Exploring mechanisms of human disease through structurally resolved protein interactome networks
Jishnu
Das
ab,
Robert
Fragoza†
bc,
Hao Ran
Lee†
ab,
Nicolas A.
Cordero†
b,
Yu
Guo
bc,
Michael J.
Meyer
abd,
Tommy V.
Vo
bc,
Xiujuan
Wang
ab and
Haiyuan
Yu
*ab
aDepartment of Biological Statistics and Computational Biology, Cornell University, Ithaca, NY 14853, USA. E-mail: haiyuan.yu@cornell.edu
bWeill Institute for Cell and Molecular Biology, Cornell University, Ithaca, NY 14853, USA
cDepartment of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853, USA
dTri-Institutional Training Program in Computational Biology and Medicine, New York, NY 10065, USA
First published on 12th September 2013
Abstract
The study of the molecular basis of human disease has gained increasing attention over the past decade. With significant improvements in sequencing efficiency and throughput, a wealth of genotypic data has become available. However the translation of this information into concrete advances in diagnostic and clinical setups has proved far more challenging. Two major reasons for this are the lack of functional annotation for genomic variants and the complex nature of genotype-to-phenotype relationships. One fundamental approach to bypass these issues is to examine the effects of genetic variation at the level of proteins as they are directly involved in carrying out biological functions. Within the cell, proteins function by interacting with other proteins as a part of an underlying interactome network. This network can be determined using interactome mapping – a combination of high-throughput experimental toolkits and curation from small-scale studies. Integrating structural information from co-crystals with the network allows generation of a structurally resolved network. Within the context of this network, the structural principles of disease mutations can be examined and used to generate reliable mechanistic hypotheses regarding disease pathogenesis.
Introduction
Over the last decade and a half, there has been a dramatic increase in the efficiency and a substantial decrease in the cost of sequencing. With the sequencing of the human genome, there was the promise of significant advances in translational medicine.1,2 However, while there has been a rapid accumulation of genomic data, the corresponding expansion in our understanding of pathogenic processes has been much slower. There are two major reasons for this. First, while there has been an explosion in the accumulation of genomic variants and disease-associated mutations, most of them have not been functionally annotated (Fig. 1A). This is reflected in the fact that while the number of single-nucleotide polymorphisms (SNPs) available from dbSNP3 and disease-associated mutations from HGMD4 have grown 3500% and 260%, respectively, over the last twelve years, the number of FDA-approved drugs has grown only 20% (Fig. 1A). Second, the difficulty in obtaining functional annotation is primarily attributable to the complex relationships between genotype and phenotype. A single gene can affect multiple traits (gene pleiotropy) and the same trait can be linked to numerous causal genes (locus heterogeneity). Furthermore, epistasis also brings additional complexity to genotype-to-phenotype relationships.5 To sidestep these complexities, numerous large-scale efforts have been undertaken to correlate sequence variants with an observable phenotype, but it has been difficult to extend the observed correlation into causation. This has often been the main critique of GWA-like studies6 and has resulted in a large fraction of phenotypes with unknown molecular mechanisms (Fig. 1B). | ||
| Fig. 1 Growth of genomic data and our understanding of pathogenesis (A) accumulation of dbSNP data, HGMD mutations, disease genes and drug targets over the past 12 years (number of dbSNP variations: ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/human_9606/chr_rpts/; number of HGMD mutations: http://www.hgmd.cf.ac.uk/ac/hahaha.php; number of disease genes: ftp://ftp.eimb.ru/omim/; number of FDA-approved drugs: http://www.fda.gov/AboutFDA/WhatWeDo/History/ProductRegulation/SummaryofNDAApprovalsReceipts1938tothepresent). (B) Distribution of OMIM phenotype entries by knowledge of molecular basis (http://www.omim.org/statistics/entry). | ||
One fundamental way to bypass the complexity of genotype-to-phenotype relationships is to directly examine the functional consequences of mutations and variants within coding regions at the protein level. Although a large number of variants are in non-coding regions, it has been shown that disease mutations and trait-associated SNPs are enriched in coding regions.7 Moreover, within the cellular environment, proteins rarely act in isolation. Interactions between proteins within the cell define major functional pathways crucial to physiological processes. The set of all interactions within the cell or the protein interactome can be represented as a network in which proteins are nodes and interactions between them are undirected edges. Thus maintenance of this network is critical to cellular function, and disease phenotypes can be viewed as perturbations to this network.8–10 Thus, the protein network can be used to gain insights into complex dependencies in pathogenic processes.8,9 It has also been shown to be useful in understanding disease sub-types and predicting disease prognosis.11,12 However, one limitation of this approach is that while such a representation is inherently two-dimensional, proteins are complex macromolecules with intricate three-dimensional structures. In this review, we outline experimental techniques used to identify protein–protein interactions and discuss recent methods developed to overlay structural information onto these interactions to construct structurally resolved protein networks. We then elucidate the importance of these networks in understanding molecular mechanisms of human disease.
