Understanding protein aggregation from the view of monomer dynamics
Lisa J.
Lapidus
*
Department of Physics and Astronomy and Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI 48824, USA. E-mail: lapidus@msu.edu
First published on 9th October 2012
Abstract
Much work in recent years has been devoted to understanding the complex process of protein aggregation. This review looks at the earliest stages of aggregation, long before the formation of fibrils that are the hallmark of many aggregation-based diseases, and proposes that the first steps are controlled by the reconfiguration dynamics of the monomer. When reconfiguration is much faster or much slower than bimolecular diffusion, then aggregation is slow, but when they are similar, aggregation is fast. The experimental evidence for this model is reviewed and the prospects for small molecule aggregation inhibitors to prevent disease are discussed.
 Lisa J. Lapidus | Lisa Lapidus obtained her PhD in atomic physics from Harvard University in 1998 then made the dramatic switch to biophysics when she joined the lab of William Eaton at the NIH. She also trained with Steven Chu at Stanford University before joining the faculty at Michigan State University in 2004. In addition to investigating intramolecular diffusion in unfolded proteins, her lab also develops new techniques to study protein folding using optical spectroscopy and microfluidics. |
Protein aggregation and disease
Protein aggregation is one of the grand problems of biophysics. It is highly complex, sensitive to initial conditions, operates on a huge range of timescales and its products range from dimeric proteins to macroscopic fibrils. Unwanted aggregation has implications in biotechnology, where large amounts of a single protein are produced in bioreactors, and human health, where aggregation-based diseases such as Alzheimer's, Parkinson's, Huntington's and Type II diabetes affect millions of people. On the other hand, there are numerous examples of functional amyloids in bacteria and yeast. While the entire process is a fascinating subject of biomolecular assembly on a massive scale, understanding the process at the earliest stages is key to preventing aggregation that causes damage. Indeed, while fibrils, the long cross-β assemblies of many individual proteins, are the best studied structures because they are large and highly stable, there is considerable evidence that small, transient oligomers are actually toxic to cells by disrupting membranes.2,3 A recent study of yeast Sup35 aggregation was able to distinguish between different species of oligomers with antibodies and found that the toxic species were less compact than non-toxic species.5Furthermore, it is increasingly understood that aggregation must be considered within the context of a cell.7 Certainly the crowded conditions in the cytoplasm will affect the likelihood that two proteins will interact, leading to aggregation, and may also affect the stability and folding pathways of some proteins.8 But a cell also contains a set of housekeeping genes to prevent, reverse or divert aggregation.9 A recent study shows that one function of chaperones, besides preventing and reversing misfolding, is to bind to small, toxic oligomers to promote assembly to larger, non-toxic oligomers, thus preserving the cell at the cost of the protein.10 The balance of protein production, chaperone activation and proteolysis has been termed proteostasis, and it appears that misfolding and aggregation of one protein can induce misfolding and aggregation of other proteins by putting the proteostasis network out of balance.11–13 A new model of this network, FoldEco, shows a cell is willing to expend large amounts of energy and discard a lot of misfolded protein to keep this balance.14 The basic implication of the proteolysis hypothesis is that an imbalance of this network leads to disease such as Alzheimer's and Parkinson's. That many of these aggregation-based diseases typically happen late in life and the fact that aggregation seems to be a generic process for all proteins suggests that aging causes proteostasis to break down, leading to disease of the most aggregation-prone proteins. There is evolutionary pressure on protein sequences to avoid aggregation and for the network to rescue misfolded and aggregated proteins but only over the reproductive lifetime of an organism, far short of the actual lifetime of modern humans.15,16
Nevertheless, the fundamental heart of this entire problem of non-functional aggregation is the physical process in which two or more proteins clump together in a non-specific and uncontrolled way, and a basic question is why some sequences aggregate and others don't. Several groups have attempted to rationalize the relationship between primary sequence and aggregation propensity,17 producing, for example, the Tango18 and the Zyggregator19 algorithms that calculate a number of properties of the sequence, including hydrophobicity, charge and secondary structure propensity, to create an aggregation metric. Generally sequences that have high hydrophobicity, low net charge and high β-sheet propensity are more likely to aggregate. While these algorithms are successful at predicting aggregation-prone sequences or regions of sequences, like protein structure prediction algorithms these methods do not describe the pathway of protein assembly in any detail such as when or how monomers adopt structures that are likely to stick to other monomers.
