 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrials and tribulations of ‘omics data analysis: assessing quality of SIMCA-based multivariate models using examples from pulmonary medicine
Åsa M. Wheelock
*ab and
Craig E. Wheelock
*bc
aRespiratory Medicine Unit, Department of Medicine, and Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden. E-mail: asa.wheelock@ki.se
bHikari Bio AB, Stockholm, Sweden
cDepartment of Medical Biochemistry and Biophysics, Division of Physiological Chemistry II, Karolinska Institutet, Stockholm, Sweden. E-mail: craig.wheelock@ki.se
First published on 20th August 2013
Abstract
Respiratory diseases are multifactorial heterogeneous diseases that have proved recalcitrant to understanding using focused molecular techniques. This trend has led to the rise of ‘omics approaches (e.g., transcriptomics, proteomics) and subsequent acquisition of large-scale datasets consisting of multiple variables. In ‘omics technology-based investigations, discrepancies between the number of variables analyzed (e.g., mRNA, proteins, metabolites) and the number of study subjects constitutes a major statistical challenge. The application of traditional univariate statistical methods (e.g., t-test) to these “short-and-wide” datasets may result in high numbers of false positives, while the predominant approach of p-value correction to account for these high false positive rates (e.g., FDR, Bonferroni) are associated with significant losses in statistical power. In other words, the benefit in decreased false positives must be counterbalanced with a concomitant loss in true positives. As an alternative, multivariate statistical analysis (MVA) is increasingly being employed to cope with ‘omics-based data structures. When properly applied, MVA approaches can be powerful tools for integration and interpretation of complex ‘omics-based datasets towards the goal of identifying biomarkers and/or subphenotypes. However, MVA methods are also prone to over-interpretation and misuse. A common software used in biomedical research to perform MVA-based analyses is the SIMCA package, which includes multiple MVA methods. In this opinion piece, we propose guidelines for minimum reporting standards for a SIMCA-based workflow, in terms of data preprocessing (e.g., normalization, scaling) and model statistics (number of components, R2, Q2, and CV-ANOVA p-value). Examples of these applications in recent COPD and asthma studies are provided. It is expected that readers will gain an increased understanding of the power and utility of MVA methods for applications in biomedical research.
 Åsa M. Wheelock | Associate Professor Åsa M. Wheelock heads the Pulmonomics research group at the Respiratory Medicine Unit, Karolinska Institute. Her research interests can be broadly defined as probing the proteomes of various sub-compartments of the lung to unravel the pathological mechanisms of a range of inflammatory lung diseases and pulmonary toxicants, focusing equal efforts on translational proteomics applications and methodology development in the fields of quantitative intact proteomics and multivariate modeling. Åsa Wheelock is also the CEO and founder of Hikari Bio AB, a spin-off company that has developed the time-resolved fluorescence-based CuTEDGE technology for in-gel protein detection and quantification. |
 Craig E. Wheelock | Associate Professor Craig E. Wheelock heads a research group at the Karolinska Institute that employs a combination of non-targeted metabolomics using high-resolution mass spectrometry (HRMS) and targeted lipidomics to investigate disease mechanisms. We perform molecular-based phenotyping to obtain increased knowledge about the mechanisms underpinning disease. Our specific interests center on producing quantitative molecular fingerprints of clinical sub-phenotypes of asthma and COPD with the goal of introducing improved methods for diagnosis, prognosis and the prediction of therapeutic responses. Towards that end, we apply an integrated systems-based approach to disease understanding and the development of predictive models in respiratory disease. |
Introduction
Despite extensive study, little is known about the pathogenesis of many chronic lung diseases and effective therapeutic interventions for chronic obstructive pulmonary disease (COPD), asthma, and idiopathic pulmonary fibrosis (IPF) are still lacking.1 Worldwide 235 million people suffer from asthma, which is the most common chronic disease among children, and COPD is expected to become the 3rd leading cause of global mortality by 2020.2–4 These statistics suggest that complex multifactorial respiratory diseases have proven recalcitrant to traditional reductionist approaches that focus on a small number of endpoints (e.g., genes or proteins) for elucidating disease mechanisms.1 A number of researchers have highlighted the potential of systems medicine-based approaches in investigating respiratory diseases including asthma,5–7 cystic fibrosis,8 COPD,9 and pulmonary hypertension.10 The application of ‘omics-based science (e.g., genomics, transcriptomics, proteomics and metabolomics) has provided a wealth of information and data related to disease, which has recently been reviewed in detail.11 However, the “system” component requires that methods move beyond data acquisition to data integration and interrogation, which can be challenging. One useful approach involves the application of multivariate statistical methods, which have the advantage of being able to integrate numerical and categorical information from multiple ‘omics datasets with e.g., clinical diagnostic and/or phenotype data, while simultaneously not requiring significant computational or bioinformatics expertise. This Opinion piece is not intended to serve as a comprehensive review of the field or a tutorial; readers interested in more in-depth discussions of these topics are directed to a number of reviews and recent applications cited throughout this paper. The purpose of this Opinion is to present an argument for the need of multivariate statistical methods in ‘omics-based data analysis and to propose distinct metrics for evaluating the quality of multivariate models.Statistical challenges in the analysis of ‘omics datasets