High-throughput experimental toolkit for interactome mapping
There are two ways in which protein interactome networks are determined – literature-curation of small-scale studies and high-throughput (HT) experiments. In literature curation, interaction data are collected from thousands of small-scale studies each of which focuses on one or a few proteins and their interactions. On the other hand, HT experiments are much larger in scale and are typically set up as an unbiased screen of a large space. The repertoire of techniques used to determine these networks using such experiments is referred to as interactome mapping.13Interactome mapping can generate binary interactions and co-complex associations.14,15 The former represents direct biophysical interactions between two proteins while the latter merely denotes membership of a complex and can often include indirect associations. There are several widely-used databases – BioGrid,16 IntAct,17 HPRD,18 iRefWeb,19 DIP,20 MINT,21 MIPS22 and VisAnt23 – that curate both categories of interactions for humans and other model organisms. However, it has been shown that the same degree of confidence cannot be associated with all interactions and those that have been validated by only one assay typically tend to be of lower quality than those that are validated by two or more assays.14,24,25 Numerous hypothesis-driven studies rely on specific interactions to design downstream experiments. Using low-quality or erroneous interactions could lead to incorrect hypotheses and futile downstream experiments. To address this, we built a repository of high-quality protein interactome networks – HINT.15 HINT also distinguishes between interactions curated from small-scale studies and those obtained from high-throughput experiments. This is essential because it has been shown that small-scale studies often contain sampling biases that make networks generated using them unsuitable for global topological analyses.14,15 In this review, we discuss five major high-throughput assays that can be used to generate binary interactome networks. To construct structurally resolved networks, it is essential for the interactions to be binary because the concept of interaction interface does not apply to indirect associations.
Yeast two-hybrid (Y2H) (Fig. 2A) was developed by Stanley Fields and Ok-Kyu Song as a genetic system to identify protein–protein interactions.26 The assay relies on the split functionality of particular eukaryotic transcription factors, for example Gal4, in which the transcription factor is split into two parts: a sequence-specific DNA-binding domain (DB) and a transcriptional-activation domain (AD). Protein–protein interactions are tested by fusing a “bait” protein X to the DB and fusing a “prey” protein Y to the AD. Each fusion protein is then expressed in haploid strains of yeast of opposite mating type. Upon mating, if protein X and Y interact, transcription factor activity will be reconstituted, allowing for downstream reporter gene expression and diploid yeast growth on selective media. The original system has undergone numerous technical modifications to make it amenable to high throughput with improved assay precision and sensitivity.27,28
 | ||
Fig. 2 Schematic representations of high-throughput assays used to generate binary interactome networks. (A) Yeast two-hybrid (Y2H). (B) Protein fragment complementation assays (PCA). (C) Luminescence-based mammalian interactome mapping (LUMIER). (D) Well-based nucleic acid programmable protein array (wNAPPA). (E) Mammalian protein–protein interaction trap (MAPPIT). (F) A high-quality reference human binary interactome comprising ∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 interactions generated from several large-scale interactome mapping efforts and thousands of small-scale studies. 000 interactions generated from several large-scale interactome mapping efforts and thousands of small-scale studies. | ||
Protein complementation assay (PCA) (Fig. 2B) is another popular approach for testing protein–protein interactions using mammalian cells. Similar to Y2H, in PCA, a fluorescent protein such as yellow fluorescent protein (YFP) (or an enzyme such as TEM-1 β-lactamase) is split into N- and C-terminal domains then fused to a bait protein X and a prey protein Y. If X and Y interact, YFP activity is reconstituted which can be observed by fluorescent microscopy or in high-throughput by using a plate reader.29 Unlike Y2H though, detectable protein–protein interactions are not limited to the nucleus. Thus, PCA can serve as a suitable assay for probing protein interactions at their native localizations in intact, living cells.
In luminescence-based mammalian interactome (LUMIER) (Fig. 2C) a bait protein X is fused to renilla or firefly luciferase enzyme and then co-expressed with a FLAG-tagged prey protein Y in mammalian HEK293T cells. Interaction between proteins X and Y can then be assayed by anti-FLAG immunoprecipitation of protein Y. Luciferase bioluminescence is then measured to detect whether protein Y was pulled down with X.30 Recent modifications allow LUMIER to be carried out in a high-throughput fashion using 96-well plates while also offering an improved quantitative readout.31
Well-based nucleic acid programmable protein array (wNAPPA) (Fig. 2D) is an in vitro assay, which begins with two expression vectors that encode for an anti-glutathione-S-transferase (GST) tagged protein X and a hemagglutinin (HA) tagged protein Y, respectively, which are anchored in a GST antibody-coated plate well. In vitro transcription and translation of chimeric proteins X and Y is then triggered by introducing rabbit reticulocyte lysate to the wells. Translated GST-tagged protein X will then bind to the GST antibodies coated in the well. A washing step then follows in which protein Y will remain in the well post-wash only if it interacts with protein X. The presence of protein Y – and therefore an interaction between proteins X and Y – is then detected by attaching horseradish peroxidase (HRP)-conjugated secondary antibodies specific to HA tagged protein Y and then measuring HRP-induced chemiluminescence.32
Mammalian protein–protein interaction trap (MAPPIT) (Fig. 2E) is based upon JAK-STAT signaling pathways. In JAK-STAT signaling, ligand-bound cytokine receptor complexes will reorganize themselves, in turn activating tethered Janus kinases (JAKs). Activated JAKs then phosphorylate tyrosine residues along the tails of the receptor complex which then serve as docking sites for signal transducer and activator of transcription (STAT) proteins. Receptor tail-docked STATs are next phosphorylated and activated by JAKs which then migrate to the nucleus to trigger STAT-dependent reporter gene activity. MAPPIT instead though uses a modified receptor complex in which the complex is split into two fragments: (1) a membrane-bound receptor that still permits JAK2 activation with mutated tyrosine residues to prevent STAT3 docking and (2) a receptor tail fragment containing STAT3 binding sites. Fragments 1 and 2 are then fused to bait protein X and prey protein Y. If proteins X and Y interact, JAK2 will activate STAT3 in trans, leading to STAT3-dependent reporter gene activity.33
Numerous studies have also tried to predict protein interactions based on machine-learning approaches34 or known co-crystal structures.35–37 However, only those predictions that have been experimentally validated can be considered high quality. Thus, by combining data from several large-scale interactome mapping efforts24,28,38,39 (that use the above techniques) with thousands of small-scale studies, a high-quality reference human binary interactome comprising ∼40000 interactions (Fig. 2F) can be generated and denotes the first step towards producing a structurally resolved network.