Aggregation is often thought to be a process by which proteins adopt specific, non-native, conformations that allow bimolecular association that is difficult to reverse (see ref. 20 for an excellent review of the field). Most analyses of aggregation kinetics indicate that the critical nucleus for the first step of aggregation is a monomer, though there may be a second critical nucleus for initiating fibril formation. Some proteins appear to aggregate from native like conformations,21,22 but most of the proteins and peptides associated with aggregation-based disease are either intrinsically disordered or have intrinsically disordered regions (i.e. Aβ, α-synuclein, IAPP and prion). There are a number of studies that have found structured intermediates as the aggregation precursor, but the difficulty in determining short-lived (less than a few milliseconds) intermediates suggests that some of these precursors could be subsets of the unfolded state under folding conditions.23–25 Furthermore, the dynamic stability of these states is usually unknown so it is unclear if there really is a single aggregation-competent precursor for most proteins. If aggregation results from an ensemble of kinetically accessible conformations then the reconfiguration time between these conformations must affect the rate of aggregation and perhaps the morphology of the resulting aggregates as well.
There are numerous papers on the structure and kinetics of the later, slower phases of aggregation as well as discussion as to which phases are toxic in aggregation-based diseases. This review will focus on the earliest steps in aggregation, bimolecular association and the formation of low molecular weight oligomers, because regardless of how aggregation progresses and which step is toxic, all aggregation must start from these first steps and prevention of bimolecular association will certainly prevent subsequent disease. In particular I present the unusual hypothesis that aggregation is kinetically controlled by reconfiguration of the protein monomers.
The simplest models of aggregation based on reconfiguration are shown in Fig. 1. The unfolded protein ensemble under aggregating conditions is M and M*, where M* are aggregation competent conformations (i.e. they have solvent exposed hydrophobic residues). The diffusive reconfiguration rate between these two states is k1 ≈ k−1 because the reconfiguration is primarily driven by diffusion and has no significant energetic barriers between the two states. The reconfiguration rate can be estimated by assuming it is the rate to diffuse one point on the chain across the chain diameter, k1 ∼ 4D/(2RG)2, where D is the intramolecular diffusion coefficient and RG is the radius of gyration of the chain, and have been observed to range from ∼104–107 s−1 (see below for details of experimental observations). Since there is no stable native structure, this rate will primarily be determined by the total hydrophobicity and hydrophobic pattern in the sequence. Two M* monomers may come into close contact by the bimolecular diffusion-controlled rate, kbi. While both monomers are in the encounter complex two things may happen, they may make stabilizing bimolecular interactions that lead to an oligomeric state O or one or both monomers may reconfigure to M which makes stabilizing bimolecular interactions difficult and the complex comes apart. The rate of forming O is slow compared to the other rates and may depend on many factors such as large-scale structural rearrangement within the complex (scheme i) and/or the addition of another monomer (scheme ii). This scheme produces three kinetic regimes. When k1 ≫ kbi reconfiguration is so high that the chain does not reside in M* long enough for the encounter complex to make stabilizing interactions. When k1 ≪ kbi the likelihood of two M* coming together is very unlikely so the encounter complex is rarely made. But when these rates are the same, the encounter complex forms fairly often and proceeds to the oligomeric state without coming apart.
![Kinetic models (i–iii) describing the early phases of aggregation. For all schemes, kbi = 1 × 105 s−1, kO = 100 s−1 and k1 as marked on the plots. (a) Solution to scheme (i), in which O forms by conformational change of [M*M*] and k−1= k1. (b) Solution to scheme (ii), in which O forms by the addition of a third monomer, M* to [M*M*] and k−1 = 0.5k1. (c) Solution to schemes (ii) and (iii) for knuc = 1 s−1 and kf = 100 s−1.](/image/article/2013/MB/c2mb25334h/c2mb25334h-f1.gif) | ||
| Fig. 1 Kinetic models (i–iii) describing the early phases of aggregation. For all schemes, kbi = 1 × 105 s−1, kO = 100 s−1 and k1 as marked on the plots. (a) Solution to scheme (i), in which O forms by conformational change of [M*M*] and k−1= k1. (b) Solution to scheme (ii), in which O forms by the addition of a third monomer, M* to [M*M*] and k−1 = 0.5k1. (c) Solution to schemes (ii) and (iii) for knuc = 1 s−1 and kf = 100 s−1. | ||
There is some evidence to support this model from other groups in the field. Early simulations of fibril formation showed that dimers formed in a two-step process whereby hydrophobic contacts form rapidly and then backbone hydrogen bonds zip up the structure through a longer relaxation process, thereby kinetically trapping some conformations.26 Experiments on mutants of superoxide dismutase 1 (SOD1) also support a two-step aggregation model, albeit with the preformed amyloid.27 An encounter complex as described above has been observed computationally for Aβ42 which finds that the second, accommodation, step reduces the free energy of the dimer.28,29 Analysis of polyglutamine aggregation also supports the formation of disordered dimers over homogeneous nucleation of β-sheet fibrils from monomers.30,31 Measurements of polyQ with different residues interrupting the sequence also show that the exact monomer conformational ensemble affects the aggregation kinetics.31 Even the elongation of Aβ16–22 fibrils show a two-step “dock and lock” mechanism in which a disordered monomer joins the fibril before adopting the cross-β conformation.32
Fig. 1a shows the solutions of kinetic scheme (i) using kbi = 1 × 105 s−1, a reasonable rate for a solution of ∼100 μM proteins of average molecular weight in water, and a range of k1. Qualitatively, the rate of aggregation is slower for very low and very high k1, but the range over which aggregation is most rapid is not quite k1= kbi. However, if the formation of O requires the addition of another M*, as shown in scheme (ii), then the solutions are in better quantitative agreement with observation and the range over which aggregation is rapid is only when k1 and kbi are within an order of magnitude. The addition of more monomers to create O raises the critical value of k1 with maximal aggregation propensity. Finally if the rate k−1 is slightly lower than k1 (k−1 = 0.5k1) in scheme (ii) then maximal aggregation occurs for k1 = 105 s−1 (Fig. 1b). Also, obviously, increasing kbi due to increased concentration (such as in a cell), increases the k1 with maximal aggregation propensity. The rate of forming O is assumed to be slower than reconfiguration, but an increased kO does not affect the overall trend of maximal oligomerization occurring when k1 ∼ kbi. It is also possible that reconfiguration within the encounter complex is slower than k−1 because bimolecular association with another monomer presents a drag on reconfiguration. Solution of scheme (ii) in which the escape rate from [M*M*] is 0.1k1 increases the oligomer formation rate but the trend of maximal k1 is unchanged.