The discrepancies between the number of variables analyzed (e.g., mRNA, proteins, metabolites) and the number of study subjects constitutes a major statistical challenge in large-scale ‘omics investigations. These “short-and-wide” dataset structures are not conducive to traditional univariate statistical methods (e.g., t-test), because the repeated hypothesis testing results in a high numbers of potential false positives. The most common way to correct for these high false positive rates is to apply some form of p-value correction, such as the False Discovery Rate (FDR) described by Benjamini–Hochberg,12 the q-value described by Storey,13 or Bonferroni correction.14,15 While the benefits of a reduction of false positives have made these methods the standard in the field for e.g., transcriptomics,16 the downsides in terms of loss of statistical power to detect true positives are rarely discussed.17–20 In a previous example,21 we showed that FDR p-value correction reduced the statistical power from 95% to 9.3% in an mRNA microarray dataset; the penalty for removing ∼800 potential false positives was a loss of 86% of the putative true positives.Part of the problem is that we apply t-statistics in a fashion for which it was not designed. The application of univariate statistics becomes a balancing act between the acceptable levels of true- and false positives.22 Maximizing the statistical power, rather than minimizing the false positive rate, may be desirable for downstream analyses (e.g., pathway mapping, enrichment analysis23). Another downside to univariate approaches is that each variable is evaluated in isolation. Given the high degree of inter-dependency between the molecules in a biological system, further accentuated when analyzed in a single large-scale experiment, statistical methods that incorporate the co-variance inherent in ‘omics analyses are more appropriate. Accordingly, multivariate statistical analysis (MVA) represents a viable complement to univariate approaches and is being increasingly employed in the study of respiratory diseases including asthma,24–30 COPD,31–37 pulmonary hypertension38 and sarcoidosis39 (Interested readers are directed to these papers as examples of the application of MVA to the interrogation of ‘omics-based datasets.). It is therefore important that the extended scientific community gains understanding of these methods, as well as the metrics necessary for evaluation of MVA experiments, in order to ensure publication of rigorous and accurate results.
Introduction to multivariate statistics
Multivariate statistics can be described as a group of tools for reducing the dimensionality of large datasets to render the visualization and interpretation more manageable (Fig. 1A).40,41 The relationship between the study subjects (observations) and collected data (variables) is described. The SIMCA-based workflow described in this opinion piece involves three main applications of MVA: (1) data overview or quality control (QC), (2) identification of subsets of biomarkers for discrimination between groups (e.g., healthy and COPD), and (3) correlation modeling between two data blocks (X and Y; e.g., lung function parameters and ‘omics data). These MVA methods provide projections of the dominating trends in the multidimensional dataset onto a few representative virtual variables, termed latent variables or principal components (PC; Fig. 1A; see Table 1 for a glossary of terms). The number of PCs can be quite large, but generally the first couple of PCs are sufficient to describe the trends of interest in the data. The new coordinates for the study subjects (observations) are referred to as scores, and the influence (weight) of the original data variables are referred to as loadings. Traditionally the observations and variables are displayed in separate plots, with the scores plot visualizing the group separation of the subjects (Fig. 1B), and the loadings plot visualizing the relationship of the original variables (e.g., genes, proteins) to the scores (i.e., subjects; Fig. 1C). The results of a single MVA can be referred to as a model in the sense that they provide a statistical description or model of the relationships present in the original data. A significant difference in multivariate relative to univariate methods is that all variables are analyzed in a single “test” (i.e., model), thereby reducing the problems associated with multiple hypothesis testing. In addition, both numerical and categorical data can be incorporated into a single model. This gives the added benefit that the covariance or interrelatedness (e.g., synergy of a subset of clinical biomarkers in classifying different asthma phenotypes) in the dataset is taken into consideration in the model construction.![Fundamentals of a multivariate analysis workflow, exemplified by OPLS modeling of the difference in exosomal miRNA profiles between mild intermittent asthmatics and healthy individuals at baseline (adapted from ref. 24). (A) Multivariate models reduce the dimensionality of the data and focus the information of interest into a couple of latent variables, similar to how the shadows of the multi-dimensional cloud of variable spots can be projected onto a plane. This process is exemplified by the red dot, which has been projected onto the plane as a blue dot (dashed arrow). The cumulative projections result in the formation of the model. (B) OPLS scores plot visualizing the separation of the subjects. The model was constructed with 1 predictive + 1 orthogonal component, resulting in a clear separation between the groups along the predictive component (x-axis; R2 = 0.76, Q2 = 0.62, p[CV-ANOVA] = 0.003). Within-group variation is displayed in the orthogonal direction (y-axis). HC: healthy controls, AC: subjects with asthma (C) loadings plot showing the influence of the miRNA variables on the group separation. MiRNAs located distally along the x-axis are important for between group separation (shown in red), while miRNAs located distally along the y-axis contribute to within group variance. (D) In order to identify the subset of miRNAs with the highest potential as biomarkers, variable selection using a combination of Variable Influence in Projection (VIP) and p(corr) was performed (see text and Table 1 for explanation of terms). Based on the resulting Q2 (predictive power) and CV-ANOVA values, iterations of variable selection using VIP > 1.0 and |p(corr)| > 0.5 as inclusion criteria were applied until the optimal model of 16 miRNA (red triangles) was acquired. Downstream pathway analysis revealed that the majority of these miRNAs are associated with regulation of IL-13.24](/image/article/2013/MB/c3mb70194h/c3mb70194h-f1.gif) | ||
| Fig. 1 Fundamentals of a multivariate analysis workflow, exemplified by OPLS modeling of the difference in exosomal miRNA profiles between mild intermittent asthmatics and healthy individuals at baseline (adapted from ref. 24). (A) Multivariate models reduce the dimensionality of the data and focus the information of interest into a couple of latent variables, similar to how the shadows of the multi-dimensional cloud of variable spots can be projected onto a plane. This process is exemplified by the red dot, which has been projected onto the plane as a blue dot (dashed arrow). The cumulative projections result in the formation of the model. (B) OPLS scores plot visualizing the separation of the subjects. The model was constructed with 1 predictive + 1 orthogonal component, resulting in a clear separation between the groups along the predictive component (x-axis; R2 = 0.76, Q2 = 0.62, p[CV-ANOVA] = 0.003). Within-group variation is displayed in the orthogonal direction (y-axis). HC: healthy controls, AC: subjects with asthma (C) loadings plot showing the influence of the miRNA variables on the group separation. MiRNAs located distally along the x-axis are important for between group separation (shown in red), while miRNAs located distally along the y-axis contribute to within group variance. (D) In order to identify the subset of miRNAs with the highest potential as biomarkers, variable selection using a combination of Variable Influence in Projection (VIP) and p(corr) was performed (see text and Table 1 for explanation of terms). Based on the resulting Q2 (predictive power) and CV-ANOVA values, iterations of variable selection using VIP > 1.0 and |p(corr)| > 0.5 as inclusion criteria were applied until the optimal model of 16 miRNA (red triangles) was acquired. Downstream pathway analysis revealed that the majority of these miRNAs are associated with regulation of IL-13.24 | ||
| PCA | Principal component analysis; unsupervised MVA method suitable for data overview and identification of outliers. |
| OPLS | Orthogonal projections to latent structures; supervised MVA method suitable for biomarker (variable) selection or classification of 2 groups. |
| Model | The plane (or hyperplane) to which the data are projected, consisting of as many dimensions as principal components extracted (Fig. 1A). |
| Principal component | The coordinates of the original observations following reduction in dimensionality to a few latent variables (Fig. 1A). |
| Data block | Group of variables or data (e.g., proteomics, metabolomics) included in a MVA model construction. |
| Scores | New values representing the observations (subjects) in the model plane. Each subject is represented as a single point in the scores plot (Fig. 1B). |
| Loadings | New values representing the variables (e.g., proteins) in the model plane. Each original variable is represented as a point in the loadings plot (Fig. 1C). |
| Hotellings T2 | Multivariate generalization of the 95% confidence interval, can be utilized to identify outliers (Fig. 1B). |
| p(corr) | Loadings scaled as a correlation coefficient (ranging from −1.0 to 1.0) between the model and original data. |
| DModX | Distance to model X; the distance of a given observation to the model plane. Useful for detection of moderate outliers. |
| R2 | The fraction of the original data explained by the model (R2 = 1.0 explains 100% of the data). Measure of the overall fit of the model. |
| Q2 | The fraction of the original data explained by the cross-validated model. Measure of the ability of the model to predict a new dataset. |
| CV-ANOVA | Cross-validated analysis of variance; provides a p-value indicating the level of significance of group separation in OPLS analyses. Based on a cross-validated model. |
| VIP | Variable importance in the projection; ranking of the original variables according to their individual contribution to the model. |
| SUS | Shared and unique structures; plot comparing the scaled loadings (p(corr)) from two OPLS models, visualizing the shared patterns of variable contribution along the diagonals, and the unique features of variable contribution along the respective axes (Fig. 2). |
There are multiple statistics packages capable of performing MVA including both freeware: the R Project for Statistical Computing (http://www.r-project.org), MetaboAnalyst,42 Multibase (Numerical Dynamics), IFRNOPLS;43,44 as well as commercial sources: STATISTICA (StatSoft), Unscrambler (CAMO Software) and SIMCA (MKS Umetrics). Our discussion of MVA is based upon the software SIMCA v.13, which is a commercial software with a user-friendly interface commonly used in biomedical research.
Types of MVA methods
There are multiple forms of multivariate statistics, which are beyond the scope of this opinion piece (e.g., factor analysis, linear discriminant analysis, canonical correlation analysis, artificial neural networks). Instead, these discussions are focused on the two most common applications for ‘omics data in biomedical research: (1) principal components analysis (PCA) and (2) orthogonal projections to latent structures (OPLS). MVA methods can be broadly grouped into supervised and unsupervised approaches. Interested readers are directed to a number of in-depth reviews and books on MVA.40,41,45–50 Unsupervised approach means that no information on group identity (e.g., diagnosis) is used to construct the model. The data are analyzed as belonging to a single block of observations and variables, referred to as the X-data block. The dominating trends of group separation inherent in the data are highlighted in the resulting model. In the ‘omics field, PCA is the most commonly used unsupervised MVA method. Unsupervised methods can be utilized for identifying strong subgroupings in the data. However, it is challenging to connect the observed group separation back to the original variables in a PCA model, which is essential for model interpretation (e.g., biomarker identification). The primary strength and utility of PCA is therefore in assessing the quality and homogeneity of the dataset (e.g., outlier identification). Accordingly, PCA is often the first step in an MVA workflow.51For biomarker discovery or hypothesis testing, supervised methods are of greater utility. Supervised means that group identity is defined, focusing the analysis on extracting the variables important for group separation (i.e., the hypothesis), or for finding correlations between two data blocks. The data are divided into two separate blocks: the X-block containing the predictor variables (e.g., putative diagnostic protein biomarkers, inflammatory lipids [eicosanoids]), and the Y-block containing the response variables (e.g., clinical parameters used for diagnosis: FEV1, methacholine challenge, BMI, etc.). Partial least squares (PLS), a commonly used supervised MVA method, is primarily useful for performing multivariate correlation analysis between two defined data blocks. As with PCA, the variables in each block have to display a similar trajectory in order to identify subgroupings; strong confounding or opposing variables will weaken or even obscure the underlying group separation or correlation. For example, including smoking and non-smoking subjects in an MVA model investigating COPD will only highlight effects due to smoking, and not the underlying disease.
Orthogonal projections to latent structures (OPLS) is an extension of PLS where the variance of interest (e.g., diagnosis) is separated from the variance that is unrelated (orthogonal) to the defined Y-block variables (i.e., hypothesis).52,53 This results in a rotation of the standard PLS model so that the variance important for the defined group separation is focused into the predictive components (x-axis; Fig. 1B and C), and variance unrelated to the tested hypothesis is filtered into orthogonal components (y-axis; Fig. 1B and C). The strength of OPLS in biomarker discovery is that the information of interest is focused into a single component, making it easier to link to the experimental variables (e.g., proteins, metabolites), as well as to evaluate the predictive power of a sub-set of biomarkers.16 The power of supervised methods, particularly OPLS, can however be a double-edged sword. If a sufficient number of orthogonal components are extracted, an OPLS analysis invariably results in group separation that is convincing by visual inspection of the scores plot. Such overfitting of MVA models can be compared with introducing polynomial fitting to a standard curve with an inherent linear relationship; if sufficient terms are introduced, a correlation of R2 = 1.0 can always be achieved (see example in ref. 21). However, the utility of such a model for prediction is limited because the predictive power is lost.
Parameters necessary for evaluating model quality
Due to the risk of visual over-interpretation of MVA results, the methods employed in an MVA experiment need to be explicitly described. In particular, there are a number of parameters that are vital to interpreting the model quality. Reporting of the parameters discussed below should be considered essential for any presentation of results from an MVA-based study. First and foremost, the number of components employed in model construction is related to the degree of overfitting, and should be provided. The R2 value, which indicates how well the model explains the dataset, and the cross-validated correlation (termed Q2 in SIMCA) should be reported. Cross-validation involves partitioning the subjects into subsets, and fitting the model after randomly excluding one subset at a time from the analysis. Q2 is the correlation based on averaging the results from repeated iterations of cross-validation, and represents a measure of the predictive power of the model (i.e., how well the model is expected to fit additional cohorts). In an ideal model the R2 and Q2 should be similar, meaning that each of the subjects contribute equally and uniformly to the observed group separation. In reality Q2 is always lower than R2; however, if Q2 is substantially lower than R2 then the robustness of the model is poor, implying overfitting. For supervised methods, the cross-validated ANOVA (CV-ANOVA) p-value can be calculated as a measure of significance for the observed group separation, with the distinct advantage of providing a familiar p-value metric of the model.54There are a couple of additional tools available in the SIMCA software that are useful for MVA-based quality control. The Hotelling's T2, corresponding to a multivariate generalization of the 95% confidence interval, can be utilized to identify outliers. This region is visualized by the large circle shown in SIMCA-generated scores plots for both PCA and OPLS models (Fig. 1B). If the data are normally distributed, then 95% should fall inside the Hotelling's T2 circle. In a second QC step, a PCA can be performed for each data block and study group separately, and investigated using the distance to model X (DModX) function to identify moderate outliers. The DModX function is the relative standard deviation (RSD) for each row and provides a measurement of the distance to the model (the plane onto which the original variables are projected; Fig. 1A). For a model that is representative for all included subjects, the DModX values should be fairly equal for all samples.
Many multivariate methods are strongly scale-dependent and as such any scaling should be described. If no scaling is applied, the most abundant variables may drive the separation, similar to the effect that a couple of high abundance data points have in a standard curve. Scaling to unit variance (UV) and mean centering (default in SIMCA) are commonly applied to ensure that large relative alterations in low abundance biomolecules exert the same influence as high abundance biomolecules. If no signal-to-noise (S/N) filtering is performed prior to the MVA analysis, UV scaling can result in variables with abundances close to the limit of detection (LOD) exerting an artificially high influence on the model (i.e., the noise is inflated). The “pareto” scaling option available in SIMCA is a viable alternative for ‘omics datasets with large dynamic ranges. Pareto scaling only partly removes the differences in abundances, and the risk of close-to-LOD variables driving the separation is less pronounced. A normal distribution is not a requirement for MVA methods. However, these methods extract the maximum variance in the data and the lack of appropriate data transformation (e.g., log transformation) may result in the skewed variance dominating the model. Accordingly, evaluation of skewedness and normality of the data along with any transformation should be reported.
Interpretation of MVA results
Group classification and biomarker selection using OPLS
One of the major strengths of supervised methods, particularly OPLS, is its application in variable selection. Variable selection is an essential step in identifying and evaluating the performance of subsets of variables for classification of patient subgroups (e.g., biomarker discovery). In MVA the question of which variables are of interest, corresponding to determining significance in univariate statistics, is not trivial. General rules for where to apply the cutoff in the continuous variable ranking, such as p < 0.05 in univariate statistics, have not yet been established. The use of a Variable Influence on Projection (VIP; also referred to as Variable Importance in Projection) score > 1.0 is common in publications. VIP is a metric that summarizes the importance of each variable in driving the observed group separation.41 However, VIP > 1.0 only implies that the variable contributes more than average to the model, and the VIP > 1.0 cutoff results in selection of up to 50% of the variables. In addition, the VIP score is a relative ranking term that changes with each iteration of variable selection, rendering it somewhat of a moving target. It is therefore often difficult to determine the optimal model based solely upon VIP values. An alternative and complementary parameter is the p(corr) value. p(corr) is the loadings (Fig. 1C) scaled as a correlation coefficient, thereby standardizing the range from −1.0 to 1.0. The p(corr) values remain stable during iterative variable selection and are comparable between models. There is no consensus on what p(corr) cutoff represents significance, but an absolute p(corr) > 0.4–0.5 is often used.24–26,31,55 For variable selection, we recommend the use of a combination of p(corr) and VIP (Fig. 1D). A constant p(corr) can be used as a cutoff point for variable selection if the aim is to maximize the statistical power. Alternatively, if the goal is to select a subset of biomarkers, several iterations of variable selections can be performed as long as the Q2 and CV-ANOVA p-value continue to increase.Overfitting is an inherent risk in OPLS analysis, and determining the appropriate number of components is essential, but not always trivial. The default automatic fitting in SIMCA extracts the maximal number of significant components, which in most cases results in an overfitted model. The result is an inflated R2, but a lowered Q2 because the overfitting occurs at the expense of the predictive power. The optimal number of components is at the break point where Q2 decreases with the addition of more components. The CV-ANOVA p-value can be used as a complement to the Q2 for determining the optimal number of components (an increasing p-value due to addition of a component implies overfitting).
Comparison of multiple groups using OPLS
The goal of biomarker discovery studies is often to determine the selectivity for the condition of interest (e.g., COPD) relative to related and/or confounding conditions (e.g., smoking). However, it can be difficult to interpret OPLS analyses that directly compare multiple groups because groups with many similar, but few distinguishing, characteristics may not separate. For example in COPD studies, habitual smoking generally causes the majority of alterations in protein- and mRNA expression patterns, and thus will confound any specific alterations due to COPD if analyzed together.31 The optimal study design in OPLS is achieved by limiting each model to comparison of two groups, which subsequently are analyzed using shared-and-unique-structures (SUS) plots using the p(corr) values.56 Fig. 2 shows a SUS-plot comparing the protein variables from a COPD study examining the respective contribution of ‘healthy smokers vs. healthy never-smokers’ relative to ‘smokers with COPD vs. healthy never-smokers’.31 Given that both models have the same baseline point (healthy never-smokers), the effect of smoking alone vs. smoking and COPD combined are extracted in the plot. The proteins that are of equal importance for the two models cluster along the diagonal (blue boxes) and are of little use as biomarkers. Proteins altered only due to COPD, but not due to smoking, are located along the y-axis (orange boxes; unique structures that only contribute to the COPD model). The latter, featuring high p(corr) values in the COPD model and low p(corr) values for the smoker model, represent putative biomarkers and/or proteins important in the disease development independent of smoking. The 19 proteins that were previously identified to be altered in a female-dominated sub-phenotype of COPD are all located in this region.31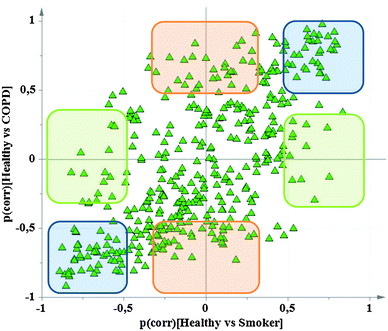 | ||
| Fig. 2 Shared and Unique Structures (SUS) plot analysis exemplified using proteomics data from a study examining gender differences in COPD phenotypes (adapted from ref. 31). The SUS plot compares the protein variables contributing to the model separating ‘healthy’ vs. ‘smokers’ (x-axis; 1 + 1 components; R2 = 0.98, Q2 = 0.84, p(CV-ANOVA) = 1.2 × 10−4) with that of the model separating ‘healthy’ vs. ‘COPD’ (y-axis; 1 + 1 components; R2 = 0.98, Q2 = 0.81, p(CV-ANOVA) = 0.012). In both models, smoking is likely to be the most prominent cause of alterations in protein abundances. The protein variables that are altered in a similar fashion regardless of COPD diagnosis are clustered along the diagonal. Accordingly, the variables in the upper right and lower left corners are not useful as selective biomarkers of COPD (blue boxes). Conversely, proteins located along the axes are specifically altered in ‘smokers’ (green boxes) and ‘COPD patients’ (orange boxes) respectively. Accordingly, the latter are good candidates for COPD biomarkers. The 19 proteins that were previously identified to be altered in a female-dominated sub-phenotype of COPD are all located in the region highlighted in orange.31 | ||
Summary
As the field of systems biology and its application to respiratory medicine continues to expand, it will be useful to gain an increased understanding of the tools employed in a systems medicine experiment and how to evaluate and interpret the results. Many of these approaches, including MVA, provide powerful and novel ways to extract information on the relationship between variables that would not be observed with univariate statistics; however these methods can be obfuscating for non-specialists. An MVA analysis should be treated identically to any other experimental step in a research pipeline, with hypothesis generation and reporting of data handling in a distinct methods section. In particular, all MVA experiments should report data preprocessing (including normalization, transformation and scaling), and model statistics (number of components, R2 and Q2, and the CV-ANOVA p-value for supervised analyses). We propose that these parameters should be considered as the minimal reporting standards for PCA- and OPLS-based workflows, and be required for publication purposes. Visual interpretation of MVA models, especially supervised models, is not appropriate and potentially misleading unless accompanied by the necessary model statistics. Adherence to these minimal reporting standards is an important step towards greater use and acceptance of these approaches. The field would also greatly benefit from an in-depth tutorial or protocol-like paper on application of MVA in ‘omics data processing towards the goal of biomarker discovery. In association with increased acquisition of ‘omics datasets, it is expected that applications of MVA methods will become widely spread and considered part of a normal biomedical research workflow.Acknowledgements
ÅMW was funded by Swedish Heart-Lung Foundation and the Karolinska Institute. CEW was funded by a fellowship from the Centre for Allergy Research (Cfa) and the Karolinska Institute.References
- W. Wu and N. Kaminski, Chronic lung diseases, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2009, 1(3), 298–308 CrossRef PubMed.
- European Respiratory Society, European Lung White Book, European Respiratory Society Journals Ltd., Huddersfield, 2003 Search PubMed.
- W. H. Organization, Chronic Respiratory Disease, Available from: www.who.int/respiratory/en/.
- A. D. Lopez and C. C. Murray, The global burden of disease, 1990–2020, Nat. Med., 1998, 4, 1241–1243 CrossRef CAS PubMed.
- S. J. Szefler and A. Dakhama, New insights into asthma pathogenesis and treatment, Curr. Opin. Immunol., 2011, 23(6), 801–807 CrossRef CAS PubMed.
- A. Dahlin and K. G. Tantisira, Integrative systems biology approaches in asthma pharmacogenomics, Pharmacogenomics, 2012, 13(12), 1387–1404 CrossRef CAS PubMed.
- C. Auffray, I. M. Adcock, K. F. Chung, R. Djukanovic, C. Pison and P. J. Sterk, An integrative systems biology approach to understanding pulmonary diseases, Chest, 2010, 137(6), 1410–1416 CrossRef PubMed.
- S. M. Studer and N. Kaminski, Towards systems biology of human pulmonary fibrosis, Proc. Am. Thorac. Soc., 2007, 4(1), 85–91 CrossRef PubMed.
- A. Agusti, P. Sobradillo and B. Celli, Addressing the complexity of chronic obstructive pulmonary disease: from phenotypes and biomarkers to scale-free networks, systems biology, and P4 medicine, Am. J. Respir. Crit. Care Med., 2011, 183(9), 1129–1237 CrossRef PubMed.
- F. Ahmad, H. C. Champion and N. Kaminski, Toward systems biology of pulmonary hypertension, Circulation, 2012, 125(12), 1477–1479 CrossRef PubMed.
- C. E. Wheelock, V. M. Goss, D. Balgoma, B. Nicholas, J. Brandsma, P. J. Skipp, S. Snowden, A. D'Amico, I. Horvath, A. Chaiboonchoe, H. Ahmed, S. Ballereau, C. Rossios, K. F. Chung, P. Montuschi, S. J. Fowler, I. M. Adcock, A. D. Postle, S. E. Dahlén, A. Rowe, P. J. Sterk, C. Auffray and R. Djukanovic, Application of 'omics technologies to biomarker discovery in inflammatory lung diseases, the U-BIOPRED Study Group, Eur. Respir. J., 2013 Search PubMed , [Epub ahead of print].
- Y. Benjamini and Y. Hochberg, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing, Journal of the Royal Statistical Society Series B (Methodological), 1995, 57, 289–300 Search PubMed.
- J. Storey, A direct approach to false discovery rates, Journal of the Royal Statistical Society, Series B (Methodological), 2002, 64(3), 479–498 CrossRef PubMed.
- C. Bonferroni, Teoria statistica delle classi e calcolo delle probabilit `a, Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze, 1936, 8, 3–62 Search PubMed.
- R. Miller, Simultaneous statistical inference, Springer Verlag, 1981 Search PubMed.
- L. Shi, G. Campbell, W. D. Jones, F. Campagne, Z. Wen and S. J. Walker, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models, Nat. Biotechnol., 2010, 28(8), 827–838 CrossRef CAS PubMed.
- S. Senn and F. Bretz, Power and sample size when multiple endpoints are considered, Pharm. Stat., 2007, 6(3), 161–170 CrossRef PubMed.
- S. Nakagawa, A farewell to Bonferroni: the problems of low statistical power and publication bias, Behav. Chem. Ecol., 2004, 15, 1044–1045 CrossRef.
- P. Z. Schochet, Technical Methods Report: Guidelines for Multiple Testing in Impact Evaluations, Mathematica Policy Research, Inc., 2008, Contract No.: NCEE 20084018 Search PubMed.
- R. J. Feise, Do multiple outcome measures require p-value adjustment?, BMC Med. Res. Methodol., 2002, 2, 8 CrossRef PubMed.
- D. Diez, A. M. Wheelock, S. Goto, J. Z. Haeggstrom, G. Paulsson-Berne and G. K. Hansson, et al. The use of network analyses for elucidating mechanisms in cardiovascular disease, Mol. BioSyst., 2010, 6(2), 289–304 RSC.
- J. P. Ioannidis, R. Tarone and J. K. McLaughlin, The false-positive to false-negative ratio in epidemiologic studies, Epidemiology, 2011, 22(4), 450–456 CrossRef PubMed.
- P. Khatri, M. Sirota and A. J. Butte, Ten years of pathway analysis: current approaches and outstanding challenges, PLoS Comput. Biol., 2012, 8(2), e1002375 CAS.
- B. Levänen, N. R. Bhakta, P. Torregrosa Paredes, R. Barbeau, S. Hiltbrunner, J. L. Pollack, C. M. Sköld, M. Svartengren, J. Grunewald, S. Gabrielsson, A. Eklund, B. M. Larsson, P. G. Woodruff, D. J. Erle and Å. M. Wheelock, Altered microRNA profiles in bronchoalveolar lavage fluid exosomes in asthmatic patients, J. Allergy Clin. Immunol., 2013, 131(3), 894–903, DOI:10.1016/j.jaci.2012.11.039.
- S. L. Lundstrom, J. Yang, H. J. Kallberg, S. Thunberg, G. Gafvelin and J. Z. Haeggstrom, et al. Allergic asthmatics show divergent lipid mediator profiles from healthy controls both at baseline and following birch pollen provocation, PLoS One, 2012, 7(3), e33780 Search PubMed.
- S. L. Lundstrom, B. Levanen, M. Nording, A. Klepczynska-Nystrom, M. Skold and J. Z. Haeggstrom, et al. Asthmatics exhibit altered oxylipin profiles compared to healthy individuals after subway air exposure, PLoS One, 2011, 6(8), e23864 Search PubMed.
- P. D. Blanc, I. H. Yen, H. Chen, P. P. Katz, G. Earnest and J. R. Balmes, et al. Area-level socio-economic status and health status among adults with asthma and rhinitis, Eur. Respir. J., 2006, 27(1), 85–94 CrossRef CAS PubMed.
- E. J. Saude, I. P. Obiefuna, R. L. Somorjai, F. Ajamian, C. Skappak and T. Ahmad, et al. Metabolomic biomarkers in a model of asthma exacerbation: urine nuclear magnetic resonance, Am. J. Respir. Crit. Care Med., 2009, 179(1), 25–34 CrossRef CAS PubMed.
- E. J. Saude, C. D. Skappak, S. Regush, K. Cook, A. Ben-Zvi and A. Becker, et al. Metabolomic profiling of asthma: diagnostic utility of urine nuclear magnetic resonance spectroscopy, J. Allergy Clin. Immunol., 2011, 127(3), 757–764 CrossRef CAS PubMed.
- N. Larsson, S. Lundstrom, R. Pinto, G. Rankin, M. Karimpour and A. Blomberg, et al. Lipid mediator profiles differ between lung compartments in asthmatic and healthy humans, Eur. Respir. J., 2013 DOI:10.1183/09031936.00209412.
- M. Kohler, A. Sandberg, S. Kjellqvist, A. Thomas, R. Karimi, S. Nyrén, A. Eklund, M. Thevis, C. M. Sköld and Å. M. Wheelock, Gender differences in the bronchoalveolar lavage cell proteome of patients with chronic obstructive pulmonary disease, J. Allergy Clin. Immunol., 2013, 131(3), 743–751, DOI:10.1016/j.jaci.2012.09.024.
- J. Yorke, S. H. Moosavi, C. Shuldham and P. W. Jones, Quantification of dyspnoea using descriptors: development and initial testing of the Dyspnoea-12, Thorax, 2010, 65(1), 21–26 CrossRef CAS PubMed.
- J. Smith, P. Albert, E. Bertella, J. Lester, S. Jack and P. Calverley, Qualitative aspects of breathlessness in health and disease, Thorax, 2009, 64(8), 713–718 CrossRef CAS PubMed.
- K. Roy, J. Smith, U. Kolsum, Z. Borrill, J. Vestbo and D. Singh, COPD phenotype description using principal components analysis, Respir. Res., 2009, 10, 41 CrossRef PubMed.
- P. R. Burgel, J. L. Paillasseur, D. Caillaud, I. Tillie-Leblond, P. Chanez and R. Escamilla, et al. Clinical COPD phenotypes: a novel approach using principal component and cluster analyses, Eur. Respir. J., 2010, 36(3), 531–539 CrossRef PubMed.
- B. K. Ubhi, K. K. Cheng, J. Dong, T. Janowitz, D. Jodrell and R. Tal-Singer, et al. Targeted metabolomics identifies perturbations in amino acid metabolism that sub-classify patients with COPD, Mol. BioSyst., 2012, 8(12), 3125–3133 RSC.
- B. K. Ubhi, J. H. Riley, P. A. Shaw, D. A. Lomas, R. Tal-Singer and W. MacNee, et al. Metabolic profiling detects biomarkers of protein degradation in COPD patients, Eur. Respir. J., 2012, 40(2), 345–355 CrossRef CAS PubMed.
- S. Duri, R. C. Molthen and C. D. Tran, Discriminating pulmonary hypertension caused by monocrotaline toxicity from chronic hypoxia by near-infrared spectroscopy and multivariate methods of analysis, Anal. Biochem., 2009, 390(2), 155–164 CrossRef CAS PubMed.
- E. Silva, S. Souchelnytskyi, K. Kasuga, A. Eklund, J. Grunewald and Å. M. Wheelock, Quantitative intact proteomics investigations of alveolar macrophages in sarcoidosis, Eur. Respir. J., 2013, 41(6), 1331–1339, DOI:10.1183/09031936.00178111.
- R. Madsen, T. Lundstedt and J. Trygg, Chemometrics in metabolomics–a review in human disease diagnosis, Anal. Chim. Acta, 2010, 659(1–2), 23–33 CrossRef CAS PubMed.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold, J. Trygg, C. Wikström and S. Wold, Multi- and megavariate data analysis, Umetrics AB, 2006 Search PubMed , http://books.google.se/books?id=B-1NNMLLoo8C&printsec=copyright#v=onepage&q&f=false.
- J. Xia, R. Mandal, I. V. Sinelnikov, D. Broadhurst and D. S. Wishart, MetaboAnalyst 2.0–a comprehensive server for metabolomic data analysis, Nucleic Acids Res., 2012, 40, W127–W133 CrossRef CAS PubMed.
- E. K. Kemsley and H. S. Tapp, OPLS filtered data can be obtained directly from non-orthogonalized PLS1, J. Chemom., 2009, 23, 263–264 CrossRef CAS.
- E. K. Kemsley and H. S. Tapp, Notes on the practical utility of OPLS, TrAC, Trends Anal. Chem., 2009, 28, 1322–1327 CrossRef PubMed.
- J. Trygg, J. Gullberg, A. I. Johansson, P. Jonsson and T. Moritz, Chemometrics in Metabolomics, in Plant Metabolomics (Biotechnology in Agriculture and Forestry 57), ed. K. Saito, R. A. Dixon and L. Willmitzer, Springer Verlag, 2006 Search PubMed.
- J. Trygg, E. Holmes and T. Lundstedt, Chemometrics in metabonomics, J. Proteome Res., 2007, 6(2), 469–479 CrossRef CAS PubMed.
- A. L. Boulesteix and K. Strimmer, Partial least squares: a versatile tool for the analysis of high-dimensional genomic data, Briefings Bioinf., 2007, 8(1), 32–44 CrossRef CAS PubMed.
- J. Beyene, D. Tritchler, S. B. Bull, K. C. Cartier, G. Jonasdottir and A. T. Kraja, et al. Multivariate analysis of complex gene expression and clinical phenotypes with genetic marker data, Mol. Genet. Epidemiol., 2007, 31(suppl 1), S103–S109 Search PubMed.
- B. K. Lavine and J. Workman, Jr., Chemometrics. Analytical chemistry, 2013, 85(2), 705–714 CrossRef CAS PubMed.
- J. F. Hair, Multivariate data analysis, Upper Saddle River, NJ: Prentice Hall, 7th edn, 2010, xxviii, p. 785 Search PubMed.
- G. M. Kirwan, E. Johansson, R. Kleemann, E. R. Verheij, A. M. Wheelock and S. Goto, et al. Building multivariate systems biology models, Anal. Chem., 2012, 84(16), 7064–7071 CrossRef CAS PubMed.
- J. Trygg and S. Wold, Orthogonal Projections to Latent Structures (OPLS), J. Chemom., 2002, 16(3), 119–128 CrossRef CAS.
- R. C. Pinto, J. Gottfries and J. Trygg, Advantages of orthogonal inspection in chemometrics, J. Chemom., 2012, 26(6), 231–235 CrossRef CAS.
- L. Eriksson, J. Trygg and S. Wold, CV-ANOVA for significance testing of PLS and OPLS (R) models, J. Chemom., 2008, 22(11–12), 594–600 CrossRef CAS.
- J. Cohen, What I have learned (so far), Am. Psychol., 1990, 45(12), 1304–1312 CrossRef.
- S. Wiklund, E. Johansson, L. Sjostrom, E. J. Mellerowicz, U. Edlund and J. P. Shockcor, et al. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models, Anal. Chem., 2008, 80(1), 115–122 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2013 |