Structurally resolved interactome networks
The reference interactome network has been widely used to try and understand the molecular basis of human disease.8 Numerous methods have been used to predict disease-associated genes,40 most of which rely heavily on a global “guilt-by-association” principle.41 Thus, if a particular gene is associated with a disease, the assumption is that all the interacting partners of the protein encoded by that gene are also associated with that disease. Such an understanding can be quite simplistic as the reference interactome is merely a two-dimensional representation and does not take into account the 3D structures of interacting proteins. Consequently, the percentage of successful predictions using such approaches is quite low.42 Since most interacting proteins share only a few of their associated disorders, it is essential to incorporate structural information regarding the location of disease mutations to make the predictions more accurate. This necessitates the construction of a structurally resolved interactome network.Over the last two decades, there have been systematic efforts to structurally classify proteins into families43,44 based on domain architecture.45 This has been used to identify domain–domain interactions of known three-dimensional complexes of interacting proteins.46,47 However, the biggest challenge in constructing a structurally resolved network from these domain–domain interactions is posed by the relatively low number of available co-crystal structures compared to the amount of available proteomic network data. Co-crystal structures are not available for >90% of available binary protein–protein interactions. Moreover, complete individual structures are available for only about 10% of interacting proteins. Mosca et al. present a comprehensive analysis that highlights the paucity of experimentally determined crystal structures compared to the number of known binary interactions.48 Thus, it is essential to build structural models both to model individual proteins49 and infer interaction interfaces.
Dr Gerstein and his colleagues took the first step in this direction and used sequence similarity to compare interacting proteins with known co-crystal complexes. The authors constructed a structurally resolved yeast protein interactome to gain insight into evolutionary rates of network hubs with distinct types of interaction interfaces.50 Schuster-Bockler and Bateman focused on using a sequence-based homology approach to analyze the sites of disease-associated mutations with respect to protein interaction interfaces. Their work indicated that only about 4% of these mutations could interfere with protein–protein interactions.51 Prieto et al. built a repository of unified structural domain–domain interactions by systematically comparing six main structural domain–domain interaction sources that are based on Protein Data Bank (PDB) structures.52 The first structurally resolved human-virus protein–protein interaction network constructed by Franzosa and Xia showed that it is common for viruses to mimic host binding interfaces even without structural similarity to the human counterparts.53
Recently, we constructed a high-quality structurally resolved human binary protein interactome network using either co-crystal structures in the PDB or a homology-based interaction interface domain inference method.54 A comprehensive list of 62663 Mendelian mutations in 3949 protein-coding genes associated with 3453 clinically distinct disorders was curated from Online Mendelian Inheritance in Man (OMIM) and Human Gene Mutation Database (HGMD), and then mapped to the structurally resolved interactome (Fig. 3A). We found that in-frame mutations are significantly enriched within interacting domains of disease-associated proteins. Furthermore, we observed that the likelihood of two in-frame mutations on the corresponding interacting domains of interacting proteins to cause the same disorder is significantly higher than that of corresponding pairs on non-interacting domains (Fig. 3B). In addition, we saw that in-frame mutation pairs on different interaction interfaces tend to cause different disorders than those on the same interface (Fig. 3C).54 These results help explain locus heterogeneity and gene pleiotropy, respectively – the alteration of specific interactions by mutations at the corresponding interface plays an important role in the pathogenesis of many disease genes. This also helps us refine the traditional guilt-by-association principle – mutations at different structural loci on the same protein can cause different diseases through disruption of separate interactions (Fig. 3D). We also used our interface inference approach to generate structurally resolved interactome networks for several other model organisms and established a database of high-quality structurally resolved protein–protein interactions, INstruct.55 Mosca et al. also used a similarly motivated structural alignment approach to infer interaction interfaces for networks in humans and eight other model organisms. Their results also suggested that structural annotation of pathways could help rationalize the mechanism of action of disease mutations.48 Thus, using structurally resolved interactome networks, it is now possible to gain insights at the molecular level into protein function and its alteration.