Once in O diffusive reconfiguration is too slow to be relevant to subsequent structural changes, usually leading to a fibrillar state. The simplest model of fibril nucleation and propagation is shown in scheme (iii) and Fig. 1c for illustration requires a slow nucleation rate (knuc), in which the oligomer undergoes a conformational change to a nascent fibril, and a more rapid fibrillation rate (kf), in which oligomers add to the end of a growing fibril. Alternatively the fibril could grow from adding monomeric M* instead, which results in a slightly more gradual fibril formation but little change in the rates. However the real process could easily be more complicated. The process would obviously be dependent on protein sequence, solution conditions and other factors that have been shown to affect morphology. It is likely that the sequence effects that determine the monomeric reconfiguration rate also affect rates of nucleation or fibrillation, leading to the much larger difference in lag times and rise times between aggregation-prone and well-behaved proteins that are typically observed. For example, α-synuclein will fibrillize in several days while an IDP such as aaVLEA has never been observed to form fibrils at all, yet the reconfiguration rate differs only by 10-fold.
At first glance this model seems unlikely to be realistic because reconfiguration times for proteins are conventionally thought to be 10–100 ns, much faster than bimolecular diffusion times.33 This estimate is based on measurements of intramolecular diffusion in disordered peptides,1,34 but more recent studies, outlined below, have observe a much larger range. While the physical basis for the sequence dependence of reconfiguration is still unknown, it could be that both the overall hydrophobicity and hydrophobic pattern of the sequence are the determining factors. For all of these sequences, the measurements are made on a broad ensemble of conformations in water that should be determined, at least in part, by hydrophobic interactions between different parts of the chain. Thus a sequence that can make large hydrophobic clusters between distant points on the chain may be less diffusive than one that has hydrophobic residues distributed evenly along the sequence, which would in turn be less diffusive than a sequence with very few hydrophobes.
Observation of reconfiguration rates
A number of methods have been shown to measure intramolecular diffusion with close-range quenching,35–37 but this review will focus on the method of Trp-Cys quenching, described in Fig. 2.34 This method is not strictly diffusion-limited (q ≫ kD−) under all conditions but resolution of the observed rate into a diffusion-limited and reaction-limited rate allows an estimation of the equilibrium distribution of intramolecular distances (P(r)).1,38,39 Since this method uses only naturally occurring amino acids easily mutated into most proteins, my lab and others have employed it on a wide variety of sequences to develop the model described above. Fig. 3a shows some of the diffusion coefficients that have been measured, covering about 3 orders of magnitude in range. These measurements can be roughly split into three groups outlined below.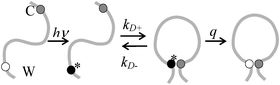 | ||
| Fig. 2 Determination of the rate of contact formation between the probe, tryptophan (W), and the quencher, cysteine (C), within an unfolded protein. Pulsed optical excitation leads to population of the lowest excited triplet state of tryptophan. Tryptophan contacts cysteine in a diffusion-limited process with rate kD+, and then either diffuses away or is quenched by the cysteine. The observed rate of Trp triplet decay is given by where kD+ is the rate of diffusion of the two ends towards each other, kD− is the rate of diffusion away and q is the rate of quenching. If q ≫ kD−, the observed lifetime reduces to kobs ≈ kD+. More generally, eqn (1) can be rearranged to give where kR is the reaction-limited rate and kD+ is the diffusion-limited rate,1T is the temperature and η is the viscosity of the solvent.The reaction-limited and diffusion-limited rates are given by Szabo Schulten and Schulten theory which describes the dynamics of unstructured peptides as diffusion on a one-dimensional potential of mean force that is related to the distribution (P(r)) of Trp-Cys distances r1 where q(r) is the rate of quenching at r (which has been determined experimentally by Lapidus et al.4), a is the point of closest approach (typically 4 Å), lc is the contour length of the chain between Trp and Cys (and, hence, their maximum separation), and D is the effective intramolecular diffusion coefficient. P(r) could be given by a simple polymer model such as a wormlike chain, by an empirical distribution based on experimental measurements or by a molecular dynamics simulation. Generally, both diffusion-limited and reaction-limited rates are inversely proportional to the average chain volume and the diffusion-limited rate is directly proportional to the diffusion coefficient,6 | ||
