royal Society of Chemistry 2013 |