 | ||
| Fig. 3 Structurally resolved interactome networks and human disease. (A) Construction of a structurally resolved interactome network onto which disease mutations are mapped. (B) Percentage of mutation pairs on two proteins that cause the same disease. (C) Percentage of mutation pairs on the same protein that cause different diseases. (D) A higher resolution of the guilt-by-association principle – mutations at different structural loci on the same protein that cause different diseases [(B) and (C) are adapted from ref. 54]. | ||
Towards a mechanistic understanding of human disease
Human disease can be viewed as a rewiring of the reference interactome through loss or gain of interactions.8 Zhong et al. experimentally showed that disease mutations could alter the underlying interactome by edge-specific changes i.e., altering specific interactions or node-specific changes i.e., leading to complete loss of protein products.10 One example they demonstrated was the disruption of the homodimeric CBS interaction (i.e., interaction of CBS with itself) by a homocystinuria associated P145L mutation (Fig. 4A). On the other hand, a homocystinuria associated P49L mutation did not disrupt the interaction – the interaction was “pseudo wild-type” (Fig. 4A). Upon examining the interface of this protein–protein interaction (which can be obtained from the co-crystal with PDB id: 1JBQ56) using our structurally resolved network approach, we found that the P145L mutation is within the interface whereas the P49L mutation is outside the interface. We also showed that each of the three distinct colorectal cancer associated mutations on the interaction interface of MLH1 (I68N, I107R and Y293D) with PMS2 disrupted the interaction while any of the three other colorectal-cancer associated mutations (N338S, Y561H and R725C) outside the interface did not disrupt the interaction54 (Fig. 4B). In this case, the interface was inferred using our homology-based interaction interface inference method.54 These studies further establish the view that mutations at the interface can disrupt specific interactions leading to human disease.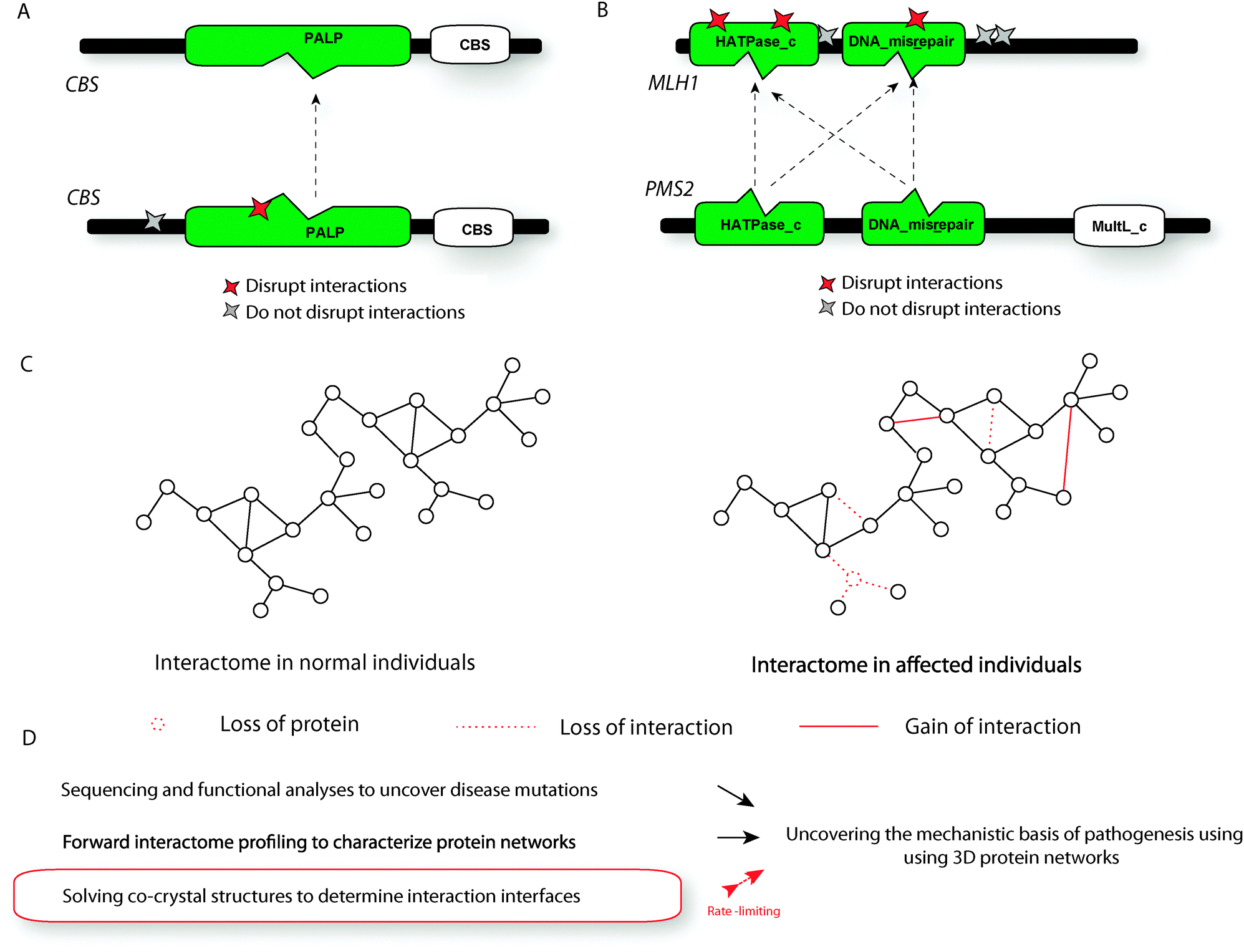 | ||
| Fig. 4 Functional consequences of human disease mutations. (A) Illustration of the interface of the CBS homodimer as obtained from a co-crystal and the location of mutations that do/do not disrupt the interaction. (B) Illustration of the predicted interface of the MLH1–PMS2 interaction and the location of mutations that do/do not disrupt the interaction. (C) Schematic representation of changes caused by disease mutations to the interactome network. (D) Summary of the pipeline used to construct 3D interactome networks to understand disease pathogenesis. | ||
In general, there are three kinds of possible changes to the interactome network – loss of a protein (and all its interactions), loss of a specific interaction, and gain of a specific interaction (Fig. 4C).10 To be able to truly understand human disease, it is necessary to experimentally analyze relationships between the structural loci of mutations and each of these alteration types at a proteomic scale. Since the vast majority of interactions do not have corresponding co-crystal structures, it is also necessary to develop better computational models that help us accurately determine the structural locations of mutations. Combining co-crystal structures with these computational models will help generate a comprehensive atlas of protein–protein interactions that is of ubiquitous importance in understanding pathogenic processes.57,58 Such an atlas can be generated by integrative methods that incorporate both experimental and computational approaches and is likely to be highly successful in elucidating the mechanistic basis of human disease caused by rewiring of the underlying protein interactome network.