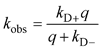
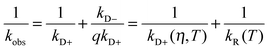
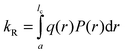
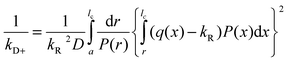
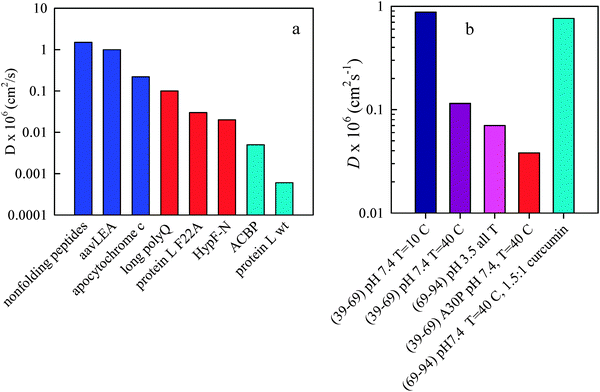 | ||
| Fig. 3 Measured diffusion coefficients. (a) Diffusion coefficients measured for a variety of sequences in water at 20 °C. The dark blue bars belong to the fast reconfiguration regime, red bars to the middle regime and cyan bars to the slow regime. (b) Diffusion coefficients of α-synuclein measured in various solution conditions, with the mutation A30P (red bar) and with the aggregation inhibitor, curcumin (cyan bar). The numbers in parenthesis indicate the position within the sequence of Trp (39 or 94) and Cys (69). | ||
Fast reconfiguration regime
The fastest coefficients (∼1.5 × 10−6 cm2 s−1) (and the first sequences that were measured) are for unstructured peptides with repeating motifs such as Gly–Ser or Ala–Gly–Gln.34,35,40 Also in this group are intrinsically disordered proteins, an unsurprising result because these sequences have little stable structure. For example, aaVLEA (D = 1.0 × 10−6 cm2 s−1), an IDP that appears to prevent aggregation during desiccation, has a very low hydrophobicity.41 It also has a large number of charged residues (39%), although the net charge is only −3.9 at pH 7. The charges alternate rapidly between positive and negative throughout the sequence. Therefore it should be unfavorable for the chain to make stable, long range interactions because two residues distant in sequence are just as likely to be repelled as attracted. Another IDP that has been measured is apocytochrome c.38 Its diffusion coefficient is 5 times slower (D = 0.2 × 10−6 cm2 s−1) than aaVLEA and likely close to the border with the aggregation-prone middle regime (see below). This difference may reflect the fact that once a heme is bound to this sequence, it becomes foldable. It is likely that other IDPs that fold upon binding to some ligand would fall in the range between aaVLEA and apocytochrome c. Generally these chains are highly expanded and well modeled by freely jointed or wormlike chains with excluded volume.1,42 Reconfiguration rates are in the range of 107–108 s−1. None of the sequences in this group have been shown to aggregate, although it is hard to be quantitative about this conclusion since aggregation might be possible on timescales that are impossible to measure in the lab. However, it is safe to say that aggregation is extremely difficult.Slow reconfiguration regime
The slowest two sequences, protein L and ACBP, are both proteins that fold on the millisecond timescale and which were measured after dilution of denaturant to near aqueous conditions using a microfluidic mixer.43,44 These diffusion coefficients are surprisingly slow, 6 × 10−10 and 5 × 10−9 cm2 s−1, respectively, but they are only slow in the absence of denaturant (with 6 M guanidine hydrochloride, most sequences have D ∼ 10−6 cm2 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6,45). Protein L is 10 times slower than ACBP which could reflect differences in sequence and also stability (ACBP is much less stable than protein L). Molecular dynamics simulations of each of these sequences reveal highly compact ensembles, RG ∼ 10–20 Å, yielding reconfiguration rates of 5 × 104 s−1 for protein L and 2.5 × 105 s−1 for ACBP.39,43 These proteins also do not easily aggregate. Although aggregation conditions were determined for protein L it required either low pH or 25% trifluoroethanol, a cosolvent known to promote aggregation in many sequences, conditions that are far from physiological.46 It is likely that the protonation of several residues at low pH would cause the chain to become more expanded and diffusive, increasing the reconfiguration rate. TFE may act the same way upon the chain.
6,45). Protein L is 10 times slower than ACBP which could reflect differences in sequence and also stability (ACBP is much less stable than protein L). Molecular dynamics simulations of each of these sequences reveal highly compact ensembles, RG ∼ 10–20 Å, yielding reconfiguration rates of 5 × 104 s−1 for protein L and 2.5 × 105 s−1 for ACBP.39,43 These proteins also do not easily aggregate. Although aggregation conditions were determined for protein L it required either low pH or 25% trifluoroethanol, a cosolvent known to promote aggregation in many sequences, conditions that are far from physiological.46 It is likely that the protonation of several residues at low pH would cause the chain to become more expanded and diffusive, increasing the reconfiguration rate. TFE may act the same way upon the chain.
The aggregation-prone middle regime
The sequences in the middle of the measured range are all known to aggregate. For example, the N-terminal domain of the human maturation factor (HypF-N) is known as one of the fastest proteins to aggregate in vitro, with experiments typically using trifluorethanol (TFE) to promote aggregation.47–49 In 3% TFE, which probably acts as a mild denaturant while causing aggregation in about 20 minutes, D ∼ 2 × 10−8 cm2 s−1 (note that this coefficient is lower than that reported in reference,45 which was an extrapolation to 0 M denaturant). As with protein L, TFE likely acts as a good solvent for the protein, making it more expanded and diffusive. The protein may reconfigure more slowly under physiological conditions but is still close to the middle regime to make aggregation somewhat likely.Polyglutamine (polyQ) peptides longer than ∼30 residues are known to aggregate and people with more than 30 Gln repeats in the larger Huntington protein are prone to Huntington's disease, with the age of onset inversely related to the total number of repeats. Measurements of shorter peptides showed that this sequence is very stiff based on the turnover in the reaction-limited rate at ∼10 residues,50 while measurements and simulation of longer peptides revealed compact structures.31,51–54 Fitting the short peptide measurements to a wormlike chain model revealed an extremely high persistence length (13 Å) but later analysis with a coarse-grained protein model showed that at long lengths the intrinsic stiffness is counterbalanced by strong attractive interactions,55 in agreement with FRET and FCS measurements of the longer lengths. Thus, unlike other disordered peptides, the diffusion coefficient of polyQ decreases with length. An extrapolation of the measured diffusion coefficient at short lengths to 40–60 residues, lengths known to promote Huntington's disease, gives D ∼ 10−7 cm2 s−1. The radius of gyration of a 30 residue polyQ peptide is estimated to be 25 Å56 yielding a reconfiguration rate of 1.6 × 106 s−1.
A point mutation of protein L, F22A increases the diffusion coefficient by almost two orders of magnitude and anecdotally is known to aggregate in water.39 Thus it appears that intramolecular diffusion is exquisitely sensitive to sequence. Using a polymeric model which accounts for preferential intramolecular interactions, we find that phenylalanine 22 in protein L preferentially makes contacts non-local in sequence, making the chain less diffusive, while alanine 22 will preferentially make more local contacts, keeping it more diffusive.38 Surprisingly, MD simulations find that RG for the F22A mutant is only 5% larger than the wildtype,39 leading to a reconfiguration rate of 1 × 106 s−1.
Solvent effects on reconfiguration and aggregation
Recently my lab has sought to investigate the role of solution conditions using the protein α-synuclein, a protein that aggregates in Parkinson's disease.57 This protein is intrinsically disordered in vitro. Though it may adopt a helical tetramer structure in vivo,58 unfolding appears to be irreversible outside the cell and aggregation only seems to originate from the monomer. Furthermore, it is well demonstrated that the aggregation propensity varies widely with solution conditions, with aggregation rates increasing with increasing temperature or decreasing pH. Fig. 3b shows the diffusion coefficient measured at low temperature at pH 7 is fast, in the regime of IDPs while diffusion coefficients at high temperature and at low pH are much lower. Measurement of reaction-limited rates, which give an estimate of the volume of the chain, show that at low pH or high temperature (40 °C), the chain is much more compact than at pH 7, 10 °C. Therefore k1 decreases from ∼8 × 106 s−1 to ∼8 × 105 s−1, into the aggregation-prone middle regime. Also, the mutation A30P, which is known to accelerate Parkinson's disease, decreases the reconfiguration rate by four-fold at all temperatures. The diffusion coefficients were measured for different loops within the sequence and remain constant along the entire length of the chain. They are also much slower than any rates measured of fragments,59 suggesting the entire chain is required to slow down the dynamics globally.Aggregation inhibitors
One possible therapeutic strategy for preventing or treating aggregation-based diseases is to find a small molecule that can inhibit aggregation. Many naturally occurring molecules, mostly polyphenols and flavenoids, have been investigated, and many have been found to prevent fibrillation,60–69 but since it is thought that small oligomers are the toxic species to cells, it's not clear if that type of assay is particularly useful. Other molecules, such as epigallocatechin gallate, have been shown to prevent the conversion of oligomer to fibril and instead promote the formation of large, non-toxic oligomers.70 But this sort of inhibitor does not prevent the initial aggregation event from occurring, and if the disease is caused by low molecular weight oligomers that transiently form before the promotion of large oligomers, or if the disease is caused by the loss of the monomer, this strategy would not prevent disease. Thus an ideal inhibitor would bind to monomers and prevent aggregation at the earliest steps. Some studies have found small molecules such as these. For example, human acylphosphatase, a model system for studying aggregation in vitro, binds strongly to heparin sulfate and enhances aggregation propensity, including oligomer formation.71Following the model laid out here, an inhibitor could prevent the first stage of aggregation by either shifting the reconfiguration rate to the fast or slow regimes. In shifting to the fast regime, the protein would become more like a typical IDP with a large volume and rapid intramolecular diffusion. Alternatively, an inhibitor could slow down diffusion to the rate of well-behaved foldable proteins. Such a molecule would nucleate hydrophobic clustering by distant parts of the chain, making the chain more compact and less diffusive. However, if it is an IDP, this slower chain is not going to ever fold and the chance to make stable bimolecular associations over long periods of time (seconds to minutes) is still quite high. Thus whether such a molecule really prevents aggregation in a cell depends in detail on how the chaperone network treats such a chain. Since it is dynamically similar to an unfolded or misfolded foldable protein, it could be tagged for proteolysis, but if chaperones were to attempt to undo the “misfolded” conformation aggregation would probably still occur because it will never fold. Furthermore, repeatedly engaging chaperones might disrupt proteostasis more than the protein without the inhibitor. Thus understanding the full cellular response is necessary when designing a therapeutic.
To further test this model, my lab has begun to investigate the effect of small molecule aggregation inhibitors on intramolecular diffusion in α-synuclein. As a first step we chose curcumin, a naturally occurring polyphenol in the spice turmeric, which has been shown to inhibit aggregation in both α-synuclein as well as the Alzheimer's peptide. Using optical changes in both the curcumin and the Trp mutated into the α-synuclein, we found that curcumin bound strongly to the monomeric protein. Both fibrilization and oligomerization were inhibited and intramolecular diffusion increased, particularly at high temperatures where aggregation was more likely (see Fig. 3b). Thus it appears that curcumin bound to α-synuclein changes the conformation ensemble by making the chain less compact and more diffusive. With the reconfiguration time increased at high temperatures, aggregation was unlikely because k1 ≫ kbi.72
Conclusions
The model presented in this paper represents a significantly different approach to aggregation and aggregation–inhibition. Instead of searching for aggregation-competent structures, which has proven extraordinarily difficult, future studies could search for aggregation-competent dynamics by measuring reconfiguration of the monomer. Drug design could be directed to tailoring interactions between the monomeric protein and the drug such that reconfiguration is speeded up or slowed down out of the middle regime without causing any unintended damage to the cell or its proteostasis network. Finally, these ideas could impact the concept of proteostasis because chaperones and proteolytic machinery might be tuned to interact with reconfiguration rates in the middle regime.Acknowledgements
I wish to thank all the members of the Lapidus lab whose hard work, patience and ingenuity made development of this “crazy hypothesis” possible, particularly Vijay Singh, Yujie Chen, Steven Waldauer, Basir Ahmad and Srabasti Acharya. This work has been supported by the National Science Foundation grants NSF MCB-0825001, NSF EF-0623664 and NSF DBI-0754570, and by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.References
- L. J. Lapidus, P. J. Steinbach, W. A. Eaton, A. Szabo and J. Hofrichter, J. Phys. Chem. B, 2002, 106, 11628–11640 CrossRef CAS.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS.
- L. J. Lapidus, W. A. Eaton and J. Hofrichter, Phys. Rev. Lett., 2001, 87, 258101 CrossRef CAS.
- R. Krishnan, J. L. Goodman, S. Mukhopadhyay, C. D. Pacheco, E. A. Lemke, A. A. Deniz and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11172–11177 CrossRef CAS.
- V. R. Singh, M. Kopka, Y. Chen, W. J. Wedemeyer and L. J. Lapidus, Biochemistry, 2007, 46, 10046–10054 CrossRef CAS.
- A. Gershenson and L. M. Gierasch, Curr. Opin. Struct. Biol., 2011, 21, 32–41 CrossRef CAS.
- H.-X. Zhou, G. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS.
- R. Sabate, N. de Groot and S. Ventura, Cell. Mol. Life Sci., 2010, 67, 2695–2715 CrossRef CAS.
- B. Mannini, R. Cascella, M. Zampagni, M. van Waarde-Verhagen, S. Meehan, C. Roodveldt, S. Campioni, M. Boninsegna, A. Penco, A. Relini, H. H. Kampinga, C. M. Dobson, M. R. Wilson, C. Cecchi and F. Chiti, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12479–12484 CrossRef CAS.
- T. Gidalevitz, V. Prahlad and R. I. Morimoto, Cold Spring Harbor Perspect. Biol., 2011, 3, a009704 CrossRef.
- S. L. Lindquist and J. W. Kelly, Cold Spring Harbor Perspect. Biol., 2011, 3, a004507 CrossRef.
- M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Cold Spring Harbor Perspect. Biol., 2011, 3, a010454 CrossRef.
- E. T. Powers, D. L. Powers and L. M. Gierasch, Cell Rep., 2012, 1, 265–276 CrossRef CAS.
- C. M. Dobson, Semin. Cell Dev. Biol., 2004, 15, 3–16 CrossRef CAS.
- M. Vendruscolo, J. Zurdo, C. E. MacPhee and C. M. Dobson, Philos. Trans. R. Soc., A, 2003, 361, 1205–1222 CrossRef CAS.
- V. Castillo, R. Grana-Montes, R. Sabate and S. Ventura, Biotechnol. J., 2011, 6, 674–685 CrossRef CAS.
- A.-M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS.
- G. G. Tartaglia, A. P. Pawar, S. Campioni, C. M. Dobson, F. Chiti and M. Vendruscolo, J. Mol. Biol., 2008, 380, 425–436 CrossRef CAS.
- F. Bemporad and F. Chiti, Chem. Biol., 2012, 19, 315–327 CrossRef CAS.
- F. Bemporad, A. De Simone, F. Chiti and C. M. Dobson, Biophys. J., 2012, 102, 2595–2604 CrossRef CAS.
- K. Pagano, F. Bemporad, F. Fogolari, G. Esposito, P. Viglino, F. Chiti and A. Corazza, J. Biol. Chem., 2010, 285, 14689–14700 CrossRef CAS.
- C. W. Bertoncini, Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef CAS.
- N. Rezaei-Ghaleh, K. Giller, S. Becker and M. Zweckstetter, Biophys. J., 2011, 101, 1202–1211 CrossRef CAS.
- G. W. Platt and S. E. Radford, FEBS Lett., 2009, 583, 2623–2629 CrossRef CAS.
- W. Hwang, S. Zhang, R. D. Kamm and M. Karplus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12916–12921 CrossRef CAS.
- L. Lang, M. Kurnik, J. Danielsson and M. Oliveberg, Proc. Natl. Acad. Sci. U. S. A., 2012 DOI:10.1073/pnas.1201795109.
- S.-H. Chong and S. Ham, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7636–7641 CrossRef CAS.
- S.-H. Chong and S. Ham, Phys. Chem. Chem. Phys., 2012, 14, 1573–1575 RSC.
- A. Vitalis and R. V. Pappu, Biophys. Chem., 2011, 159, 14–23 CrossRef CAS.
- R. H. Walters and R. M. Murphy, J. Mol. Biol., 2011, 412, 505–519 CrossRef CAS.
- P. H. Nguyen, M. S. Li, G. Stock, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 111–116 CrossRef CAS.
- J. Kubelka, J. Hofrichter and W. A. Eaton, Curr. Opin. Struct. Biol., 2004, 14, 76–88 CrossRef CAS.
- L. J. Lapidus, W. A. Eaton and J. Hofrichter, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7220–7225 CrossRef CAS.
- O. Bieri, J. Wirz, B. Hellrung, M. Schutkowski, M. Drewello and T. Kiefhaber, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9597–9601 CrossRef CAS.
- D. Nettels, A. Hoffmann and B. Schuler, J. Phys. Chem. B, 2008, 112, 6137–6146 CrossRef CAS.
- H. Neuweiler, A. Schulz, M. Bohmer, J. Enderlein and M. Sauer, J. Am. Chem. Soc., 2003, 125, 5324–5330 CrossRef CAS.
- Y. Chen, W. J. Wedemeyer and L. J. Lapidus, J. Phys. Chem. B, 2010, 114, 15969–15975 CrossRef CAS.
- V. A. Voelz, V. R. Singh, W. J. Wedemeyer, L. J. Lapidus and V. S. Pande, J. Am. Chem. Soc., 2010, 132, 4702–4709 CrossRef CAS.
- A. Moglich, K. Joder and T. Kiefhaber, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12394–12399 CrossRef.
- S. Chakrabortee, C. Boschetti, L. J. Walton, S. Sarkar, D. C. Rubinsztein and A. Tunnacliffe, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18073–18078 CrossRef CAS.
- M. Buscaglia, L. J. Lapidus, W. A. Eaton and J. Hofrichter, Biophys. J., 2006, 91, 276–288 CrossRef CAS.
- V. A. Voelz, M. Jäger, S. Yao, Y. Chen, L. Zhu, S. A. Waldauer, G. R. Bowman, M. Friedrichs, O. Bakajin, L. J. Lapidus, S. Weiss and V. S. Pande, J. Am. Chem. Soc., 2012, 134, 12565–12577 CrossRef CAS.
- S. A. Waldauer, O. Bakajin and L. J. Lapidus, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13713–13717 CrossRef CAS.
- Y. J. Chen, C. Parrini, N. Taddei and L. J. Lapidus, J. Phys. Chem. B, 2009, 113, 16209–16213 CrossRef CAS.
- T. Cellmer, R. Douma, A. Huebner, J. Prausnitz and H. Blanch, Biophys. Chem., 2007, 125, 350–359 CrossRef CAS.
- M. Calamai, N. Taddei, M. Stefani, G. Ramponi and F. Chiti, Biochemistry, 2003, 42, 15078–15083 CrossRef CAS.
- S. Campioni, M. F. Mossuto, S. Torrassa, G. Calloni, P. P. de Laureto, A. Relini, A. Fontana and F. Chiti, J. Mol. Biol., 2008, 379, 554–567 CrossRef CAS.
- F. Chiti, N. Taddei, F. Baroni, C. Capanni, M. Stefani, G. Ramponi and C. M. Dobson, Nat. Struct. Biol., 2002, 9, 137–143 CrossRef CAS.
- V. R. Singh and L. J. Lapidus, J. Phys. Chem. B, 2008, 112, 13172–13176 CrossRef CAS.
- S. L. Crick, M. Jayaraman, C. Frieden, R. Wetzel and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16764–16769 CrossRef CAS.
- X. L. Wang, A. Vitalis, M. A. Wyczalkowski and R. V. Pappu, Proteins, 2006, 63, 297–311 CrossRef CAS.
- J. P. Bernacki and R. M. Murphy, Biochemistry, 2011, 50, 9200–9211 CrossRef CAS.
- C. C. Lee, R. H. Walters and R. M. Murphy, Biochemistry, 2007, 46, 12810–12820 CrossRef CAS.
- J. L. Digambaranath, T. V. Campbell, A. Chung, M. J. McPhail, K. E. Stevenson, M. A. Zohdy and J. M. Finke, Proteins, 2011, 79, 1427–1440 CrossRef CAS.
- R. H. Walters and R. M. Murphy, J. Mol. Biol., 2009, 393, 978–992 CrossRef CAS.
- B. Ahmad, Y. Chen and L. J. Lapidus, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2336–2341 CrossRef CAS.
- T. Bartels, J. G. Choi and D. J. Selkoe, Nature, 2011, 477, 107–U123 CrossRef CAS.
- A. Grupi and E. Haas, J. Mol. Biol., 2011, 405, 1267–1283 CrossRef CAS.
- A. Gallardo-Godoy, J. Gever, K. L. Fife, B. M. Silber, S. B. Prusiner and A. R. Renslo, J. Med. Chem., 2011, 54, 1010–1021 CrossRef CAS.
- A. R. A. Ladiwala, J. S. Dordick and P. M. Tessier, J. Biol. Chem., 2011, 286, 3209–3218 CrossRef CAS.
- C. Lendel, C. W. Bertoncini, N. Cremades, C. A. Waudby, M. Vendruscolo, C. M. Dobson, D. Schenk, J. Christodoulou and G. Toth, Biochemistry, 2009, 48, 8322–8334 CrossRef CAS.
- A. Natalello, J. Liu, D. Ami, S. M. Doglia and A. de Marco, Proteins: Struct., Funct., Bioinf., 2009, 75, 509–517 CrossRef CAS.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef CAS.
- K. Ono, M. M. Condron, L. Ho, J. Wang, W. Zhao, G. M. Pasinetti and D. B. Teplow, J. Biol. Chem., 2008, 283, 32176–32187 CrossRef CAS.
- Y. Porat, A. Abramowitz and E. Gazit, Chem. Biol. Drug Des., 2006, 67, 27–37 CAS.
- W. Qi, A. Zhang, T. A. Good and E. J. Fernandez, Biochemistry, 2009, 48, 8908–8919 CrossRef CAS.
- J. N. Rao, V. Dua and T. S. Ulmer, Biochemistry, 2008, 47, 4651–4656 CrossRef CAS.
- M. Zhu, S. Rajamani, J. Kaylor, S. Han, F. Zhou and A. L. Fink, J. Biol. Chem., 2004, 279, 26846–26857 CrossRef CAS.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CAS.
- N. Motamedi-Shad, T. Garfagnini, A. Penco, A. Relini, F. Fogolari, A. Corazza, G. Esposito, F. Bemporad and F. Chiti, Nat. Struct. Mol. Biol., 2012, 19, 547–554 CAS , S541–S542.
- B. Ahmad and L. J. Lapidus, J. Biol. Chem., 2012, 287, 9193–9199 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |