 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In silico modeling and evaluation of Gordonia alkanivorans for biodesulfurization†
Shilpi Aggarwal, I. A. Karimi* and Gregorius Reinaldi Ivan
Department of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117576, Singapore. E-mail: cheiak@nus.edu.sg; Fax: +65-6779-1936; Tel: +65-6516-6359
First published on 3rd July 2013
Abstract
The genus Gordonia is well known for its catabolic diversity and ability to transform several compounds including the various recalcitrant polyaromatic sulfur heterocycles (PASHs) found in the fossil fuels. In fact, some strains offer the unique ability to desulfurize even benzothiophene (BT) and other thiophenic compounds, which most of the commonly studied rhodococci strains cannot. In this work, we present the first genome scale metabolic model for G. alkanivorans, a desulfurizing strain, to enable a holistic study of its metabolism and comparison with R. erythropolis. Our model consists of 881 unique metabolites and 922 reactions associated with 568 ORFs/genes and 544 unique enzymes. It successfully predicts the growth rates from experimental studies and quantitatively elucidates the pathways for the desulfurization of the commonly studied sulfur compounds, namely dibenzothiophene (DBT) and benzothiophene (BT). Using our model, we identify the minimal media for G. alkanivorans, and show the significant effect of carbon sources on desulfurization with ethanol as the best source. Our model shows that the sulfur-containing amino acids such as cysteine and methionine decrease desulfurization activity, and G. alkanivorans prefers BT over DBT as a sulfur source. It also suggests that this preference may be driven by the lower NADH requirements for BT metabolism rather than the higher affinity of the transport system for BT. Our in silico comparison of R. erythropolis and G. alkanivorans suggests the latter to be a better desulfurizing strain due to its versatility for both BT and DBT, higher desulfurization activity, and higher growth rate.
Introduction
The combustion of fossil fuels releases various pollutants such as the oxides of carbon (COx), nitrogen (NOx), and sulfur (SOx). SOx, in particular, have attracted increasingly stringent regulations due to their harmful effects on the environment and human health.1 Desulfurization is a key step in the pre-processing of fossil fuels to achieve compliance with these regulations. At present, hydrodesulfurization is the most common method for desulfurization. In addition to being expensive and energy-intensive, it requires extreme conditions of temperature and pressure to remove sulfur from certain recalcitrant compounds such as dibenzothiophene (DBT), benzothiophene (BT), and their derivatives.2 Furthermore, being non-specific in action, it leads to the undesirable hydrogenation of certain non-sulfur aromatic compounds that contribute to the lubricating property and thermal stability of the fuels. Therefore, there is a need for developing efficient, specific, and economical desulfurization methods. To this end, biodesulfurization is considered a prospective alternative.Biodesulfurization employs whole microbes or their enzymes as catalysts to remove the sulfur atom selectively from the various recalcitrant compounds present in the fossil fuels.1 Several strains of Pseudomonas, Rhodococcus, Mycobacterium, Gordonia, etc. have been studied for their ability to metabolize various polyaromatic sulfur heterocycles (PASHs) present in fossil fuels. Most desulfurization studies in the literature have used DBT as the model compound. While several rhodococci strains exhibit non-destructive desulfurization of DBT, R. erythropolis IGTS8 was the first to be identified3 and has received the most attention. However, most rhodococci are unable to show high activity for the alkyl derivatives of DBT and show no activity for BT and other thiophenic compounds. Furthermore, there are only limited biochemical and genetic studies of bacteria that exhibit desulfurization of both DBT and BT.4,5 Since fossil fuels do contain these compounds in significant amounts, it is critical to study microbes that possess activity for compounds other than DBT. Furthermore, because desulfurizing these compounds requires distinct pathways, bacterial strains that possess the associated genes for all these pathways are clearly desirable. Gordonia is an attractive genus in this regard, because its members exhibit much metabolic versatility.6
Numerous Gordonia strains exhibit higher desulfurization activities5,6 than the rhodococci and for a broader range of PASHs.5,7–11 Of them, G. alkanivorans5,7,12,13 desulfurizes DBT via the well-known 4S pathway14 that non-destructively eliminates the sulfur atom from DBT with the concomitant release of 2-hydroxybiphenyl (HBP), the sulfur free compound. The 4S pathway in G. alkanivorans is conferred by three genes namely dszA, dszB, and dszC.7 The dszABC genes of G. alkanivorans are highly similar to those from R. erythropolis. However, besides DBT, it can also specifically cleave the C–S bond in BT and other thiophenes. Because of its ability to desulfurize a wider range of PASHs, G. alkanivorans appears to offer some advantage over R. erythropolis for biodesulfurization. Moreover, G. alkanivorans strains are reported15 to show nearly 2–10 times higher desulfurization activities than the desulfurizing R. erythropolis strains. In other words, it has the greater ability to reduce the overall sulfur content of the fossil fuels.
In spite of its promise, desulfurization studies with G. alkanivorans are far more limited than those with R. erythropolis. Although it does offer higher desulfurizing activity than R. erythropolis, the activity levels are still not acceptable for commercial application. Thus, there is a need to identify and study the factors and host functions that may play key roles in controlling the extent of desulfurization by G. alkanivorans. However, the complexity of metabolic networks makes it difficult to predict or identify such host functions intuitively or using a trial-and-error experimental approach. Since cellular activities are invariably and intricately coupled, a holistic study of the various metabolic functions occurring within G. alkanivorans besides the desulfurization of PASHs is essential to understand the interactions between the various components of its metabolic network. Such a study would also allow one to compare Gordonia strains with rhodococci in a theoretical and comprehensive manner. However, no such holistic study on Gordonia exists in the literature.
Constraint-based metabolic models16 have successfully been used to perform such holistic studies to elucidate relationships among various metabolic activities both qualitatively and quantitatively. These models, constructed based on the genomic and biochemical information of an organism, clearly establish the correspondence between its gene(s), protein(s) and metabolic function(s). They are easier to build, as they require only stoichiometric rather than kinetic information about various metabolic reactions. Nevertheless, they provide an effective framework for studying genotype–phenotype relationships, interactions among various metabolic activities, and internal flux distributions associated with various metabolic activities under given environmental conditions. Such constraint-based genome-scale metabolic (GSM) models have been reconstructed and analyzed widely for several industrially important bacterial strains such as Escherichia coli,17,18R. erythropolis,19,20Saccharomyces cerevisae,21,22 and Zymomonas mobilis,23 and even mammalian cells such as mouse hydridoma.24,25 Once constructed, these models can be very useful in exploring the possible states and properties of the metabolic network of an organism.
This work reports the first in silico GSM model for G. alkanivorans. It covers the key metabolic pathways such as central metabolism, amino acids biosyntheses, nucleotide metabolism, and sulfur metabolism that describes the assimilation of sulfur into biomass. It can help in understanding the metabolic architecture of G. alkanivorans, and its host functions related to desulfurization. We validate the model using the available desulfurization and growth data in the literature,11 and use it to study the effects of various medium components such as carbon sources, amino acids, and vitamins on the desulfurization activity of G. alkanivorans. We assess the properties of its metabolic network such as flexibility and robustness using flux variability26 and gene essentiality analyses. Finally, we use flux sum analyses27 to study qualitatively and quantitatively the effect of intracellular metabolites on growth and desulfurization activity and propose several experimentally testable conditions and modifications that may help enhance the desulfurizing activity of G. alkanivorans.
Results and discussion
Reconstructed GSM of G. alkanivorans
Our initial draft model comprised 739 reactions. However, it failed to show any cell growth on substrates known to be utilized by G. alkanivorans. We found that nearly 60% of all precursor metabolites could not be synthesized by the draft model. For instance, the draft model was unable to produce amino acids such as methionine and histidine, although G. alkanivornas is known5 to synthesize them and survive without any external supply. Our draft model also showed zero growth with DBT and BT as sole sulfur sources, although G. alkanivorans is known5 to metabolize them. Therefore, we added relevant pathways and reactions based on the information available in the literature and databases.5,14,28,29 As mentioned earlier, G. alkanivorans offers the advantage of desulfurizing a wider range of PASHs such as the alkyl derivatives of BT, DBT, and thiophene. However, we could not include such alkyl derivatives in our model, as their metabolic pathways and intermediates are still unknown. Thus, there is a clear need for obtaining genetic and biochemical data for the associated metabolic pathways in G. alkanivorans.GapFind revealed 279 DEMs that could not be produced in the initial draft of our model, as they were disconnected from the rest of the network either upstream or downstream. Gapfill could identify the possible candidate reactions to restore the connectivity for only 140 of the 279 DEMs. We performed BLASTp analyses for assigning putative ORFs to the enzymes associated with these reactions. To ensure that we do not add reactions indiscriminately to our model, we used a high e cut-off of 10−30 to include only the reactions with a strong evidence of ORFs. We could locate ORFs for only 62 (∼22%) DEMs, thus we did not include other reactions.
Our final curated model consists of 881 unique metabolites and 922 reactions associated with 568 ORFs/genes and 544 unique enzymes. Of these 922 reactions, 67 account for the transport of various metabolites across the membrane, while the rest (855) account for intracellular metabolic activities. Table 1 lists the features of our GSM model. The BLASTp analyses identified possible annotations for 55 ORFs in G. alkanovirans, which are given in Table 2. However, the model still has 217 DEMs, which warrants further biochemistry studies.
| Features | Properties |
|---|---|
| Reactions in genome scale mode | 922 |
| No. of ORFs included | 568 |
| No. of enzymes included | 544 |
| Intracellular reactions | 855 |
| Transport reactions | 67 |
| Metabolites in genome scale model | 881 |
| Internal metabolites | 814 |
| External metabolites | 67 |
| E.C.No. | Enzyme name | Current annotation | NCBI accession | NE value |
|---|---|---|---|---|
| EC 1.1.1.103 | L-Threonine 3-dehydrogenase | Alcohol dehydrogenase | ZP_08767112.1 | 3.00 × 10−29 |
| EC 1.1.1.29 | Glycerate dehydrogenase | D-3-Phosphoglycerate dehydrogenase | ZP_08766341.1 | 1.00 × 10−20 |
| EC 1.1.1.81 | Hydroxypyruvate reductase | Putative oxidoreductase | ZP_08767993.1 | 3.00 × 10−22 |
| EC 1.2.7.6 | Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin) | Putative dehydrogenase | ZP_08767188.1 | 3.00 × 10−04 |
| EC 3.1.4.17 | 3′,5′-Cyclic-nucleotide phosphodiesterase | Putative LuxR family transcriptional regulator | ZP_08766296.1 | 3.00 × 10−12 |
| EC 3.2.1.122 | Maltose-6′-phosphate glucosidase | Molybdenum cofactor biosynthesis protein A | ZP_08767656.1 | 8.00 × 10−05 |
| EC 3.5.4.21 | Creatinine deaminase | Putative hydrolase | ZP_08765243.1 | 7.00 × 10−04 |
| EC 1.1.1.17 | Mannitol-1-phosphate 5-dehydrogenase | Putative phosphoribosylglycinamide formyltransferase 2 | ZP_08768014.1 | 0.0001 |
| EC 1.1.1.26 | Glyoxylate reductase | D-3-Phosphoglycetate dehydrogenase | ZP_08766341.1 | 1.00 × 10−42 |
| EC 1.1.1.36 | Acetoacetyl-CoA reductase | 3-Oxoacyl-[acyl-catrien-protein] reductase | ZP_08768232.1 | 6.00 × 10−44 |
| EC 1.1.1.60 | 2-Hydroxy-3-oxopropionate reductase | 3-Hydroxyisobutytate dehydrogenase | ZP_08765584.1 | 3 × 10−44 |
| EC 1.1.1.65 | Pyridoxine 4-dehydrogenase | Putative aldo/keto reductase | ZP_08764692.1 | 7 × 10−11 |
| EC 1.1.1.79 | Glyoxylate reductase (NADP+) | D-3-Phosphoglycerate dehydrogenase | ZP_08766341.1 | 1.00 × 10−42 |
| EC 1.1.1.83 | D-Malate dehydrogenase (decarboxylating) | 3-Isopropylmalate dehydrogenase | ZP_08766340.1 | 8.00 × 10−81 |
| EC 1.1.5.8 | Quinate dehydrogenase (quinone) | Putative non-ribosomal peptide synthetase | ZP_08767228.1 | 8.00 × 10−04 |
| EC 1.17.3.2 | Xanthine oxidase | Putative xanthine dehydrogenase | ZP_08766184.1 | 2.00 × 10−52 |
| EC 1.18.6.1 | Nitrogenase | Chromosome partitioning protein ParA | ZP_08768173.1 | 7.00 × 10−11 |
| EC 1.2.1.22 | Lactaldehyde dehydrogenase | Succinate-semialdehyde dehydrogenase | ZP_08765499.1 | 4.00 × 10−91 |
| EC 1.2.7.5 | Aldehyde ferredoxin oxidoreductase | Hypothetical protein GOALK_120_00670 | ZP_08768084.1 | 3.00 × 10−06 |
| EC 1.2.7.7 | 3-Methyl-2-oxobutarroate dehydrogenase (ferredoxin) | Putative oxidoreductase | ZP_08766668.1 | 3.00 × 10−05 |
| EC 1.21.4.3 | Sarcosine reductase | Putative acetyl-CoA acetyltransferase | ZP_08764801.1 | 7.00 × 10−05 |
| EC 1.3.1.78 | Arogenate dehydrogenase (NADP+) | Prephenate dehydrogenase & | ZP_08765663.1 | 2.00 × 10−26 |
| EC 1.3.99.10 | Isovaleryl-CoA dehydrogenase | Acyl-CoA dehydrogenase | ZP_08765489.1 | 4.00 × 10−79 |
| EC 1.4.3.21 | Primary-amine oxidase | Adenosylcobinamide kinase | ZP_08763719.1 | 0.0007 |
| EC 1.4.99.5 | Glycine dehydrogenase (cyanide-forming); | Putative ferredoxin reductase | ZP_08764700.1 | 2.00 × 10−09 |
| EC 1.7.2.2 | Nitrite reductase (cytochrome; ammonia-forming) | Ethanolamine ammonia-lyase large subunit | ZP_08767599.1 | 0.0002 |
| EC 1.7.7.2 | Ferredoxin-nitrate reductase | Putative nitrate/sulfite reductase | ZP_08764724.1 | 1.00 × 10−175 |
| EC 2.1.1.2 | Guanidinoacetate N-methyltransferase | Hypothetical protein GOALK_050_00300 | ZP_08765250.1 | 0.016 |
| EC 2.1.3.1 | Methylmalonyl-CoA carboxytransferase | Pyruvate carboxylase | ZP_08766189.1 | 5.00 × 10−13 |
| EC 2.1.4.1 | Glycine amidinotransferase | Isocitrate lyase | ZP_08765259.1 | 9.00 × 10−04 |
| EC 2.3.1.182 | (R)-Citramalate synthase | Putative 4-hydroxy-2-oxovalerate aldolase | ZP_08765376.1 | 1.00 × 10−13 |
| EC 2.3.3.10 | Hydroxymethylglutatyl-CoA synthase | 3-Oxoacyl-[acyl-carrier-protein] synthase III | ZP_08764811.1 | 6.00 × 10−07 |
| EC 2.4.2.28 | S-Methyl-5′-thioadenosine phosphorylase | Purine nucleoside phosphorylase | ZP_08766163.1 | 6.00 × 10−17 |
| EC 2.5.1.82 | Hexaphenyl diphosphate synthase [geranylgeranyl-diphosphate specific] | Putative polyprenyl diphosphate synthase | ZP_08765134.1 | 2 × 10−33 |
| EC 2.5.1.83 | Hexaphenyl-diphosphate synthase [(2E,6E)-farnesyl-diphosphate specific] | Putative polyprenyl diphosphate synthase | ZP_08765134.1 | 2.00 × 10−33 |
| EC 2.5.1.84 | All-trans-nonaphenyl-diphosphate synthase [geranyl-diphosphate specific] | Putative polyprenyl diphosphate synthase | ZP_08765134.1 | 3.00 × 10−42 |
| EC 2.7.1.100 | S-Methyl-5-thioribose kinase | Hypothetical protein | ZP_08767309.1 | 3.00 × 10−06 |
| EC 2.7.1.48 | Uridine kinase | Uracil phosphoribosyltransferase | ZP_08766161.1 | 2.00 × 10−18 |
| EC 3.1.1.17 | Gluconolactonase | Hypothetical protein | ZP_08765725.1 | 6.00 × 10−12 |
| EC 3.1.2.4 | 3-Hydroxyisobutyryl-CoA hydrolase | Hypothetical protein | ZP_08764807.1 | 3.00 × 10−66 |
| EC 3.2.1.93 | Alpha,alpha-phosphotrehalase | Alpha-glucosidase | ZP_08767019.1 | 3.00 × 10−82 |
| EC 3.2.2.1 | Purine nucleosidase | Putative ribonucleoside hydrolase | ZP_08767439.1 | 4.00 × 10−25 |
| EC 3.5.1.59 | N-Carbamoylsarcosine amidase | Putative hydrolase | ZP_08765823.1 | 200 × 10−40 |
| EC 3.5.2.10 | Creatininase | Putative creatininase family protein | ZP_08767265.1 | 7 × 10−18 |
| EC 3.5.2.15 | Cyanuric acid amidohydrolase | Hypothetical protein | ZP_08768158.1 | 0.006 |
| EC 3.5.3.9 | Allantoate deiminase | Putative M20D family peptidase | ZP_08766098.1 | 1.00 × 10−08 |
| EC 3.5.4.1 | Cytosine deaminase | Putative cytosine deaminase | ZP_08764308.1 | 2.00 × 10−67 |
| EC 3.5.4.12 | dCMP deaminase | tRNA-specific adenosine deaminase | ZP_08765661.1 | 2.00 × 10−19 |
| EC 3.5.5.1 | Nitrilase | Putative carbon–nitrogen hydrolase | ZP_08767356.1 | 1.00 × 10−11 |
| EC 4.1.2.20 | 2-Dehydro-3-deoxygluconate aldolase | Putative citrate lyase beta subunit | ZP_08765089.1 | 1.00 × 10−05 |
| EC 4.2.1.66 | Cyanide hydratase | Putative amidohydrolase | ZP_08767351.1 | 4.00 × 10−10 |
| EC 4.2.1.84 | Nitrile hydratase | Thiocyanate hydrolase gamma subunit | ZP_08768164.1 | 2 × 10−48 |
| EC 5.1.3.6 | UDP-glucuronate 4-epimerase | UDP-glucose 4-epimerase | ZP_08763819.1 | 2 × 10−17 |
| EC 6.2.1.25 | Benzoate-CoA ligase | Putative fatty-acid-CoA ligase | ZP_08763669.1 | 1.00 × 10−60 |
| EC 6.2.1.4 | Succinate-CoA ligase (GDP-forming) | Succinyl-CoA synthetase beta subunit | ZP_08766733.1 | 2.00 × 10−70 |
Model validation
We mimicked the experiments of Rhee et al.11 and compared the growth rates predicted by our model with their experimental values. From their experiments, we inferred glucose to be limiting, as its complete depletion from the medium triggered the stationary phase. So we computed and used specific uptake rates for glucose at several time points to constrain our model. We assumed unlimited supply for other medium components, as they were in excess. As seen in Fig. 1, our predicted biomass growth rates are in close agreement with the experimental data of Rhee et al.11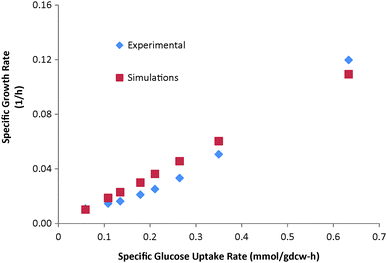 | ||
| Fig. 1 Experimental and simulated growth rates at various glucose uptake rates from Rhee et al.11 | ||
Minimal media and growth
Gordonia strains are known for their metabolic versatility6 and demand no special nutrients to the best of our knowledge. Thus, we infer that they can synthesize all the necessary precursor metabolites from simple sources in a medium. However, it is desirable to identify the essential nutrients and their alternatives based on our model.Rhee et al.11 used a rich medium (as described in Materials and methods) to study the growth of G. alkanivorans with DBT as the sole sulfur source. Using our model, we simulated growth by removing one nutrient at a time from the rich medium of Rhee et al.11 From that, we identified glucose, oxygen, an ammonium salt, a phosphorus source, and DBT to comprise the minimal medium. As alternatives, we identified BT, cysteine, and sulfate for sulfur, and glutamate for both carbon and nitrogen.
Iida et al.30 experimented with 31 carbon substrates. We used our in silico model to simulate their experiments and detect cell growth on these 31 sources. In each simulation, we specified 1 mmol per gdcw per h uptake of a different substrate as the sole carbon source along with the minimal media and maximized cell growth. Table 3 compares our model predictions with the observations of Iida et al.30 Our model predicts growth correctly for 16 of the 31 substrates. We observe both false positive and false negative results for the remaining 15 substrates. In the former, our model shows false growth, while in the latter, it fails to show growth. These errors arise, because the biochemical information on G. alkanivorans is still incomplete. Further work in this regard is warranted. Since our model lacks regulatory mechanisms, this is another source of error. However, model predictions in this case may be improved by incorporating regulatory information.
| Carbon source | Experimental utilization | In silico utilization |
|---|---|---|
| D-Galactose | + | − |
| L-Rhamnose | − | − |
| D-Ribose | + | + |
| Sucrose | + | + |
| Turanose | + | − |
| Arabitol | + | − |
| Inositol | + | + |
| Glucarate | + | − |
| Gluconate | + | + |
| D-Glucosaminic acid | + | + |
| Caprate | + | − |
| Citrate | + | + |
| 4-Aminobutyrate | − | + |
| 2-Hydroxyvalerate | + | − |
| 2-Oxoglutarate | + | + |
| Pimelate | + | + |
| Succinate | + | + |
| Benzoate | + | − |
| 3-Hydroxybenzoate | + | − |
| 4-Hydroxybenzoate | + | − |
| Phenylacetate | + | − |
| Quinate | + | − |
| L-Alanine | + | + |
| L-Aspartate | + | + |
| L-Leucine | + | − |
| L-Proline | + | + |
| L-Serine | − | + |
| L-Valine | − | − |
| Putrescine | + | + |
| Tyramine | + | − |
| Acetamide | − | − |
Gene essentiality
Most cells can withstand disturbances at the genetic as well as metabolic levels by utilizing alternative genes, enzymes, and pathways depending on their environment. However, the non-functionality of certain reactions and genes may be lethal for a cell. We studied the robustness of G. alkanivorans metabolism by assessing its ability to exhibit in silico growth in the case of gene knockouts or mutations. The utilization of a pathway, and thus the essentiality of its reactions and genes, will in general depend on medium components. We used five minimal media with alternative carbon sources of ethanol, fumarate, oxoglutarate, pyruvate, and glutamate, and evaluated the essentialities of genes and reactions for each medium. For reactions, we removed one reaction at a time by setting its flux as zero, and maximized cell growth on minimal medium. If the model could not produce cell mass, then we classified the reaction as essential, and vice versa. Similarly, for genes, if the removal of a gene prevented cell growth, then we classified that gene as essential, and vice versa. To remove a gene, we set the fluxes of all its associated reactions to zero in our in silico model. However, if a reaction was controlled by two or more isozymes, then the reaction was kept active in the absence of any one of the associated genes.We identified 116 reactions and 75 genes to be essential irrespective of the medium. As seen in Fig. 2, most essential reactions belong to the amino acids metabolism followed by nucleotides metabolism, central metabolism, and cell wall metabolism. Any reduction in their activity levels may reduce growth or prove lethal for G. alkanivorans. The difference in the numbers of essential reactions and essential genes is due to isozymes, as several reactions are catalyzed by enzymes that multiple genes encode. These reactions are essential at the metabolic level, but not the genetic level.
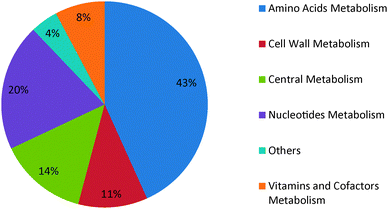 | ||
| Fig. 2 Distribution of essential reactions over various cellular subsystems in G. alkanivorans. | ||
Flux variability
Metabolic network models normally exhibit multiple alternative solutions for flux distributions. Flux variability analysis (FVA26) allows us to study the range over which a given flux can vary in alternative solutions. We performed an FVA on our model for an ethanol uptake of 10 mmol per gdcw per h and unrestricted supplies of other essential nutrients. For the maximum cell growth, we computed the minimum and maximum possible fluxes for each reaction. Nearly 85% of the reactions showed significant flux variations. The remaining 15% represent the inflexible parts of the metabolism for growth, which mainly include the biosynthesis pathways for amino acids and nucleotides. We also observed that the non-oxidative part of the pentose phosphate pathway (PPP) showed no flux variation and operated in the reverse manner, which highlights the importance of PPP for growth. Besides replenishing NADPH, PPP also produces sugars for the synthesis of nucleotides. In this case, the non-oxidative part operates in the reverse to supply ribose-5-phosphate for nucleotide biosynthesis. This is in agreement with the role of non-oxidative part of PPP for the synthesis of ribose-5-phospate for nucleotides.31 Since the nucleotide biosynthesis pathways show fixed fluxes, we can infer that their fluxes are coupled with those of PPP.Metabolite essentiality
Metabolites are the key players in cellular metabolism. Even if the net accumulation of an internal metabolite may be zero, its overall activity (consumption/generation) level inside the cell is a key indicator of its importance. FSA27 attempts to determine this level of activity for a metabolite, and the effect of changes in that level on cellular phenotypes.We studied the growth of G. alkanivorans on ethanol using our model. For a fixed uptake of 10 mmol per gdcw per h and maximum growth, we computed the base flux sum for each metabolite. Of the 814 internal metabolites, only 34% had a positive flux sum, while the remaining 66% had no activity. The former were mainly the cofactors essential for growth. When we maximized the flux sums under maximum growth, nearly 26% of the internal metabolites showed no activity. These were the dead-end metabolites that could not be eliminated from the model using the available data, observations, and procedures. Some metabolites (∼10%) showed infinite flux sum due to the presence of cycles27 in our metabolic network. As expected, cofactors showed consistent activity. When we minimized the flux sums, 24% of the metabolites seemed essential for growth. They are mostly associated with the essential reactions identified in previous studies.
We also repeated the flux sum analysis to study the effects of various metabolites on the desulfurization of DBT and BT. The desulfurization of DBT (BT) varies linearly with the flux sums of the intermediate metabolites in the 4S (BT metabolism) pathway. Thus, any attenuation in these flux sums would reduce the desulfurization exhibited by G. alkanivorans. In addition to these intermediates, NADH, oxygen, and ferricytochrome c are essential for DBT (BT) desulfurization.
Impact of media on desulfurization
Studies32–35 have shown that medium composition has a prominent effect on the desulfurizing activity of R. erythropolis. However, no such studies exist for G. alkanivorans. In the absence of such experimental work, our in silico model helps shed some light on the impact of medium components such as carbon sources, amino acids, and vitamins on the desulfurizing activities of G. alkanivorans.Carbon sources
Biodesulfurization is considered a potential alternative to the conventional hydro-desulfurization process, as it may reduce costs significantly.1 In biodesulfurization, an effective but inexpensive carbon source is required for cell growth.36 We evaluated 17 carbon sources (acetate, citrate, ethanol, formate, fructose, fumarate, gluconate, glucose, glutamate, glycerol, lactate, malate, oxaloacetate, oxoglutarate, pyruvate, and succinate). We performed 17 simulations with one carbon source at a time. Assuming an uptake of 20 mg per gdcw per h for each source, we predicted the maximum cell growth rate and corresponding desulfurization activity. Ethanol appears (Fig. 3) to be the best for both growth and desulfurization. The carbon sources as compared to ethanol (growth of 0.027 h−1 and desulfurization of 3.3 μmol HBP per gdcw per h) have the 100% basis rank as follows: fumarate (80%) > oxoglutarate (78.79%) > pyruvate (78.43%) > glutamate (78.24%) > succinate (78%) > acetate ≈ fructose ≈ glucose ≈ lactate (76.86%) > glycerol (75%) > citrate (71.88%) > oxaloacetate (69.70%) > malate (69.11%) > formate (50%). This ranking remained unchanged even for BT as the sole sulfur source.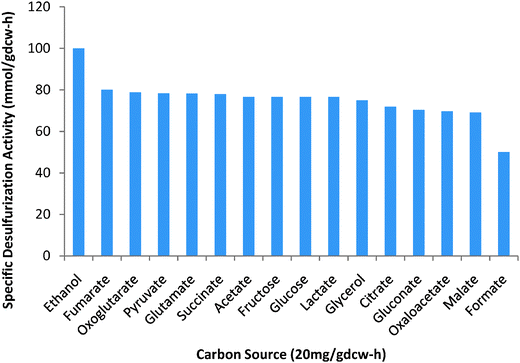 | ||
| Fig. 3 Specific desulfurizing activities for an uptake rate of 20 mg per gdcw per h of various carbon sources. | ||
As discussed by Aggarwal et al.,20 NADH production and usage could explain why ethanol is the best. For the cell to consume 1 mol DBT as a sulfur source via the 4S pathway requires 4 moles of NADH. Additionally, NADH is required for other growth related activities. The carbon nutrient is the main source of this energy. It affects the cofactor regeneration in cellular metabolism. Therefore, a carbon source that provides more NADH during its metabolism is likely to support higher desulfurization and growth. One mole of ethanol generates two additional moles of NADH. This is the highest among all 17 substrates, and thus it seems to be the best substrate for both growth and desulfurization.
Vitamins and amino acids
Yan et al.35 observed that the addition of nicotinamide and riboflavin improved desulfurization by R. erythropolis significantly. No such studies exist for G. alkanivorans. Therefore, we studied the impact of nicotinamide, riboflavin, and the twenty amino acids on the desulfurizing activity of G. alkanivorans using our model. Using a fixed uptake (1 mmol per gdcw per h) for glucose and unlimited uptake of other medium components, we obtained a growth rate of 0.17 h−1 and a desulfurization rate of 0.021 mmol per gdcw per h. Then, we added nicotinamide, riboflavin, and amino acids individually at 1 mmol per gdcw per h uptake and simulated growth again. Nicotinamide and riboflavin affected neither growth nor desulfurization. It is possible that they act at the regulatory or transcriptional rather than the metabolic level as hypothesized by Yan et al.35 Since our model does not include these effects, it is unable to show the impact of nicotinamide and riboflavin.In contrast, some amino acids did affect the growth of and desulfurization by G. alkanivorans. Fig. 4 shows the relative effects of various amino acids on desulfurization. Arginine, histidine, isoleucine, leucine, lysine, phenylalanine, tryptophan, tyrosine, and valine affected neither growth nor desulfurization. In contrast, cysteine and methionine had strong effects on desulfurization. While no desulfurization occurred in the presence of cysteine, it was reduced by 63% in the presence of methionine.
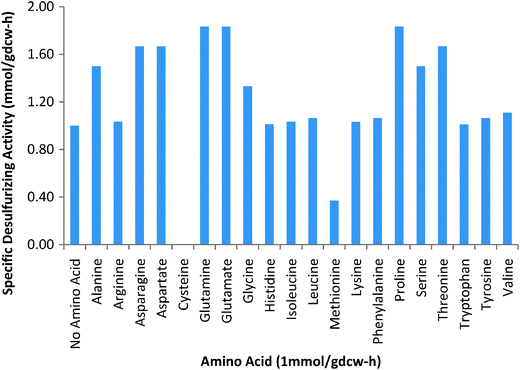 | ||
| Fig. 4 Specific desulfurizing activities for an uptake rate of 1 mmol per gdcw per h of various amino acids. | ||
The effect of cysteine is similar to what Aggarwal et al.20 showed for R. erythropolis, and we can explain as follows. Like R. erythropolis, G. alkanivorans can use cysteine as a sole sulfur source. Using cysteine is energetically less expensive than DBT, as 1 mole DBT requires additional 4 moles of NADH.20 Therefore, the cell prefers to consume cysteine rather than DBT, and no DBT desulfurization occurs.
The reduced desulfurization in the presence of methionine may be due to the inability of G. alkanivorans to produce all the sulfur-containing metabolic precursors solely from methionine. For instance, they cannot produce cysteine, L-homocysteine, coenzyme A, etc. solely from methionine, and hence need an additional sulfur source such as DBT or BT. While this may be real, but no evidence exists in the literature, it may well be a gap in our model, which prevents the use of methionine as a sole sulfur source. As with cysteine, the use of methionine is energetically more favourable than DBT. Thus, the cell uses it as much as possible first before using DBT, lowering desulfurization. This is different from what Aggarwal et al.20 observed for R. erythropolis.
Alanine, asparagine, aspartate, glutamine, glutamate, glycine, proline, serine, and threonine improved growth and desulfurization greatly. These, in contrast to cysteine and methionine, can serve as sole carbon sources as well. Thus, they supplement glucose and promote higher growth and cofactor regeneration. Since sulfur is essential for growth, higher growth leads to greater sulfur usage and higher desulfurization.
Desulfurization of BT and DBT
Several PASHs such as the thiophenic compounds (BT, DBT and their derivatives) in fossil fuels are usually recalcitrant to hydrodesulfurization. Most studies use DBT as the model sulfur compound representative of most of these PASHs. However, BT is not utilized by several DBT-desulfurizing strains,11,28,37 as it may be desulfurized via a different pathway and genes. Since G. alkanivorans can utilize both BT and DBT as sole sulfur sources,5 it is critical to study the desulfurization of BT as well. Alves et al.5 studied the desulfurization of BT and DBT, when supplied simultaneously in a glucose-based medium to G. alkanivorans. They found that BT was consumed preferentially before DBT. They opined that this was probably due to the presence of a non-specific uptake system for thiophenic compounds with a higher affinity for BT than DBT. We used our model to study this. We assumed a fixed uptake of glucose (20 mg per gdcw per h) with unlimited supplies of BT and DBT, and maximized biomass growth. The model consumed BT only with a growth rate of 0.021 h−1 and a desulfurization rate of 2.55 μmol per gdcw per h. This corresponds to the minimum in silico sulfur requirement of the cell for maximum growth at a glucose uptake of 20 mg per gdcw per h. In subsequent simulations, we gradually reduced the uptake of BT from 2.55 μmol per gdcw per h to zero. As expected, the DBT uptake gradually increased (Fig. 5) to meet the cellular demand of sulfur. Clearly, our model is in agreement with the experimental results of Alves et al.5 Since our model has no regulatory mechanism, and its uptake system has no bias for DBT or BT, we may rule out the non-specific uptake factors for explaining the preference of BT over DBT. Instead, energy usage offers a plausible explanation. 1 mole BT requires 2 moles NADH, while 1 mole DBT requires 4 moles NADH. Thus, for the cell, BT is a better sulfur source than DBT energetically.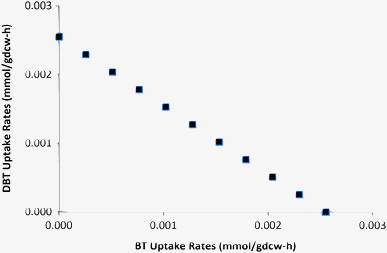 | ||
| Fig. 5 Effect of increasing the BT uptake rate on specific DBT desulfurization. | ||
Next, we examined the effects of BT and DBT on growth. We performed two simulations. In the first, we provided BT, and in the second, we provided DBT as the sole sulfur source. For both cases, we fixed their uptake rates at 20 mg per gdcw per h with unlimited supply of glucose and other nutrients. The maximum growth rate was 1.24 h−1 with BT, and 0.90 h−1 with DBT. Thus, BT promotes higher growth than DBT. This can also be explained by the lower energy requirements of BT as mentioned in the previous paragraph.
Comparison with R. erythropolis model
As mentioned earlier, R. erythropolis is the most widely studied bacteria for desulfurization, but G. alkanivorans shows desulfurization activity for a wider range of PASHs. However, it would be desirable to compare these two bacteria under the same conditions. For this, we studied the desulfurization of DBT and BT individually by the existing genome scale metabolic model of R. erythropolis20 and our reconstructed model of G. alkanivorans. We maximized cell growth for a fixed glucose uptake of 1 mmol per gdcw per h with unlimited supplies of DBT/BT and other minimal nutrients. Both R. erythropolis and G. alkanivorans can utilize DBT as the sole sulfur source and give out HBP as the desulfurized product in the medium. However, G. alkanivorans can utilize BT, but R. erythropolis cannot. This suggests that G. alkanivorans can indeed be a more suitable biocatalyst for desulfurizing the fossil fuels, which normally have a spectrum of DBT, BT, and their derivatives.Next, we performed simulations to compare the desulfurizing activities of G. alkanivorans and R. erythropolis. We maximized biomass for 1 mmol per gdcw per h uptake of glucose as the sole carbon source and unlimited supply of DBT as the sole sulfur source. We observed that the growth rate and the corresponding desulfurizing activity were higher for G. alkanivorans (0.15 h−1, 18.13 μmol HBP per gdcw per h) than R. erythropolis (0.14 h−1, 13.54 μmol HBP per gdcw per h). Note that the desulfurization activity exhibited by the two strains increases with the increase in glucose uptake rates as shown in Fig. 6. However, for any fixed value of glucose uptake, the desulfurization activity observed with G. alkanivorans is higher than that with R. erythropolis.
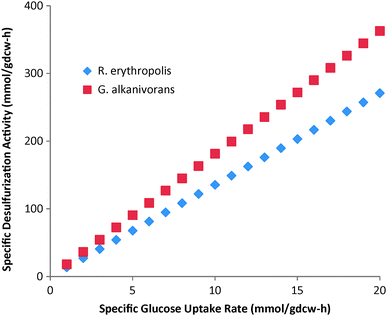 | ||
| Fig. 6 Effect of specific glucose uptake rates on specific desulfurizing activity of G. alkanivorans and R. erythropolis. | ||
We then used the two models to compute the minimum sulfur requirements (in terms of DBT) of the two strains for a unit growth rate. Supplying all the nutrients in excess, we minimized the DBT uptake for a fixed biomass growth rate of 1 h−1. G. alkanivorans needed 120 μmol per gdcw per h of DBT versus 93.90 μmol per gdcw per h for R. erythropolis. These analyses show that G. alkanivorans possesses higher desulfurization activity than R. erythropolis under the same medium conditions, thus it is likely to be a better catalyst for biodesulfurization.
Materials and methods
Model reconstruction
The reconstruction of a GSM model for an organism requires the identification and classification of its metabolite reactions and the establishment of their appropriate gene–protein-reaction (GPR) associations. It is an iterative process involving the collection and processing of diverse information about cellular metabolism, biochemistry, and various strain-specific parameters of the organism.38 We reconstructed the GSM model of G. alkanivorans in three steps: (i) constructing an initial draft model based on genome annotations, (ii) model improvement to enable cell growth, and (iii) model analysis for identifying and filling network gaps based on biochemical information.For reconstructing an initial draft model of G. alkanivorans, we annotated the genome sequence of G. alkanivorans using the tools available on the online annotation server RAST.39 We manually processed this information to establish the GPR associations and assign appropriate gene(s) to the various enzymes and their corresponding reactions in the metabolic network. We also checked all reactions for elemental balancing. Then, we cross-checked the GPR associations and the reaction directionality with the information available for G. alkanivorans in KEGG40 and MetaCyc.41 We incorporated any additional reactions or pathways that were available in MetaCyc and KEGG. We removed all the reactions that accounted for the polymerization of monomers and conversion of general class compounds such as ROH, RCOOH, etc.
After this, we identified several broken pathways, dead end metabolites (DEMs), and missing reactions in the model, which arise mainly due to the lack of metabolite connectivity and presence of gaps in the network.42 To complete and enhance our model, we employed several means. First, we looked for additional reactions based on the literature evidence and other biochemical information. Second, we used optimization-based automated procedures of GapFill and GapFind, proposed by Kumar et al.,42 to identify and restore the connectivity of the DEMs and to identify and fill the remaining network gaps. We used GAMS/CPLEX 10.043 to execute these procedures and systematically determine and eliminate these network gaps by restoring the connectivity within the metabolic network. All these required adding new reactions into the model, for which no genetic evidence is currently available. Therefore, we tried to identify and assign possible ORFs that may potentially encode for these missing functions. For this, we performed BLASTp searches between the translated set of genes associated with these additional reactions in various databases and the genome of G. alkanivorans. While we used a high e cut-off of 10−30 for most network improvement reactions, we used a low cut-off of 10−5 for some reactions to enable the essential activity of biomass generation.
Desulfurization pathway for BT
DBT is metabolized by G. alkanivorans via the widely studied ‘4S’ pathway.1,20 However, BT is utilized via a distinct pathway as shown in Fig. 7. Limited work28,37 exists on this BT-desulphurization pathway, and it does not provide any details on the stoichiometry and associated enzymology. Therefore, we took the detailed reactions from the UM-BBD database.29 As shown in Fig. 7, BT is desulfurized via a 4-step process. In the first two steps, BT is oxidized via the action of benzothiophene monooxygenase to benzothiophene-S,S-dioxide with benzothiophene-S-oxide as the intermediate. Then, benzothiophene-S,S-dioxide is further oxidized to (Z)-2-(2-hydroxyphenyl)ethenesulfinate by the benzothiophene-S,S-dioxide monooxygenase. Finally, (Z)-2-(2-hydroxyphenyl) ethenesulfinate is desulfurized to o-hydroxystyrene through the specific cleavage of the C–S bond by 2-(2′-hydroxyphenyl)benzene sulfinate desulfinase. The resulting sulfur atom is released as a sulfite moiety.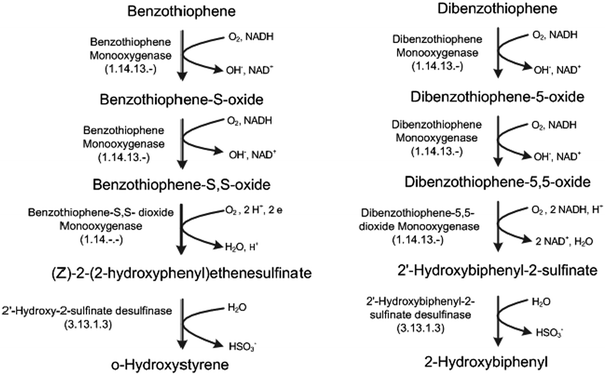 | ||
| Fig. 7 Pathways for desulfurization of benzothiophene (BT) and dibenzothiophene (DBT). | ||
As we can see from Fig. 7, the two pathways (4S vs. BT-desulfurization) use different enzymes. Not only this, they have different energy requirements in terms of reducing equivalents. The BT-desulfurization requires 2 moles of NADH per mole of BT, while the ‘4S’ pathway requires 4 moles of NADH per mole of DBT. Therefore, the former seems to be more energy-efficient than the latter.
Experimental studies
We used the experimental data of Rhee et al.11 for validating our in silico model. Rhee et al.11 isolated and studied DBT desulfurization characteristics of Gordonia sp. CYSK1. They used a minimal salt medium (MSM) consisting of 5.0 g glucose, 5.0 g K2HPO4, 1.0 g NaH2PO4, 1.0 g NH4Cl, 0.2 g MgCl2, 0.01 g CaCl2·2H2O, 1 ml of sulfur-free trace element solution dissolved in EDTA, and 1 ml of vitamin solution. The MSM was supplemented with DBT dissolved in ethanol at a concentration of 100 mM. They cultured in 250 ml Erlenmeyer flasks containing 50 ml of MSM at 30 °C on a gyratory shaker. During the culture, they measured concentration profiles of glucose via reverse-phase HPLC, DBT and HBP via GC, and cell growth via absorbance at 600 nm. They observed that the cells gave HBP as the final desulfurized product and did not re-assimilate it as a carbon source.We used the experimental data of Iida et al.30 to study the utilization of various carbon sources. Iida et al.30 studied substrate utilization patterns of several Gordonia strains.
Model analyses
For analyzing and predicting phenotypes, the constraint-based GSM models typically solve the following linear optimization problem (Flux Balance Analysis or FBA), which is based on the assumption that intracellular metabolites are at pseudo steady state:where i represents metabolites (i = 1,2,…,I), j represents reactions (j = 1,2,…,J), Z represents an appropriate cellular objective, νj (−∞ < νLj ≤ νj ≤ νUj < ∞) is the flux of reaction j, bi (−∞ < bLi ≤ bi ≤ bUi < ∞) is the flux of metabolite i, bi = 0 for each internal metabolite i, bi > 0 for the release of an external metabolite, bi < 0 for the uptake of an external metabolite, and S (i × j) is the matrix of stoichiometric coefficients. The model can be constrained further by fixing the fluxes (bi) of one or more extracellular metabolites based on their experimentally measured uptake/release rates, setting realistic bounds on various fluxes, or demanding a known maintenance energy for the organism. The solution of the FBA model gives the possible flux distributions that may represent the metabolic state of a cell under given environmental conditions.
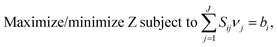
To solve the FBA model, we need a cellular objective (Z). Several cellular objectives such as maximum cell growth, minimum substrate utilization, minimum maintenance energy, etc.44 have been used in the literature. Cell growth is the most common, as microbial cells have evolved to maximize growth. It can be expressed as a synthetic reaction consuming multiple biomass precursor metabolites in some ratios, which can be determined from cell composition. In the absence of any data in the literature on the cellular composition of G. alkanivorans, we adapted information from the metabolic models of a related organism, Corynebacterium glutamicum.45,46 Such adaptation from related organisms is an established practice in the reconstruction of metabolic models.16 We used MetaFluxNet47 and GAMS/CPLEX 10.043 to solve and analyze our FBA model.
Conclusion
We have presented the first genome scale metabolic model for G. alkanivorans. We have identified 55 new genome annotations to explain some functionalities missing from its current genome annotations. Our model successfully predicts and explains the limited experimental observations reported in the literature. It suggests that ethanol may be the best carbon source among the sixteen studied in this work. Also, the model analyses show that the presence of sulfur containing amino acids, cysteine and methionine in the medium, can reduce the desulfurization activity of G. alkanivorans. Our model also confirms the experimental observations that BT is preferentially desulfurized over DBT by G. alkanivorans. However, it suggests that BT's lower energy requirements in terms of NADH rather than specific uptake mechanisms may be a better explanation for this preference. Further, our analyses show that NADH plays an important role in desulfurization, thus re-engineering G. alkanivorans for improved supply/regeneration of NADH is likely to increase desulfurization. Our model appropriately captures the inter-relationships between the various metabolic activities occurring within G. alkanivorans and can be used to study other properties of its metabolic network and devise metabolic engineering strategies for obtaining improved strains. Our comparative study of R. erythropolis and G. alkanivorans suggests that G. alkanivorans can be a better desulfurizing strain as it can desulfurize both BT and DBT, and also exhibits higher desulfurization activity.References
- M. Soleimani, A. Bassi and A. Margaritis, Biotechnol. Adv., 2007, 25, 570–596 CrossRef CAS.
- C. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- J. J. Kilbane II and K. Jackowski, Biotechnol. Bioeng., 1992, 40, 1107–1114 CrossRef.
- K. J. Kayser, L. Cleveland, H. S. Park, J. H. Kwak, A. Kolhatkar and J. J. Kilbane II, Appl. Microbiol. Biotechnol., 2002, 59, 737–745 CrossRef CAS.
- L. Alves, R. Salgueiro, C. Rodrigues, E. Mesquita, J. Matos and F. M. Gírio, Appl. Biochem. Biotechnol., 2005, 120, 199–208 CrossRef CAS.
- M. Arenskötter, D. Bröker and A. Steinbüchel, Appl. Environ. Microbiol., 2004, 70, 3195–3204 CrossRef.
- L. Alves, M. Melo, D. Mendonça, F. Simões, J. Matos, R. Tenreiro and F. M. Gírio, Enzyme Microb. Technol., 2007, 40, 1598–1603 CrossRef CAS.
- J. J. Kilbane II and J. Robbins, Appl. Microbiol. Biotechnol., 2007, 75, 843–851 CrossRef.
- S. C. C. Santos, D. S. Alviano, C. S. Alviano, M. Pádula, A. C. Leitão, O. B. Martins, C. M. S. Ribeiro, M. Y. M. Sassaki, C. P. S. Matta, J. Bevilaqua, G. V. Sebastián and L. Seldin, Appl. Microbiol. Biotechnol., 2006, 71, 355–362 CrossRef CAS.
- G. Q. Li, S. S. Li, S. W. Qu, Q. K. Liu, T. Ma, L. Zhu, F. L. Liang and R. L. Liu, Biotechnol. Lett., 2008, 30, 1759–1764 CrossRef CAS.
- S. K. Rhee, J. H. Chang, Y. K. Chang and H. N. Chang, Appl. Environ. Microbiol., 1998, 64, 2327–2331 CAS.
- M. Shavandi, M. Sadeghizadeh, A. Zomorodipour and K. Khajeh, Bioresour. Technol., 2008, 100, 475–479 CrossRef.
- L. Alves and S. M. Paixão, Bioresour. Technol., 2011, 102, 9162–9166 CrossRef CAS.
- K. A. Gray, O. S. Pogrebinsky, G. T. Mrachko, L. Xi, D. J. Monticello and C. H. Squires, Nat. Biotechnol., 1996, 14, 1705–1708 CrossRef CAS.
- G. Mohebali, A. S. Ball, B. Rasekh and A. Kaytash, Enzyme Microb. Technol., 2007, 40, 578–584 CrossRef CAS.
- M. Durot, P. Y. Bourguignon and V. Schachter, FEMS Microbiol. Rev., 2009, 33, 164–190 CrossRef CAS.
- A. M. Feist, C. S. Henry, J. L. Reed, M. Krummenacker, A. R. Joyce, P. D. Karp, L. J. Broadbelt, V. Hatzimanikatis and B. Ø. Palsson, Mol. Syst. Biol., 2007, 3, 121 CrossRef.
- J. S. Edwards and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5528–5533 CrossRef CAS.
- S. Aggarwal, I. A. Karimi and D. Y. Lee, FEMS Microbiol. Lett., 2011, 315, 115–121 CrossRef CAS.
- S. Aggarwal, I. A. Karimi and D. Y. Lee, Mol. BioSyst., 2011, 7, 3122–3131 RSC.
- I. Famili, J. Förster, J. Nielsen and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13134–13139 CrossRef CAS.
- N. C. Duarte, M. J. Herrgård and B. Ø. Palsson, Genome Res., 2004, 14, 1298–1309 CrossRef CAS.
- H. Widiastuti, J. Y. Kim, S. Selvarasu, I. A. Karimi, H. Kim, J. S. Seo and D. Y. Lee, Biotechnol. Bioeng., 2011, 108, 655–665 CrossRef CAS.
- S. Selvarasu, I. A. Karimi, G. H. Ghim and D. Y. Lee, Mol. BioSyst., 2009, 6, 152–161 RSC.
- S. Selvarasu, V. V. T. Wong, I. A. Karimi and D. Y. Lee, Biotechnol. Bioeng., 2009, 102, 1494–1504 CrossRef CAS.
- R. Mahadevan and C. H. Schilling, Metab. Eng., 2003, 5, 264–276 CrossRef CAS.
- B. K. S. Chung and D. Y. Lee, BMC Syst. Biol., 2009, 3, 117 CrossRef.
- T. Matsui, T. Onaka, K. Maruhashi and R. Kurane, Appl. Microbiol. Biotechnol., 2001, 57, 212–215 CrossRef CAS.
- J. Gao, L. B. M. Ellis and L. P. Wackett, Nucleic Acids Res., 2010, 38, D488–D491 CrossRef CAS.
- S. Iida, H. Taniguchi, A. Kageyama, K. Yazawa, H. Chibana, S. Murata, F. Nomura, R. M. Kroppenstedt and Y. Mikami, Int. J. Syst. Evol. Microbiol., 2005, 55, 1871–1876 CrossRef CAS.
- R. K. Murray, D. K. Granner and P. A. R. V. W. Mayes, Harper's Biochemistry, Prentice Hall International, London, 1996 Search PubMed.
- C. H. Del Olmo, A. Alcon, V. E. Santos and F. Garcia-Ochoa, Enzyme Microb. Technol., 2005, 37, 157–166 CrossRef CAS.
- H. Honda, H. Sugiyama, I. Saito and T. Kobayashi, J. Ferment. Bioeng., 1998, 85, 334–338 CrossRef CAS.
- H. Yan, M. Kishimoto, T. Omasa, Y. Katakura, K. I. Suga, K. Okumura and O. Yoshikawa, J. Biosci. Bioeng., 2000, 89, 361–366 CrossRef CAS.
- H. Yan, X. Sun, Q. Xu, Z. Ma, C. Xiao and N. Jun, J. Environ. Sci., 2008, 20, 613–618 CrossRef CAS.
- E. Franchi, F. Rodriguez, L. Serbolisca and F. de Ferra, Oil Gas Sci. Technol., 2003, 58, 515–520 CAS.
- S. C. Gilbert, J. Morton, S. Buchanan, C. Oldfield and A. McRoberts, Microbiology, 1998, 144, 2545–2553 CrossRef CAS.
- R. Saha, P. F. Suthers and C. D. Maranas, PLoS One, 2011, 6, e21784 CAS.
- R. K. Aziz, D. Bartels, A. Best, M. DeJongh, T. Disz, R. A. Edwards, K. Formsma, S. Gerdes, E. M. Glass, M. Kubal, F. Meyer, G. J. Olsen, R. Olson, A. L. Osterman, R. A. Overbeek, L. K. McNeil, D. Paarmann, T. Paczian, B. Parrello, G. D. Pusch, C. Reich, R. Stevens, O. Vassieva, V. Vonstein, A. Wilke and O. Zagnitko, BMC Genomics, 2008, 9, 75 CrossRef.
- M. Kanehisa and S. Goto, Nucleic Acids Res., 2000, 28, 27–30 CrossRef CAS.
- R. Caspi, H. Foerster, C. A. Fulcher, P. Kaipa, M. Krummenacker, M. Latendresse, S. Paley, S. Y. Rhee, A. G. Shearer, C. Tissier, T. C. Walk, P. Zhang and P. D. Karp, Nucleic Acids Res., 2008, 36, D623–D631 CrossRef CAS.
- V. Satish Kumar, M. S. Dasika and C. D. Maranas, BMC Bioinf., 2007, 8, 212 CrossRef.
- A. Brooke, D. Kendrick, A. Meeraus and R. Raman, GAMS a User's Guide, GAMS Development Corporation, Washington, DC, 1998 Search PubMed.
- A. P. Burgard and C. D. Maranas, Biotechnol. Bioeng., 2003, 82, 670–677 CrossRef CAS.
- K. R. Kjeldsen and J. Nielsen, Biotechnol. Bioeng., 2009, 102, 583–597 CrossRef CAS.
- Y. Shinfuku, N. Sorpitiporn, M. Sono, C. Furusawa, T. Hirasawa and H. Shimizu, Microb. Cell Fact., 2009, 8, 43 CrossRef.
- D. Y. Lee, H. Yun, S. Park and S. Y. Lee, Bioinformatics, 2003, 19, 2144–2146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of metabolites, reactions, and dead-end metabolites. See DOI: 10.1039/c3mb70132h |
| This journal is © The Royal Society of Chemistry 2013 |