Conclusion
A key bottleneck in translational medicine has been the sharp imbalance between the number of available genomic variants and the number of well-understood disease mechanisms. The complex nature of genotype-to-phenotype relationships has made functional annotation of variants an extremely challenging problem. Analyzing alterations at the proteomic level promises to offer possible solutions to these problems as human disease can be viewed as altered protein function. Since proteins mediate cellular functions by interacting with other proteins, it is necessary to examine these changes in the context of the underlying network of protein–protein interactions. A combination of high-throughput experiments and literature curation is being used to generate the reference human protein interactome network. By incorporating structural details of proteins involved in these interactions, it is possible to generate a structurally resolved network. Within the context of this network, it is possible to examine structural details of disease-causing mutations and generate mechanistic hypotheses regarding pathogenesis (Fig. 4D). Follow-up of these hypotheses is likely to uncover key functional principles underlying human disease and identify more reliable drug targets.References
- E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R. A. Gibbs, D. M. Muzny, S. E. Scherer, J. B. Bouck, E. J. Sodergren, K. C. Worley, C. M. Rives, J. H. Gorrell, M. L. Metzker, S. L. Naylor, R. S. Kucherlapati, D. L. Nelson, G. M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, D. R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H. M. Lee, J. Dubois, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R. W. Davis, N. A. Federspiel, A. P. Abola, M. J. Proctor, R. M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D. R. Cox, M. V. Olson, R. Kaul, N. Shimizu, K. Kawasaki, S. Minoshima, G. A. Evans, M. Athanasiou, R. Schultz, B. A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W. R. McCombie, M. de la Bastide, N. Dedhia, H. Blocker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J. A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D. G. Brown, C. B. Burge, L. Cerutti, H. C. Chen, D. Church, M. Clamp, R. R. Copley, T. Doerks, S. R. Eddy, E. E. Eichler, T. S. Furey, J. Galagan, J. G. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L. S. Johnson, T. A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W. J. Kent, P. Kitts, E. V. Koonin, I. Korf, D. Kulp, D. Lancet, T. M. Lowe, A. McLysaght, T. Mikkelsen, J. V. Moran, N. Mulder, V. J. Pollara, C. P. Ponting, G. Schuler, J. Schultz, G. Slater, A. F. Smit, E. Stupka, J. Szustakowski, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y. I. Wolf, K. H. Wolfe, S. P. Yang, R. F. Yeh, F. Collins, M. S. Guyer, J. Peterson, A. Felsenfeld, K. A. Wetterstrand, A. Patrinos, M. J. Morgan, P. de Jong, J. J. Catanese, K. Osoegawa, H. Shizuya, S. Choi and Y. J. Chen, Nature, 2001, 409, 860–921 CrossRef CAS PubMed.
- J. C. Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. G. Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Wang, A. Wang, X. Wang, J. Wang, M. Wei, R. Wides, C. Xiao, C. Yan, A. Yao, J. Ye, M. Zhan, W. Zhang, H. Zhang, Q. Zhao, L. Zheng, F. Zhong, W. Zhong, S. Zhu, S. Zhao, D. Gilbert, S. Baumhueter, G. Spier, C. Carter, A. Cravchik, T. Woodage, F. Ali, H. An, A. Awe, D. Baldwin, H. Baden, M. Barnstead, I. Barrow, K. Beeson, D. Busam, A. Carver, A. Center, M. L. Cheng, L. Curry, S. Danaher, L. Davenport, R. Desilets, S. Dietz, K. Dodson, L. Doup, S. Ferriera, N. Garg, A. Gluecksmann, B. Hart, J. Haynes, C. Haynes, C. Heiner, S. Hladun, D. Hostin, J. Houck, T. Howland, C. Ibegwam, J. Johnson, F. Kalush, L. Kline, S. Koduru, A. Love, F. Mann, D. May, S. McCawley, T. McIntosh, I. McMullen, M. Moy, L. Moy, B. Murphy, K. Nelson, C. Pfannkoch, E. Pratts, V. Puri, H. Qureshi, M. Reardon, R. Rodriguez, Y. H. Rogers, D. Romblad, B. Ruhfel, R. Scott, C. Sitter, M. Smallwood, E. Stewart, R. Strong, E. Suh, R. Thomas, N. N. Tint, S. Tse, C. Vech, G. Wang, J. Wetter, S. Williams, M. Williams, S. Windsor, E. Winn-Deen, K. Wolfe, J. Zaveri, K. Zaveri, J. F. Abril, R. Guigo, M. J. Campbell, K. V. Sjolander, B. Karlak, A. Kejariwal, H. Mi, B. Lazareva, T. Hatton, A. Narechania, K. Diemer, A. Muruganujan, N. Guo, S. Sato, V. Bafna, S. Istrail, R. Lippert, R. Schwartz, B. Walenz, S. Yooseph, D. Allen, A. Basu, J. Baxendale, L. Blick, M. Caminha, J. Carnes-Stine, P. Caulk, Y. H. Chiang, M. Coyne, C. Dahlke, A. Mays, M. Dombroski, M. Donnelly, D. Ely, S. Esparham, C. Fosler, H. Gire, S. Glanowski, K. Glasser, A. Glodek, M. Gorokhov, K. Graham, B. Gropman, M. Harris, J. Heil, S. Henderson, J. Hoover, D. Jennings, C. Jordan, J. Jordan, J. Kasha, L. Kagan, C. Kraft, A. Levitsky, M. Lewis, X. Liu, J. Lopez, D. Ma, W. Majoros, J. McDaniel, S. Murphy, M. Newman, T. Nguyen, N. Nguyen, M. Nodell, S. Pan, J. Peck, M. Peterson, W. Rowe, R. Sanders, J. Scott, M. Simpson, T. Smith, A. Sprague, T. Stockwell, R. Turner, E. Venter, M. Wang, M. Wen, D. Wu, M. Wu, A. Xia, A. Zandieh and X. Zhu, Science, 2001, 291, 1304–1351 CrossRef CAS PubMed.
- E. M. Smigielski, K. Sirotkin, M. Ward and S. T. Sherry, Nucleic Acids Res., 2000, 28, 352–355 CrossRef CAS PubMed.
- P. D. Stenson, M. Mort, E. V. Ball, K. Howells, A. D. Phillips, N. S. Thomas and D. N. Cooper, Genome Med., 2009, 1, 13 CrossRef PubMed.
- H. J. Cordell, Hum. Mol. Genet., 2002, 11, 2463–2468 CrossRef CAS PubMed.
- E. T. Dermitzakis and A. G. Clark, Science, 2009, 326, 239–240 CrossRef CAS PubMed.
- L. A. Hindorff, P. Sethupathy, H. A. Junkins, E. M. Ramos, J. P. Mehta, F. S. Collins and T. A. Manolio, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9362–9367 CrossRef CAS PubMed.
- M. Vidal, M. E. Cusick and A. L. Barabasi, Cell, 2011, 144, 986–998 CrossRef CAS PubMed.
- A. L. Barabasi, N. Gulbahce and J. Loscalzo, Nat. Rev. Genet., 2011, 12, 56–68 CrossRef CAS PubMed.
- Q. Zhong, N. Simonis, Q. R. Li, B. Charloteaux, F. Heuze, N. Klitgord, S. Tam, H. Yu, K. Venkatesan, D. Mou, V. Swearingen, M. A. Yildirim, H. Yan, A. Dricot, D. Szeto, C. Lin, T. Hao, C. Fan, S. Milstein, D. Dupuy, R. Brasseur, D. E. Hill, M. E. Cusick and M. Vidal, Mol. Syst. Biol., 2009, 5, 321 CrossRef PubMed.
- H. Y. Chuang, E. Lee, Y. T. Liu, D. Lee and T. Ideker, Mol. Syst. Biol., 2007, 3, 140 CrossRef PubMed.
- I. W. Taylor, R. Linding, D. Warde-Farley, Y. Liu, C. Pesquita, D. Faria, S. Bull, T. Pawson, Q. Morris and J. L. Wrana, Nat. Biotechnol., 2009, 27, 199–204 CrossRef CAS PubMed.
- M. Vidal, FEBS Lett., 2005, 579, 1834–1838 CrossRef CAS PubMed.
- H. Yu, P. Braun, M. A. Yildirim, I. Lemmens, K. Venkatesan, J. Sahalie, T. Hirozane-Kishikawa, F. Gebreab, N. Li, N. Simonis, T. Hao, J. F. Rual, A. Dricot, A. Vazquez, R. R. Murray, C. Simon, L. Tardivo, S. Tam, N. Svrzikapa, C. Fan, A. S. de Smet, A. Motyl, M. E. Hudson, J. Park, X. Xin, M. E. Cusick, T. Moore, C. Boone, M. Snyder, F. P. Roth, A. L. Barabasi, J. Tavernier, D. E. Hill and M. Vidal, Science, 2008, 322, 104–110 CrossRef CAS PubMed.
- J. Das and H. Yu, BMC Syst. Biol., 2012, 6, 92 CrossRef PubMed.
- C. Stark, B. J. Breitkreutz, A. Chatr-Aryamontri, L. Boucher, R. Oughtred, M. S. Livstone, J. Nixon, K. Van Auken, X. Wang, X. Shi, T. Reguly, J. M. Rust, A. Winter, K. Dolinski and M. Tyers, Nucleic Acids Res., 2011, 39, D698–D704 CrossRef CAS PubMed.
- S. Kerrien, B. Aranda, L. Breuza, A. Bridge, F. Broackes-Carter, C. Chen, M. Duesbury, M. Dumousseau, M. Feuermann, U. Hinz, C. Jandrasits, R. C. Jimenez, J. Khadake, U. Mahadevan, P. Masson, I. Pedruzzi, E. Pfeiffenberger, P. Porras, A. Raghunath, B. Roechert, S. Orchard and H. Hermjakob, Nucleic Acids Res., 2012, 40, D841–D846 CrossRef CAS PubMed.
- T. S. Keshava Prasad, R. Goel, K. Kandasamy, S. Keerthikumar, S. Kumar, S. Mathivanan, D. Telikicherla, R. Raju, B. Shafreen, A. Venugopal, L. Balakrishnan, A. Marimuthu, S. Banerjee, D. S. Somanathan, A. Sebastian, S. Rani, S. Ray, C. J. Harrys Kishore, S. Kanth, M. Ahmed, M. K. Kashyap, R. Mohmood, Y. L. Ramachandra, V. Krishna, B. A. Rahiman, S. Mohan, P. Ranganathan, S. Ramabadran, R. Chaerkady and A. Pandey, Nucleic Acids Res., 2009, 37, D767–D772 CrossRef CAS PubMed.
- B. Turner, S. Razick, A. L. Turinsky, J. Vlasblom, E. K. Crowdy, E. Cho, K. Morrison, I. M. Donaldson and S. J. Wodak, Database, 2010, 2010, baq023 CrossRef PubMed.
- L. Salwinski, C. S. Miller, A. J. Smith, F. K. Pettit, J. U. Bowie and D. Eisenberg, Nucleic Acids Res., 2004, 32, D449–D451 CrossRef CAS PubMed.
- L. Licata, L. Briganti, D. Peluso, L. Perfetto, M. Iannuccelli, E. Galeota, F. Sacco, A. Palma, A. P. Nardozza, E. Santonico, L. Castagnoli and G. Cesareni, Nucleic Acids Res., 2012, 40, D857–D861 CrossRef CAS PubMed.
- H. W. Mewes, A. Ruepp, F. Theis, T. Rattei, M. Walter, D. Frishman, K. Suhre, M. Spannagl, K. F. Mayer, V. Stumpflen and A. Antonov, Nucleic Acids Res., 2011, 39, D220–D224 CrossRef CAS PubMed.
- Z. Hu, J. H. Hung, Y. Wang, Y. C. Chang, C. L. Huang, M. Huyck and C. DeLisi, Nucleic Acids Res., 2009, 37, W115–W121 CrossRef CAS PubMed.
- K. Venkatesan, J. F. Rual, A. Vazquez, U. Stelzl, I. Lemmens, T. Hirozane-Kishikawa, T. Hao, M. Zenkner, X. Xin, K. I. Goh, M. A. Yildirim, N. Simonis, K. Heinzmann, F. Gebreab, J. M. Sahalie, S. Cevik, C. Simon, A. S. de Smet, E. Dann, A. Smolyar, A. Vinayagam, H. Yu, D. Szeto, H. Borick, A. Dricot, N. Klitgord, R. R. Murray, C. Lin, M. Lalowski, J. Timm, K. Rau, C. Boone, P. Braun, M. E. Cusick, F. P. Roth, D. E. Hill, J. Tavernier, E. E. Wanker, A. L. Barabasi and M. Vidal, Nat. Methods, 2009, 6, 83–90 CrossRef CAS PubMed.
- M. E. Cusick, H. Yu, A. Smolyar, K. Venkatesan, A. R. Carvunis, N. Simonis, J. F. Rual, H. Borick, P. Braun, M. Dreze, J. Vandenhaute, M. Galli, J. Yazaki, D. E. Hill, J. R. Ecker, F. P. Roth and M. Vidal, Nat. Methods, 2009, 6, 39–46 CrossRef CAS PubMed.
- S. Fields and O. Song, Nature, 1989, 340, 245–246 CrossRef CAS PubMed.
- A. J. Walhout and M. Vidal, Methods, 2001, 24, 297–306 CrossRef CAS PubMed.
- H. Yu, L. Tardivo, S. Tam, E. Weiner, F. Gebreab, C. Fan, N. Svrzikapa, T. Hirozane-Kishikawa, E. Rietman, X. Yang, J. Sahalie, K. Salehi-Ashtiani, T. Hao, M. E. Cusick, D. E. Hill, F. P. Roth, P. Braun and M. Vidal, Nat. Methods, 2011, 8, 478–480 CrossRef CAS PubMed.
- I. Remy and S. W. Michnick, Nat. Methods, 2006, 3, 977–979 CrossRef CAS PubMed.
- M. Barrios-Rodiles, K. R. Brown, B. Ozdamar, R. Bose, Z. Liu, R. S. Donovan, F. Shinjo, Y. Liu, J. Dembowy, I. W. Taylor, V. Luga, N. Przulj, M. Robinson, H. Suzuki, Y. Hayashizaki, I. Jurisica and J. L. Wrana, Science, 2005, 307, 1621–1625 CrossRef CAS PubMed.
- M. Taipale, I. Krykbaeva, M. Koeva, C. Kayatekin, K. D. Westover, G. I. Karras and S. Lindquist, Cell, 2012, 150, 987–1001 CrossRef CAS PubMed.
- N. Ramachandran, E. Hainsworth, B. Bhullar, S. Eisenstein, B. Rosen, A. Y. Lau, J. C. Walter and J. LaBaer, Science, 2004, 305, 86–90 CrossRef CAS PubMed.
- P. Ulrichts, I. Lemmens, D. Lavens, R. Beyaert and J. Tavernier, Methods Mol. Biol., 2009, 517, 133–144 CAS.
- R. Jansen, H. Yu, D. Greenbaum, Y. Kluger, N. J. Krogan, S. Chung, A. Emili, M. Snyder, J. F. Greenblatt and M. Gerstein, Science, 2003, 302, 449–453 CrossRef CAS PubMed.
- P. Aloy and R. B. Russell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5896–5901 CrossRef CAS PubMed.
- N. Tuncbag, A. Gursoy, R. Nussinov and O. Keskin, Nat. Protocols, 2011, 6, 1341–1354 CAS.
- Q. C. Zhang, D. Petrey, L. Deng, L. Qiang, Y. Shi, C. A. Thu, B. Bisikirska, C. Lefebvre, D. Accili, T. Hunter, T. Maniatis, A. Califano and B. Honig, Nature, 2012, 490, 556–560 CrossRef CAS PubMed.
- J. F. Rual, K. Venkatesan, T. Hao, T. Hirozane-Kishikawa, A. Dricot, N. Li, G. F. Berriz, F. D. Gibbons, M. Dreze, N. Ayivi-Guedehoussou, N. Klitgord, C. Simon, M. Boxem, S. Milstein, J. Rosenberg, D. S. Goldberg, L. V. Zhang, S. L. Wong, G. Franklin, S. Li, J. S. Albala, J. Lim, C. Fraughton, E. Llamosas, S. Cevik, C. Bex, P. Lamesch, R. S. Sikorski, J. Vandenhaute, H. Y. Zoghbi, A. Smolyar, S. Bosak, R. Sequerra, L. Doucette-Stamm, M. E. Cusick, D. E. Hill, F. P. Roth and M. Vidal, Nature, 2005, 437, 1173–1178 CrossRef CAS PubMed.
- U. Stelzl, U. Worm, M. Lalowski, C. Haenig, F. H. Brembeck, H. Goehler, M. Stroedicke, M. Zenkner, A. Schoenherr, S. Koeppen, J. Timm, S. Mintzlaff, C. Abraham, N. Bock, S. Kietzmann, A. Goedde, E. Toksoz, A. Droege, S. Krobitsch, B. Korn, W. Birchmeier, H. Lehrach and E. E. Wanker, Cell, 2005, 122, 957–968 CrossRef CAS PubMed.
- X. Wang, N. Gulbahce and H. Yu, Briefings Funct. Genomics, 2011, 10, 280–293 CrossRef CAS PubMed.
- S. Oliver, Nature, 2000, 403, 601–603 CrossRef CAS PubMed.
- M. Oti, B. Snel, M. A. Huynen and H. G. Brunner, J. Med. Genet., 2006, 43, 691–698 CrossRef CAS PubMed.
- A. Andreeva, D. Howorth, J. M. Chandonia, S. E. Brenner, T. J. Hubbard, C. Chothia and A. G. Murzin, Nucleic Acids Res., 2008, 36, D419–D425 CrossRef CAS PubMed.
- I. Sillitoe, A. L. Cuff, B. H. Dessailly, N. L. Dawson, N. Furnham, D. Lee, J. G. Lees, T. E. Lewis, R. A. Studer, R. Rentzsch, C. Yeats, J. M. Thornton and C. A. Orengo, Nucleic Acids Res., 2013, 41, D490–D498 CrossRef CAS PubMed.
- R. D. Finn, J. Mistry, J. Tate, P. Coggill, A. Heger, J. E. Pollington, O. L. Gavin, P. Gunasekaran, G. Ceric, K. Forslund, L. Holm, E. L. Sonnhammer, S. R. Eddy and A. Bateman, Nucleic Acids Res., 2010, 38, D211–D222 CrossRef CAS PubMed.
- A. Stein, A. Ceol and P. Aloy, Nucleic Acids Res., 2011, 39, D718–D723 CrossRef CAS PubMed.
- R. D. Finn, M. Marshall and A. Bateman, Bioinformatics, 2005, 21, 410–412 CrossRef CAS PubMed.
- R. Mosca, A. Ceol and P. Aloy, Nat. Methods, 2013, 10, 47–53 CrossRef CAS PubMed.
- U. Pieper, B. M. Webb, D. T. Barkan, D. Schneidman-Duhovny, A. Schlessinger, H. Braberg, Z. Yang, E. C. Meng, E. F. Pettersen, C. C. Huang, R. S. Datta, P. Sampathkumar, M. S. Madhusudhan, K. Sjolander, T. E. Ferrin, S. K. Burley and A. Sali, Nucleic Acids Res., 2011, 39, D465–D474 CrossRef CAS PubMed.
- P. M. Kim, L. J. Lu, Y. Xia and M. B. Gerstein, Science, 2006, 314, 1938–1941 CrossRef CAS PubMed.
- B. Schuster-Bockler and A. Bateman, Genome Biol., 2008, 9, R9 CrossRef PubMed.
- C. Prieto and J. De Las Rivas, Proteins, 2010, 78, 109–117 CrossRef CAS PubMed.
- E. A. Franzosa and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10538–10543 CrossRef CAS PubMed.
- X. Wang, X. Wei, B. Thijssen, J. Das, S. M. Lipkin and H. Yu, Nat. Biotechnol., 2012, 30, 159–164 CrossRef CAS PubMed.
- M. J. Meyer, J. Das, X. Wang and H. Yu, Bioinformatics, 2013, 29, 1577–1579 CrossRef CAS PubMed.
- M. Meier, M. Janosik, V. Kery, J. P. Kraus and P. Burkhard, EMBO J., 2001, 20, 3910–3916 CrossRef CAS PubMed.
- D. Devos and R. B. Russell, Curr. Opin. Struct. Biol., 2007, 17, 370–377 CrossRef CAS PubMed.
- R. Mosca, T. Pons, A. Ceol, A. Valencia and P. Aloy, Curr. Opin. Struct. Biol., 2013 DOI:10.1016/j.sbi.2013.07.005 [Epub ahead of print].
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |