Probing cell–cell communication with microfluidic devices
Feng
Guo
a,
Jarrod B.
French
b,
Peng
Li
a,
Hong
Zhao
b,
Chung Yu
Chan
a,
James R.
Fick
c,
Stephen J.
Benkovic
*b and
Tony Jun
Huang
*a
aDepartment of Engineering Science and Mechanics, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: junhuang@psu.edu; Fax: +1 814-865-9974; Tel: +1 814-863-4209
bDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: sjb1@psu.edu; Fax: +1 814-863-0735; Tel: +1 814-865-2973
cPenn State Hershey Medical Group, 1850 East Park Avenue, Suite 112, State College, PA 16803, USA
First published on 10th July 2013
Abstract
Intercellular communication is a mechanism that regulates critical events during embryogenesis and coordinates signalling within differentiated tissues, such as the nervous and cardiovascular systems. To perform specialized activities, these tissues utilize the rapid exchange of signals among networks that, while are composed of different cell types, are nevertheless functionally coupled. Errors in cellular communication can lead to varied deleterious effects such as degenerative and autoimmune diseases. However, the intercellular communication network is extremely complex in multicellular organisms making isolation of the functional unit and study of basic mechanisms technically challenging. New experimental methods to examine mechanisms of intercellular communication among cultured cells could provide insight into physiological and pathological processes alike. Recent developments in microfluidic technology allow miniaturized and integrated devices to perform intercellular communication experiments on-chip. Microfluidics have many advantages, including the ability to replicate in vitro the chemical, mechanical, and physical cellular microenvironment of tissues with precise spatial and temporal control combined with dynamic characterization, high throughput, scalability and reproducibility. In this Focus article, we highlight some of the recent work and advances in the application of microfluidics to the study of mammalian intercellular communication with particular emphasis on cell contact and soluble factor mediated communication. In addition, we provide some insights into likely direction of the future developments in this field.
Introduction
Cell–cell communication is of fundamental importance in directing cellular functions.1–3 The intercellular exchange of information allows the regulation of cell proliferation, apoptosis, differentiation, response to emergent stimulation, and more. Evidence of impaired intercellular communication is present in diseases such as cancer, autoimmune disorders, and diabetes.2,3 Thus, revealing the underlying mechanism of intercellular communication is essential to understand and treat such diseases as well as being of interest for related health research such as tissue engineering, stem cell regenerative therapy and cancer diagnosis. However, the mechanisms of intercellular communication still remain poorly understood, mostly due to the extreme complexity of intercellular communication networks in multicellular systems. There are a large number of technical challenges inherent to the investigation of the spatial and temporal communication within highly organized groups of cells.Many efforts have been made to exploit different approaches for the study of intercellular communication. Direct in vivo studies possess the power to maintain the native microenvironment during investigations of cell–cell communication, but are limited by the expense and complexity of imaging systems required to carry out such experiments.4 Importantly, when using in vivo approaches it is extremely difficult to accurately discriminate signals of interest from other signals and background noise. In vitro platforms are important complementary approaches for the investigation of intercellular communication as they can provide simplified experimental conditions and data interpretation. Conventional in vitro techniques are often inadequate to obtain higher spatial and temporal resolutions which are critical to better understanding many cell–cell communication questions. In contrast, emerging microfluidic platforms excel in manipulating fluids and cells with the resolution that cannot be matched by existing approaches and have potential to advance our knowledge on cell–cell communication in vitro.5
In moving from macro to micro-scale, microfluidic cell culture provides a more in vivo like microenvironment under which experiments can be performed at physiologically relevant time and length-scales.5–11 Unlike static media in Petri dishes and well plates, which experience uncontrollable convectional mixing, microfluidics offers precise control of dynamic perfusion and the extracellular chemical microenvironments through sophisticated manipulation of small fluidic volumes in microchannels.12,13 Similarly, while conventional cell culture methods suffer from random cell loading, microfluidics allow precise control over cell spatial arrangement with the capability of single cell manipulation.14,15 By combining these functionalities to tune the spatial and temporal parameters, microfluidics enable quantitative study of specific communication events in a highly defined microenvironment. Moreover, the behaviour of cells during the communication process can be temporally characterized under varied and repeatable conditions. Furthermore, intercellular communication signals (soluble factors or micro-vesicles, for example) can be isolated for later analysis, or measured and quantified in situ. To date, many microfluidic systems have been developed to investigate different types of intercellular communication. These range from the study of general population behaviours, to single cells interactions, and to the quantification of signalling events. In this Focus article, we will summarize some of the recent successes in the application of microfluidics to the study of cell–cell communication in mammalian systems. We will highlight particular advances relating to the study of direct contact and soluble factor mediated communication and will provide our perspective on the future of microfluidics in the cell–cell communication field.
Intercellular population communication
Multicellular organisms rely on intercellular communication to coordinate both development and environmental responses across diverse cell populations. The study of cell-to-cell communication, an inherently multi-cellular process, can provide insights into collective behaviours and communication patterns of the cell population. In a multi-cellular system, intercellular communication is highly dependent on physical contact and soluble factor regulation. There is high demand for platforms that can provide spatial control of the cell populations, as well as spatial and temporal manipulation of cellular secretion perfusion. In this section, we discuss microfluidic approaches for addressing conventional problems in the study of intercellular population communication. By addressing the technical problems of conventional cell culture methods, we will be able to further understand the mechanics and dynamics of intercellular communication.Gap junctions are nano-scale protein channels, and directly connect the cytoplasm of two cells. Intercellular communications through gap junctions occur by the diffusion of small molecules between neighbouring coupled cells. Altered gap junction communication is associated with numerous pathophysiological states, including cancer, cardiac, neurological, and skin diseases.2 To study gap junction based communication, cells must be in contact with each other. Since the employment of microfluidics enables the precise spatial control of cell seeding and medium loading, it is a good way to study gap junction based communication. Spatial and density effects on this type of intercellular communication can be studied with high temporal resolution. Sun et al. investigated the role of gap junctions in cell collective behaviour using a straight channel microfluidic device.16 Collective behaviour is an emergent response from a group of individuals lacking central coordination, for example, bees swarming, fish schooling, or birds flocking.17 Similarly, mammalian cells coordinate their responses during complex multicellular processes by sensing variations in the concentration of signalling molecules that drive processes such as wound-healing and cell migration. The behaviour of mammalian cells, however, is commonly associated with a dependence of signalling pathways and time-dependent gene expression.18 Thus, understanding intercellular communication of mammalian cells demands sensitive measurements of the cellular response to signalling molecules. In their investigation of the collective behaviour of fibroblasts, Sun et al. first cultured fibroblast cells in the microfluidic channel with different cell densities before exposing the cells to flow of an ATP solution over the cells (Fig. 1A).16 When cells are exposed to an external flow of ATP, the change in calcium ion level in the cells in response to the stimulant can be quantified using a calcium-sensitive fluorescent dye. The fluorescent images of fibroblast cells under the same flow conditions show that more cells are activated in the high-density colony than in the low-density colony (Fig. 1B). Furthermore, the quantitative analysis of ATP stimulation (Fig. 1C and D) indicated that cells in the dense colony can respond to reach threshold intensity faster than cells in the low density colony, independent of flow direction. In addition, the calcium response in the high density colony is highly correlated, while this is not the case in the low density colony. Conversely, when cells are seeded in a hydrogel matrix to disrupt the direct contacts, the correlation of the calcium response disappears. Taken together, these experiments implicate gap junction-based intercellular communication among cultured fibroblasts.
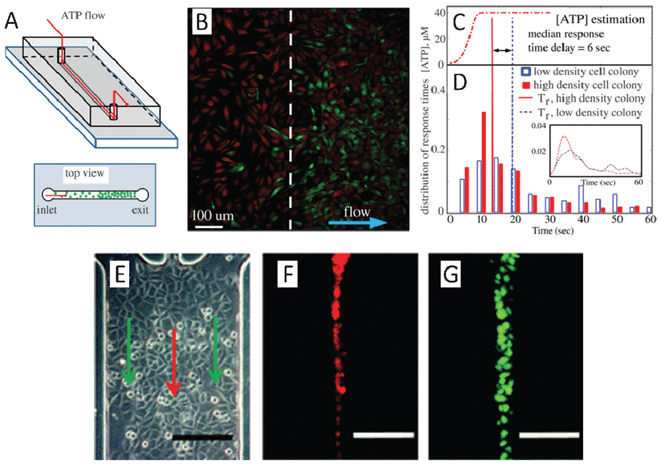 | ||
| Fig. 1 (A)–(D) Quantification of spatiotemporal variation in the collective behaviour of fibroblasts. (A) Schematic of the microfluidic device used to investigate the role of gap junctions in the collective behaviour of cells. (B) A calcium fluorescent image captured while ATP solution flows through fibroblast cells of non-uniform spatial density (Red shows cell locations; Green describes Fluo-4 calcium indicator). (C) The temporal profile of ATP concentration estimated from fluorescent intensity of the fluorescein solution recorded above the cell monolayer in the same flow conditions for both high and low density populations of cells. (D) Quantitative analysis of ATP stimulation response of individual cells in colonies with different densities. (E)–(G) Hydrodynamic flow based dye loading over confluent cells. (E) Three inlet channels merge into a main channel to form a three-stream laminar flow as indicated by the arrows. (F) The red fluorescent solution (DiI dye) is well defined in a stream with sharp concentration boundaries, minimizing the transverse diffusion between flow streams. (G) Cells in a specific column are exposed to membrane permeable fluorescence dyes (calcein/AM) under continuous flow. Using this experimental setup, dye transfer dynamics via gap junction can be quantitatively monitored by time-lapse fluorescent microscopy and the extent of dye spread and effective diffusivities can be evaluated (scale bar: 200 μm). Images reproduced from ref. 16 and 19. | ||
In addition to the ability to manipulate cell seeding densities, the chemical environment can also be controlled in microfluidic devices by exploiting the properties of the laminar flow.19–27 Several studies have made use of this property to better characterize gap junction-based intercellular communication.19–25 The biophysical properties of a gap junction, such as selectivity, permeability, and regulatory and gating mechanisms, are emerging as important topics of investigation. It is difficult, however, to perform quantitative characterizations of gap junctions in conventional macro-scale platforms due to the lack of precise spatial control. One way to overcome this limitation is the use of the laminar flow in microfluidic devices, where multiple streams with distinct chemical composition can be formed. Moreover, the position and the widths of each stream can be adjusted accordingly with micrometre resolution.28 Chen and Lee used this method to selectively deliver dye into a subset of cells in a column within a culture chamber of fully confluent cells.19 In their device, three inlet channels merge into a main channel to form a three-stream laminar flow (Fig. 1E). The dye solution is well defined in a stream with sharp concentration boundaries that minimizes the transverse diffusion between flow streams (Fig. 1E–F). Cells in a particular column are exposed to membrane permeable fluorescent dyes (calcein/AM) under continuous flow (Fig. 1G). The dynamics of the dye transfer process occurring via gap junction to neighbouring cells can be quantitatively monitored by time-lapse fluorescent microscopy. The extent of dye spread and effective diffusivities can then be evaluated. The Hua group also employed a multi-stream laminar flow to selectively expose confluent cultured cells with different dyes and inhibitors.21 This allowed them to precisely characterize the diffusion coefficient of four different dyes using a theoretical diffusion model. The device that this group employed can screen multiple inhibitors in parallel in a single cell preparation, demonstrating its potential for high throughput. Furthermore, by replacing the confluent cell culture with patterned arrays of cells made by micro-printing, they were also able to demonstrate and characterize dynamic dye transfer in single cells.23
Gap junction-mediated intercellular communication depends on the direct contact between cells, whereas signalling through secreted molecules allow cells to communicate without contacting each other.12 Within a population of cells, the soluble factors responsible for the signals are produced by a source cell at certain rate (molecules per second) and diffused in the three dimensional extracellular environment. Those ligands can bind to cell-surface receptors of the same cell, or homotypic cells (autocrine), or may actuate other nearby heterotypic cells (paracrine). Soluble factor signalling is involved in regulating many biological events such as embryonic stem cell pluripotency, mammalian embryogenesis, tumour formation and metastasis.2 However, in the conventional cell culture platforms, spontaneously occurring spatial fluctuations (temperature, solute concentration, or dissolved gas concentration) lead to rapid convection and mass transfer, which complicates measurements of the soluble factors. The convective mixing causes secreted molecules to rapidly diffuse over the entire volume. New technologies are in great demand to help understand the mechanism underlying soluble factor communication in various biological events. As a result of micro-scale channel design and the absence of a liquid/air interface, random convective mixing becomes insignificant in microfluidic devices.29 Thus, microfluidic devices show potential for addressing this problem in the study of soluble signalling communication processes. Experimental control of soluble factors is the key to understanding the spatially confined, closed-loop nature of these processes. By altering transportation of signalling molecules, one can control perfusion (diffusion, convection, and reaction), and in turn modulate the activity of autocrine or paracrine loops to discover the underlying mechanism.30–32
One way in which microfluidics can be employed to study soluble factor regulation is with user-defined, single-layer microfluidic devices. Song et al. used a unidirectional perfusion microfluidic device to study the effect of embryonic germ cells on cancer cells.33 They cultured human embryonic germ cell and ovarian cancer cell (SKOV3) populations into the single microfluidic channel and separated them with a barrier structure. The device offered a slow and continuous flow allowing the control of the direction of flow of molecules secreted by the embryonic germ cells towards the cancer cell colony. They found that the apoptotic signals in the SKOV3 culture area decreased along the flow of medium and demonstrated that human embryonic germ cells can induce ovarian cancer cell apoptosis. Similarly, soluble factor mediated intercellular population communication can be investigated by culturing two populations of cells in different chambers and establishing flow across the chambers independently and serially.34 While these approaches enable investigations not possible with macro-scale devices, they are still limited by a lack of precise spatial control.
To expand the functionalities of devices, certain on-chip components can be incorporated into cell culture microfluidic devices for the investigation of soluble factor communication. Pneumatic valves and pumps, such as those developed by the Quake group,35 enable precise and sophisticated on-chip liquid manipulation. These types of devices open a promising avenue for various chemical, biochemical, and biomedical applications. Multilayer soft lithography pneumatic micro-valves and pumps can be integrated into microfluidic cell culture chips and provide a fully automatic and high-throughput cell culture system.36 Within such a device, the fluidic volume can be precisely and dynamically removed, diluted, trapped, transported, and replaced from region to region. The accuracy of the spatial-temporal flow control shows great potential to control convection, diffusion, and reaction, offering digital regulation of the cellular microenvironment. Thus, these on-chip valves and pumps can be used to develop further advanced perfusion chips for the investigation of the autocrine and paracrine loops.37–40 For example, the identification of unknown diffusion loops commonly relies on density-dependent phenotype assays. However, such loops may not display density-dependent phenotypes although they are sufficiently active in isolated cells. Recently, Blagovic et al. developed a perfusion chip to identify the existence of a diffusible signalling loop participating in the differentiation of mouse embryonic stem cells (mESCs) into neuroectodermal processors that was previously undiscovered by conventional assays.37 This double-layer device contains multiple mirror-symmetry designed cell culture chambers, closed valves, and active bubble traps (Fig. 2A–B). The mESCs can be selectively seeded into well-defined culture environments and attached in the absence of flow through the digital operation of on-chip microvalves. Cell-secreted factors can be removed to down regulate diffusible signalling. Multiple chambers can run side-by-side with or without cell-secreted factors so that effects of diffusible signalling can be isolated from confounding factors such as shear, nutrient depletion, and microsystem effects. By comparing cell growth and differentiation with different diffusible signalling conditions, the authors found that soluble factor signalling drives neuroectodermal commitment of mESCs through both fibroblast growth factor 4-dependent and -independent pathways. Another approach that also successfully utilized micro-valves provided different mechanical stimulation and defined flow environments to study cell–cell communication in suspension.41 In general, on-chip valving methods have been demonstrated as a versatile, quantitative, and high-throughput method in many cell–cell and cell–microenvironment studies.42–45
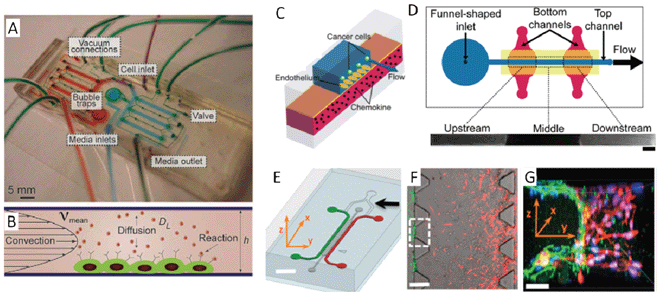 | ||
| Fig. 2 (A)–(B) Perfusion chip integrated with on-chip valves and pumps for the investigation of autocrine and paracrine loops. (A) The image shows a valve-based microfluidic perfusion device in which the upper pneumatic control layer is in green, while the culture chambers are in red and blue. (B) The schematic describes this device which can fine tune the convection, diffusion, and reaction. (C)–(D) Microfluidic vasculature enables the study of cancer and endothelial cell interactions under controlled conditions. (C) The schematic shows that the construction of the microfluidic vasculature which is made by sandwiching a thin, porous polyester membrane between the top and bottom PDMS layers. Chemokines diffuse through the membrane and activate the endothelium from the basal face while the cancer cells flow through the top channel. (D) The top view of the microfluidic device showing the funnel-shaped inlets, flow direction in the top channel, and the design of region-specific chemokine stimulation. When both lower channels were filled with fluorescent dye solution, HDMECs overlying intersecting locations were labelled with the dye. The lower panel shows the spatially-restricted remainder of the fluorescent dye at 5.5 h after initial treatment (scale bar: 800 μm). (E)–(G) Three-dimensional hydrogel matrix microfluidic device for tumour cell intravasation and endothelial barrier function studies. (E) Schematic of a microfluidic device to model the tumour–vasculature interface in three-dimensions. Fibrosarcoma and endothelial cells are separately loaded into the channels illustrated with red or green colours, respectively. The endothelial cells can be cultured to form a monolayer of artificial vasculature. (F) Fibrosarcoma cells (red) invade through the three-dimensional hydrogel matrixes (gray) toward the endothelium (green). The white dashed square indicates an interface region of the three-dimensional hydrogel matrix (scale bar: 300 μm). (G) A three-dimensional constructed image of the tumour cells invading and adhering to the endothelium in the intravasation process (scale bar: 30 μm). Images reproduced from ref. 37, 53 and 69. | ||
Instead of discriminating between the contact-based and soluble factor-based communication, many physiological and pathological processes result from the combination of the two types of communications. An example of this is cancer cell intravasation, the entry of cancer cells into blood vessels, which is a key step in cancer metastasis.46 This complex process involves the interaction between cancer cells and endothelial layers of blood vessels mediated by both contact-based and soluble factor communication. A systematic understanding of the underlying mechanism of cancer cell intravasation is critical for the development of effective cancer treatments. However, the interactions between the tumour cells and vascular endothelial cells occur in a very complicated microenvironment. The cellular milieu contains a diverse mixture of diffusion factors, biophysical forces and cell–matrix interactions, making it challenging to study and quantitatively measure the intravasation process in vivo.46 While using planar microfluidics can overcome some of these difficulties, it is impossible to reconstitute the in vivo process with conventional two dimensional monolayer cell culture models. Methods that are able to provide a controlled three-dimensional environment must therefore be employed to study this process.
Commercially available track-etched membranes (polycarbonate, polyethylene terephthalate, or other materials) can be integrated into two poly(dimethylsiloxane) (PDMS) modes to form three-dimensional on-chip structures for the study of diverse cellular processes.47–52 Using such a method, the Takayama group created an artificial microfluidic vasculature to study the intravascular adhesion of metastatic breast cancer cells.53 The microfluidic vasculature is comprised of a PDMS/membrane/PDMS structure (Fig. 2C). A funnel shaped inlet is built and connected to the main channel on the top PDMS layer. The main channel on the top PDMS layer contains a confluent monolayer of human dermal microvascular endothelial cells (HDMECs) on a polyester membrane (Fig. 2D). The membrane allows small bio-molecules to diffuse through certain intersections between the top channel and individual bottom channels. When both lower channels were filled with cell staining fluorescent dye solution, HDMECs overlying intersecting locations become labelled with dyes. The lower panel of Fig. 2D shows a spatially-restricted image of the remaining fluorescent dye approximately 5.5 h after initial treatment. By activating endothelia over certain intersecting locations with different effector proteins, such as chemokines, serial tumour–endothelial cell interactions can be reproduced with different metastasis-supporting potential in a method similar to the way they occur physiologically. The authors find that a particular chemokine, called CXCL12, acts through a particular receptor, CXCR4, on the endothelium to promote the adhesion of circulating breast cells, suggesting that this CXCL12–CXCR4 signalling may help to limit breast cancer metastasis. This work demonstrates that this microfluidic vasculature can mimic organ-specific localization and polarization of signalling molecules under variable flow conditions, offering a spatially-restricted stimulation of the endothelium for the study of the mechanism of complex cancer cell metastasis.
Another approach to mimic in vivo conditions involves the incorporation of microfluidic devices into a three-dimensional hydrogel matrix.54–69 This method provides an ideal means of regulating soluble factors, cell–cell interactions, and cell–matrix interactions. Moreover, the local distribution of secreted soluble factors from cells embedded in three-dimensional extracellular matrix hydrogels accumulates similarly to real tissues.57,61 This three-dimensional microfluidic device allows the creation of a microenvironment that approximates the in vivo environment, provides spatial and temporal control of micro-scale fluid behaviours, and enables high resolution, real-time imaging. These capabilities make this approach well suited to study the communication of cancer cells during the intravasation process. Recently, Zervantonakis et al. developed a microfluidic device to recreate the tumor–vascular interface in three-dimensions to test the hypothesis that carcinoma cell intravasation is regulated by signalling molecules from interacting cells and cellular interactions with macrophages.69 The device contains two independently addressable microchannels connected via a three-dimensional extracellular matrix hydrogel (Fig. 2E), for seeding the tumor and endothelial cells. The endothelial cells form a continuous monolayer on the three-dimensional extracellular matrix–endothelial channel interface while the tumour cells migrate to the endothelial barrier under the regulation of externally applied growth-factor or paracrine signals produced by other cells (Fig. 2F). The dynamic tumour cell intravasation process can be monitored with high-resolution and time-lapse microscopy (Fig. 2G). The spatially resolved endothelial permeability was measured and demonstrated that endothelial barriers impair correct signalling with macrophages via secretion of tumour necrosis factor alpha, a cytokine known to regulate immune cells. Further experimentation with highly invasive fibrosarcoma cells also revealed that endothelial barrier impairment is associated with the higher number and faster dynamics of tumour and endothelial cell interactions.
In this section we highlighted the recent developments in microfluidic approaches to intercellular population communication studies. Many major advances are currently being made using such microfluidic tools as laminar flow, on-chip valving and pumping, and incorporation of membrane or hydrogel, along with an assortment of other exciting technological developments. These devices provide local cell density control, spatial cell population operation, small fluidic volume manipulation and real-time, in situ imaging capability. With these technologies, researchers are able to investigate gap junction and soluble factor communication in a more controllable and quantitative manner than previously possible.
Intercellular communication at single cell level
Studying intercellular communication in a group of cells is indispensable and provides a rich source of information on global impacts. Studying single cells, however, will help to reveal otherwise unattainable details and mechanisms by reducing the complexity of the communication of the system. In addition, high-throughput single cell studies will shed light on the heterogeneity of global intercellular communications. In a two-cell system (homotypic or heterotypic cell pairs), intercellular communication can occur by many different mechanisms such as direct physical contact, diffusion of soluble factors, electrical signal transmission, and transduction of mechanical cues within the extracellular matrix. The techniques to study single cells are more demanding as they require the precision to manipulate the individual cells. In this section, we will discuss several recent advances for probing cell–cell communication at the single cell level utilizing different micromanipulation technologies.One means to manipulate single cells in suspension is microfluidic hydrodynamic trapping.70–73 Suspended cells flow along a path of least fluidic resistance and cells will be carried to trap or bypass channels. Once a trap is occupied by one cell, fluidic resistance is increased and subsequent cells will be sequentially directed to the next trap. To date, many efforts have been made to employ this approach in simplifying intercellular communication and unravelling the underlying mechanisms of this phenomenon. Specifically, the Lee group developed a method for trapping multiple cell pairs for the study of intercellular communication by functional gap junctions.25 They later improved the functionality of their approach to a single-cell level co-culture platform for the study of intercellular communication between mouse embryonic stem cells (mESC) and mouse embryonic fibroblast cells (MEF).71 By combining hydrodynamic trapping with a semi-isolated chamber, their device can perform highly efficient cell pairing using minimal physical restraints for highly reliable, long-term characterization. As shown in Fig. 3A, a single MEF cell array was created by trapping each individual cell in small junctions located in the bottom of the chambers. Those cells were incubated so that they could migrate away from the trapping junction to the culture chamber. Then, in a similar manner, the mESCs can be trapped at the same position. Thus, the distance between a mESC and a MEF can be tuned by adjusting the incubation time of the MEF cell. In a single operation, this platform yields greater than 50% efficiency in the heterotypic pairing of single cells. Within the first 10 h of visualization of the MEF–mESC pair interaction, migration behaviour dependant on cell-to-cell distance can be classified into three patterns. These include Type 1, where the MEF and mESC pair showed a correlated migration with a relatively close initial intercellular distance (less than 80 μm); Type 2, where the MEF and mESC did not show any clear migration with intermediate initial intercellular distance (between 80 μm to 260 μm), and Type 3, where the MEF randomly migrated and the mESC did not migrate with far initial intercellular distance (more than 260 μm). Overall, the hydrodynamic trapping method can efficiently trap and group cells or particles in a large-scale array format. One consideration is that the trapping array formation is flow velocity dependent and the application of inappropriate flow will lead to mechanical cell damage. With optimized chip design and proper flow control, however, this method offers an easy and high-throughput means to study intercellular communication.
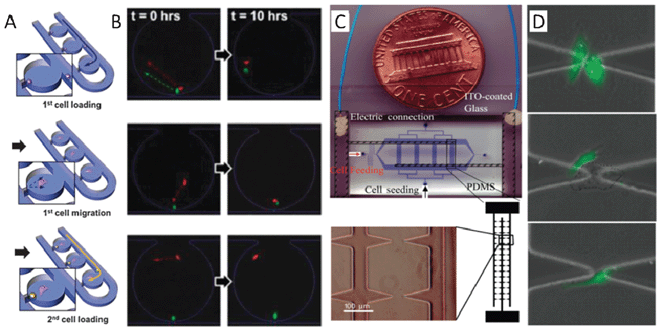 | ||
| Fig. 3 (A)–(B) One-to-one cell pairing and interaction studies with a hydrodynamic trapping microfluidic device. (A) A schematic showing the process of creating a heterotypic cell pair in isolated chambers by the hydrodynamic trapping method. A single mouse embryonic fibroblast cell (MEF) array is created by trapping each individual cell in small junctions located in the bottom of the chambers. Those cells are incubated so that they migrate away from the trapping junction toward the culture chamber. Then, mouse embryonic stem cells (mESC) can be trapped in the same way, but at different initial intercellular distances. MEF cell migration distance can be obtained through tuning of the incubation time of the MEF cell. (B) The three types of distance- and intercellular communication-dependent migration behaviour. Type 1 (top panel): the MEF and mESC show correlated migrations with close initial intercellular distance; Type 2 (middle panel): the MEF and mESC did not show any clear migration with intermediate initial intercellular distance; Type 3 (bottom panel): the MEF showed random migrations and the mESC did not migrate with far initial intercellular distance. (C)–(D) Homotypic or heterotypic cell grouping and interaction investigation in a dielectrophoretic-based microfluidic device. (C) The fabrication of a dielectrophoretic-based microfluidic device includes a PDMS channel network (blue) and a glass substrate patterned with an array of sharp, triangular ITO electrodes (bottom panel). The electric field can be sharply focused around the tips of the triangular electrodes so that the cells are pulled down and positioned at each tip. Single cells can be trapped with approximately 77% efficiency at the electrode tips. (D) Images of a patterned HUVEC–HUVEC pair (top panel), a HUVEC–A549 pair (middle panel), and a single HUVEC cell (bottom panel) after culturing for 4 h. Images reproduced from ref. 71 and 81. | ||
Another means to aid in the study of communication at the single cell level is dielectrophoresis, which can manipulate and pattern cells by exerting force using a non-uniform electric field.74–80 Once merged with microfluidics it provides a combination of microenvironment control and the ability to precisely arrange single cells. The Levchenko group utilized this platform to analyse communication mechanisms during the tumour microvascularization process.81 The device was fabricated by bonding a PDMS channel onto a glass slide patterned with an array of sharply triangular indium-tin-oxide electrodes (Fig. 3C). The electric field can be sharply focused around the tips of the triangular electrodes so that the cells can be pulled down and positioned at each tip and single cells can be trapped at the electrode tips with an approximately 77% rate of success. This device allows patterning of many homotypic or heterotypic cell pairs with specific initial locations and enables precise regulation of soluble factors as well as the alternation of the cell culture medium (Fig. 3D). Note that a special non-conductive cell medium is required in the manipulation process because the conductive molecules will destroy the non-uniform electric field distribution. In addition, the cells must be maintained in a low conductivity medium with careful control over the field as the electrical current may be damaging to the cells. Through the dynamic characterization of pairing endothelial cells (HUVEC cells) and endothelial tumour cells (A549 cells, human lung cancer cells), the authors found that cell migration speed and migration patterns were strongly affected by the secreted factors, collagen, and vascular endothelial growth factor (VEGF). After blocking the VEGF-mediated receptor, the movement of A549–HUVEC pairs was not greatly affected, though movement of single HUVECs and HUVEC–HUVEC pairs changed dramatically. Interestingly, VEGF secretion by cancer cells did not have a significant effect on cell migration. This work directly demonstrates the significance of intercellular communication for cancer cell growth and metastasis. This approach provides a controllable, flexible and effective means for high throughput intercellular communication studies.
Other technologies such as optical, magnetic, and acoustic cell manipulation have also been integrated into microfluidics in order to manipulate single cells. In general, optical methods, such as optical tweezers, are good at precisely handling single cells even at subcellular resolution.82–84 Pascoal et al. studied the nanotube-mediated cell–cell communications using optical tweezers.83 By focusing optical tweezers on the membrane of a living cell, the plasma membrane can be pulled to form membrane nanotubes with 200 nanometre diameters and 100 micrometre lengths. These membrane nanotubes from one cell can be stretched to make contact with adjacent cells, forming intercellular electrical communication via connexions. Optical tweezers can also be used to manipulate cells in three-dimensions, to precisely control the attachment of single cells to a matrix or to engineer interactions between two or three cells. However, this technology is limited by throughput (i.e., the number of cells in the cell assembly) and its complexity and can also potentially cause damage to cells due to the high power density. Large numbers of heterotypic cell pairs can also be created using a magnetic pattern array method. Felton et al. produced thousands of magnetic traps, each designed to accommodate only two cells and confine them at these sites for co-culturing.85 The two types of cells labelled with ferromagnetic nanowires can be attached to the array sites through magnetic dipole interactions, and cultured for further cell interaction studies. This magnetic method also can perform high throughput cell stretching for mechanotransduction studies.86 The drawback of this technology is the requirement of magnetic labelling of the cells. The emerging acoustic approaches87–96 may provide a means to overcome some of these limitations by allowing micro-scale manipulation in a non-invasive, non-contact manner and may offer new opportunities for probing single cell intercellular communication.
Perspectives
In the previous sections, we examined some recent advances in the development of microfluidic devices for the investigation of cell–cell communication. Those devices address several different challenges faced in macro-scale cell culture systems. The microfluidic systems provide simplified conditions and the ability to quantitatively measure signals for investigation of intercellular communication from whole cell populations to the single cells level. Overall, in designing and constructing these devices, researchers have sought to push the boundaries of our understanding of intercellular communication and derive benefits from these studies that will, ultimately, help to diagnose97–99 and treat disease.100It is essential to recognize that there are many types of signals that make up intercellular communication. Each individual cell can communicate with others through physical contact, diffusion of soluble factors, electrical signal transmission, and transduction of mechanical cues within the extracellular matrix. Challenges still remain in precisely isolating a specific signal from the vast background of communication information constantly being produced. However, the tools provided by microfluidic approaches have begun to demonstrate their utility in this endeavour. Moreover, we have begun to realize that each type of intercellular communication may involve numerous types of signals. For example, soluble factor communication can be mediated by cytokines, hormones, growth factors, and other bio-molecules.12 Some of the signalling molecules have been well studied, but most of them may still remain unknown due to the limitations of the current, conventional method of study. New microfluidic approaches are needed to aid in the discovery of new signalling molecules and to identify their functions. Recently, the combination of mass spectrometry and microfluidic devices has been employed to characterize signalling molecules and metabolites, offering a glimpse into the unknowns of soluble factor signalling.101 Approaches such as these are necessary in order to fully reveal how the temporal and spatial control of communication by soluble factors determines the fate of individual cells. The incorporation of in-line bioanalytical technologies, such as mass spectrometry or nuclear magnetic resonance (see below), with microfluidic devices holds great promise for the future of cell–cell communication studies.
Physical interactions and mechanical forces also coordinate many physiological activities and pathological processes such as the intravasation and extravasation of cancer cells and early embryonic development. Transduction of mechanical signals into cellular biochemical responses allows neighbouring cells to communicate with one another.102–105 However, it is challenging to precisely isolate the intercellular mechanical stimulus from the non-cellular environment and provide a controllable mechanical stimulus to a small number of target cells without influencing neighbouring cells.106 To effectively study the effect of mechanical forces and their physiological responses, these issues need to be considered during experimental design. The ideal device would allow for three-dimensional manipulations of cells in order to construct cell assemblies in suspension or in a matrix with precise control over cell position, contact, distance, and various mechanical stimuli. In addition, non-contact manipulation would be preferential in order to avoid the cell damage that may result from physical handling. Such devices should also allow for the performance of high-throughput assays within a well-defined chemical environment.
In addition to considerations of varying signal type, isolation and quantification of signals is an important element to consider. Experiments conducted with such devices will deepen our understanding of intercellular communication and its relationship to organism-scale functionality and disease development. An example of the evolution of such devices is the quantitative characterization of the distance dependence of cell–cell interactions with the single cell barcode chip.107 This technology, developed by the Heath group, incorporates a multiplex sandwich-type enzyme-linked immunoassay with microfluidics to enable statistical analysis of proteins from interacting cell pairs (Fig. 4A). The cells were loaded into many small chambers to form cell pairs, cultured for a defined period of time, and lysed in sealed chambers under the control of pneumatic valves. The secreted, cytoplasmic, and membrane proteins from the interacting cell pairs can then be captured and characterized. This assay allows the study of the dependence of protein expression level on cell–cell distance. After incubating individual cell pairs for 6 h, the average protein levels in each of 500 cell pairs is calculated for each distance range and then normalized to the single cell data from the same chip. The authors determined that all of the proteins studied were inhibited at short cell–cell distances and three of the proteins (IL-6, p-EGFR, and p-ERK) were activated at larger cell–cell distances (Fig. 4B). While microfluidic approaches such as these have yielded some successes in the identification and quantification of known communication signals, new microfluidic systems and improved techniques are still in great demand in order to define and characterize the many still unknown communication signals.
 | ||
| Fig. 4 (A)–(B) Single cell barcode chip for quantitative investigation of distance-dependent cell–cell interactions. (A) A microfluidic-based, multiplex, quantitative protein assay allows statistical analysis of proteins from interacting cell pairs. The cells can be loaded into many small chambers to form cell pairs, cultured, and, after a defined time, lysed at sealed chambers. The secreted, cytoplasmic, and membrane proteins from the interacting cell pair can then be detected in situ. This device can provide information on the number and position of cells, as well as the fluorescence intensity from each antibody-based protein assay. (B) This curve illustrates that the cell signalling activity is dependent on the cell–cell distance. After incubating individual cell pairs for 6 h, the average protein level in each of the 500 cell pairs is calculated for each distance range and then normalized to the single cell data from the same chip. At small cell–cell distance all proteins show inhibitory activity, while at larger cell–cell distances, three proteins (IL-6, p-EGFR, and p-ERK) are activating. (C)–(E) A microfluidic system for on-chip detection of circulating microvesicles and glioblastoma cancer therapeutic response. (C) A schematic showing the integrated microfluidic system for on-chip detection of circulating microvesicles. This system allows the detection and concentration measurement of MNP-targeted microvesicles as well as in-line μNMR characterization so that the abundant microvesicles produced by human glioblastoma cells can be analysed. (D) High-magnification image of many microvesicles on the cell surface and (E) transmission electron microscopy image of microvesicles targeted with magnetic nanoparticles via a CD63-specific antibody. The functionalized magnetic nanoparticles can selectively bind with target circulating microvesicles (black dots and indicated by an arrow). Images reproduced from ref. 107 and 111. | ||
Circulating microvesicles (cMVs), fragments of cell plasma membrane ranging from 50 nm to 100 nm, are considered as important cell communication mediators.108 Many reports have shown that they can contain various kinds of cell contents, such as messenger RNAs, microRNAs, proteins, and lipids.109 Although much evidence has indicated the role of microvesicles in cell–cell communication, the exact mechanism of such communication is still unclear.110 Microfluidic technologies are clearly an ideal fit for this type of investigation. For example, Shao and colleagues developed an integrated microfluidic system coupled with on-chip microfiltration and miniaturized micro nuclear magnetic resonance (μNMR) for the characterization of microvesicles.111 They used this device to profile microvesicles released by glioblastoma cancer cells directly from blood samples of patients by determining the expression levels of marker proteins (Fig. 4C). It is noteworthy that the authors used this platform to monitor drug treatment effects both in vitro and in vivo. In their design, glioblastoma multiform (GBM) microvesicles are first labelled with magnetic nanoparticles (MNP) coated with antibodies that target the protein markers. cMVs are passed through a chaotic mixer and a membrane filter where the labelling occurs. At the outlet of the microfluidic chamber the MNP–cMV complexes are injected into the in-line μNMR through a microcoil for signal acquisition and quantitative readouts of the expression levels of protein biomarkers (Fig. 4D–E). Notably, the expression levels of certain proteins can serve as signatures of GBM formation allowing for effective discrimination of GBM-derived cMVs from the host cells. Furthermore, this protein typing assay has been demonstrated to allow the evaluation of drug treatment efficacy.
As discussed in this Focus article, the employment of microfluidics as a tool for cell–cell communication study has provided many new opportunities to advance our knowledge in this field. To date, the investigation of intercellular population communication with microfluidic devices has enabled better control over the microenvironment. In addition, single cell intercellular communication studies have now become possible with advanced microfluidic manipulation technology. The development of new technologies to identify and quantitatively characterize various communication signals are highly desirable and will surely lead to exciting new advances for this field. Finally, to facilitate the adoption of microfluidic devices by the biomedical research community, future device development should emphasize precision, robustness, throughput, and reproducibility.
Acknowledgements
The authors would like to thank Mr Justin Kiehne and Dr Ray Pugh's kind help and discussion. This research was supported by the National Institutes of Health (NIH) Director's New Innovator Award (1DP2OD007209-01), National Science Foundation and the Penn State Center for Nanoscale Science (MRSEC). Components of this work were conducted at the Penn State node of the NSF-funded National Nanotechnology Infrastructure Network (NNIN).References
- S. N. Bhatia, U. J. Balis, M. L. Yarmush and M. Toner, FASEB J., 1999, 13, 1883–900 CAS.
- N. M. Kumar and N. B. Gilula, Cell, 1996, 84, 381–8 CrossRef CAS.
- T. Pawson, Nature, 1995, 373, 573–80 CrossRef CAS.
- C. A. Staton, S. M. Stribbling, S. Tazzyman, R. Hughes, N. J. Brown and C. E. Lewis, Int. J. Exp. Pathol., 2004, 233–48 CrossRef CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–11 CrossRef CAS.
- H.-W. Wu, C.-C. Lin and G.-B. Lee, Biomicrofluidics, 2011, 5, 13401 CrossRef.
- D. N. Breslauer, P. J. Lee and L. P. Lee, Mol. BioSyst., 2006, 2, 97–112 RSC.
- M. L. Kovarik, P. C. Gach, D. M. Ornoff, Y. Wang, J. Balowski, L. Farrag and N. L. Allbritton, Anal. Chem., 2012, 84, 516–40 CrossRef CAS.
- K. Gupta, D.-H. Kim, D. Ellison, C. Smith, A. Kundu, J. Tuan, K.-Y. Suh and A. Levchenko, Lab Chip, 2010, 10, 2019–31 RSC.
- E. W. K. Young and D. J. Beebe, Chem. Soc. Rev., 2010, 39, 1036–48 RSC.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–54 CrossRef CAS.
- L. Przybyla and J. Voldman, Annu. Rev. Anal. Chem., 2012, 5, 293–315 CrossRef CAS.
- J. Y. Park, S. Takayama and S.-H. Lee, Integr. Biol., 2010, 2, 229–40 RSC.
- C. Yi, C.-W. Li, S. Ji and M. Yang, Anal. Chim. Acta, 2006, 560, 1–23 CrossRef CAS.
- J. Nilsson, M. Evander, B. Hammarström and T. Laurell, Anal. Chim. Acta, 2009, 649, 141–57 CrossRef CAS.
- B. Sun, J. Lembong, V. Normand, M. Rogers and H. A. Stone, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7753–8 CrossRef CAS.
- T. S. Deisboeck and I. D. Couzin, BioEssays, 2009, 31, 190–7 CrossRef.
- J. C. Conrad, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7591–2 CrossRef CAS.
- S. Chen and L. P. Lee, Integr. Biol., 2010, 2, 130–8 RSC.
- F. Wang, H. Wang, J. Wang, H.-Y. Wang, P. L. Rummel, S. V. Garimella and C. Lu, Biotechnol. Bioeng., 2008, 100, 150–8 CrossRef CAS.
- C. Bathany, D. Beahm, J. D. Felske, F. Sachs and S. Z. Hua, Anal. Chem., 2011, 83, 933–9 CrossRef CAS.
- P. Chen, P. Chen, X. Feng, W. Du and B.-F. Liu, Anal. Bioanal. Chem., 2013, 405, 307–14 CrossRef CAS.
- N. Ye, C. Bathany and S. Z. Hua, Lab Chip, 2011, 11, 1096–101 RSC.
- J. Sun, Y. Zheng, X. Feng, W. Du and B.-F. Liu, Anal. Chim. Acta, 2012, 721, 104–9 CrossRef CAS.
- P. J. Lee, P. J. Hung, R. Shaw, L. Jan and L. P. Lee, Appl. Phys. Lett., 2005, 86, 223902 CrossRef.
- D. Ahmed, C. Y. Chan, S.-C. S. Lin, H. S. Muddana, N. Nama, S. J. Benkovic and T. J. Huang, Lab Chip, 2013, 13, 328–31 RSC.
- D. Ahmed, X. Mao, J. Shi, B. K. Juluri and T. J. Huang, Lab Chip, 2009, 9, 2738–41 RSC.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Nature, 2001, 411, 1016 CrossRef CAS.
- H. Yu, I. Meyvantsson, I. A. Shkel and D. J. Beebe, Lab Chip, 2005, 5, 1089–95 RSC.
- V. N. Goral, Y.-C. Hsieh, O. N. Petzold, J. S. Clark, P. K. Yuen and R. A. Faris, Lab Chip, 2010, 10, 3380–6 RSC.
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. M. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–8 RSC.
- M. Domenech, H. Yu, J. Warrick, N. M. Badders, I. Meyvantsson, C. M. Alexander and D. J. Beebe, Integr. Biol., 2009, 1, 267–74 RSC.
- X. Song, B. Kong and D. Li, Biotechnol. Lett., 2008, 30, 1537–43 CrossRef CAS.
- R. D. Lovchik, N. Tonna, F. Bianco, M. Matteoli and E. Delamarche, Biomed. Microdevices, 2010, 12, 275–82 CrossRef.
- M. A. Unger, H.-P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–6 CrossRef CAS.
- A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–63 CrossRef.
- K. Blagovic, L. Y. Kim and J. Voldman, PLoS One, 2011, 6, e22892 CAS.
- D. Ellison, A. Munden and A. Levchenko, Mol. BioSyst., 2009, 5, 1004–12 RSC.
- T.-H. Hsu, J.-L. Xiao, Y.-W. Tsao, Y.-L. Kao, S.-H. Huang, W.-Y. Liao and C.-H. Lee, Lab Chip, 2011, 11, 1808–14 RSC.
- S. H. Hong, M. Hegde, J. Kim, X. Wang, A. Jayaraman and T. K. Wood, Nat. Commun., 2012, 3, 613 CrossRef.
- T. Xu, W. Yue, C.-W. Li, X. Yao and M. Yang, Lab Chip, 2010, 10, 2271–8 RSC.
- J. Lii, W.-J. Hsu, H. Parsa, A. Das, R. Rouse and S. K. Sia, Anal. Chem., 2008, 80, 3640–7 CrossRef CAS.
- W. Liu, L. Li, X. Wang, L. Ren, X. Wang, J. Wang, Q. Tu, X. Huang and J. Wang, Lab Chip, 2010, 10, 1717–24 RSC.
- C.-H. Zheng, L. Zhao, G.-E. Chen, Y. Zhou, Y.-H. Pang and Y.-Y. Huang, Anal. Chem., 2012, 84, 2088–93 CrossRef CAS.
- J.-W. Huang, H.-J. Pan, W.-Y. Yao, Y.-W. Tsao, W.-Y. Liao, C.-W. Wu, Y.-C. Tung and C.-H. Lee, Lab Chip, 2013, 13, 1114–20 RSC.
- D. Wirtz, K. Konstantopoulos and P. C. Searson, Nat. Rev. Cancer, 2011, 11, 512–22 CrossRef CAS.
- H. J. Kim, J. Q. Boedicker, J. W. Choi and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18188–93 CrossRef CAS.
- A. Y. Hsiao, Y.-S. Torisawa, Y.-C. Tung, S. Sud, R. S. Taichman, K. J. Pienta and S. Takayama, Biomaterials, 2009, 30, 3020–7 CrossRef CAS.
- S. H. Ma, L. A. Lepak, R. J. Hussain, W. Shain and M. L. Shuler, Lab Chip, 2005, 5, 74–85 RSC.
- Y.-S. Torisawa, B. Mosadegh, G. D. Luker, M. Morell, K. S. O'Shea and S. Takayama, Integr. Biol., 2009, 1, 649–54 RSC.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–8 CrossRef CAS.
- V. V. Abhyankar and D. J. Beebe, Anal. Chem., 2007, 79, 4066–73 CrossRef CAS.
- J. W. Song, S. P. Cavnar, A. C. Walker, K. E. Luker, M. Gupta, Y.-C. Tung, G. D. Luker and S. Takayama, PLoS One, 2009, 4, e5756 Search PubMed.
- I. K. Zervantonakis, C. R. Kothapalli, S. Chung, R. Sudo and R. D. Kamm, Biomicrofluidics, 2011, 5, 13406 CrossRef.
- H. Tekin, J. G. Sanchez, C. Landeros, K. Dubbin, R. Langer and A. Khademhosseini, Adv. Mater., 2012, 24, 5543–7 CrossRef CAS.
- J. Fukuda, A. Khademhosseini, Y. Yeo, X. Yang, J. Yeh, G. Eng, J. Blumling, C.-F. Wang, D. S. Kohane and R. Langer, Biomaterials, 2006, 27, 5259–67 CrossRef CAS.
- B. G. Chung, K.-H. Lee, A. Khademhosseini and S.-H. Lee, Lab Chip, 2012, 12, 45–59 RSC.
- H. Aubin, J. W. Nichol, C. B. Hutson, H. Bae, A. L. Sieminski, D. M. Cropek, P. Akhyari and A. Khademhosseini, Biomaterials, 2010, 31, 6941–51 CrossRef CAS.
- D. R. Albrecht, V. L. Tsang, R. L. Sah and S. N. Bhatia, Lab Chip, 2005, 5, 111–8 RSC.
- S. Chung, R. Sudo, P. J. Mack, C.-R. Wan, V. Vickerman and R. D. Kamm, Lab Chip, 2009, 9, 269–75 RSC.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–59 CrossRef CAS.
- S. Ladet, L. David and A. Domard, Nature, 2008, 452, 76–9 CrossRef CAS.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Y. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–62 CrossRef CAS.
- Y. Du, E. Lo, S. Ali and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9522–7 CrossRef CAS.
- T. Liu, B. Lin and J. Qin, Lab Chip, 2010, 10, 1671–7 RSC.
- J. H. Yeon, H. R. Ryu, M. Chung, Q. P. Hu and N. L. Jeon, Lab Chip, 2012, 12, 2815–22 RSC.
- D.-H. T. Nguyen, S. C. Stapleton, M. T. Yang, S. S. Cha, C. K. Choi, P. A. Galie and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6712–7 CrossRef CAS.
- A. P. Wong, R. Perez-Castillejos, J. Christopher Love and G. M. Whitesides, Biomaterials, 2008, 29, 1853–61 CrossRef CAS.
- I. K. Zervantonakis, S. K. Hughes-Alford, J. L. Charest, J. S. Condeelis, F. B. Gertler and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13515–20 CrossRef.
- J.-P. Frimat, M. Becker, Y.-Y. Chiang, U. Marggraf, D. Janasek, J. G. Hengstler, J. Franzke and J. West, Lab Chip, 2011, 11, 231–7 RSC.
- S. Hong, Q. Pan and L. P. Lee, Integr. Biol., 2012, 4, 374–80 RSC.
- A. Skelley, O. Kirak, H. Suh, R. Jaenisch and J. Voldman, Nat. Methods, 2009, 6, 147–52 CrossRef CAS.
- W.-H. Tan and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1146–51 CrossRef CAS.
- R. S. Thomas, H. Morgan and N. G. Green, Lab Chip, 2009, 9, 1534–40 RSC.
- C.-T. Ho, R.-Z. Lin, W.-Y. Chang, H.-Y. Chang and C.-H. Liu, Lab Chip, 2006, 6, 724–34 RSC.
- M. Gel, Y. Kimura, O. Kurosawa, H. Oana, H. Kotera and M. Washizu, Biomicrofluidics, 2010, 4, 022808–8 Search PubMed.
- M. Suzuki, T. Yasukawa, H. Shiku and T. Matsue, Biosens. Bioelectron., 2008, 24, 1049–53 CrossRef.
- D. R. Albrecht, T. B. Underhill, G. H. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–75 CrossRef CAS.
- J. Voldman, Annu. Rev. Biomed. Eng., 2006, 8, 425–54 CrossRef CAS.
- S. V. Puttaswamy, S. Sivashankar, R.-J. Chen, C.-K. Chin, H.-Y. Chang and C. H. Liu, Biotechnol. J., 2010, 5, 1005–15 CrossRef CAS.
- Z. Yin, D. Noren, C. J. Wang, R. Hang and A. Levchenko, Mol. Syst. Biol., 2008, 4, 232–6 CrossRef.
- H. Zhang and K.-K. Liu, J. R. Soc. Interface, 2008, 5, 671–90 CrossRef CAS.
- P. Pascoal, D. Kosanic, M. Gjoni and H. Vogel, Lab Chip, 2010, 10, 2235–41 RSC.
- W. Timp, U. Mirsaidov, P. Matsudaira and G. Timp, Lab Chip, 2009, 9, 925–34 RSC.
- E. J. Felton, C. R. Copeland, C. S. Chen and D. H. Reich, Lab Chip, 2012, 12, 3117–26 RSC.
- P. Tseng, J. W. Judy and D. Di Carlo, Nat. Methods, 2012, 9, 1113–9 CrossRef CAS.
- J. Shi, S. Yazdi, S.-C. S. Lin, X. Ding, I.-K. Chiang, K. Sharp and T. J. Huang, Lab Chip, 2011, 11, 2319–24 RSC.
- X. Ding, S.-C. S. Lin, M. I. Lapsley, S. Li, X. Guo, C. Y. Chan, I.-K. Chiang, L. Wang, J. P. McCoy and T. J. Huang, Lab Chip, 2012, 12, 4228–31 RSC.
- X. Ding, S.-C. S. Lin, B. Kiraly, H. Yue, S. Li, I.-K. Chiang, J. Shi, S. J. Benkovic and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11105–9 CrossRef CAS.
- S. Li, X. Ding, F. Guo, Y. Chen, M. I. Lapsley, S.-C. S. Lin, L. Wang, J. P. McCoy, C. E. Cameron and T. J. Huang, Anal. Chem., 2013, 85, 5468–74 CrossRef CAS.
- X. Mao and T. J. Huang, Lab Chip, 2012, 12, 4006–9 RSC.
- J. Shi, H. Huang, Z. Stratton, Y. Huang and T. J. Huang, Lab Chip, 2009, 9, 3354–9 RSC.
- X. Ding, J. Shi, S.-C. S. Lin, S. Yazdi, B. Kiraly and T. J. Huang, Lab Chip, 2012, 12, 2491–7 RSC.
- J. Shi, X. Mao, D. Ahmed, A. Colletti and T. J. Huang, Lab Chip, 2008, 8, 221–3 RSC.
- S.-C. S. Lin, X. Mao and T. J. Huang, Lab Chip, 2012, 12, 2766–70 RSC.
- J. Shi, D. Ahmed, X. Mao, S.-C. S. Lin, A. Lawit and T. J. Huang, Lab Chip, 2009, 9, 2890–5 RSC.
- Y. Zhao, Z. S. Stratton, F. Guo, M. I. Lapsley, C. Y. Chan, S.-C. S. Lin and T. J. Huang, Lab Chip, 2013, 13, 17–24 RSC.
- X. Mao and T. J. Huang, Lab Chip, 2012, 12, 1412–6 RSC.
- P. Li, Z. S. Stratton, M. Dao, J. Ritz and T. J. Huang, Lab Chip, 2013, 13, 602–9 RSC.
- P. Neuži, S. Giselbrecht, K. Länge, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–32 CrossRef.
- S. Mao, J. Zhang, H. Li and J.-M. Lin, Anal. Chem., 2013, 85, 868–76 CrossRef CAS.
- S. S. Hur, J. C. Del Álamo, J. S. Park, Y.-S. Li, H. A. Nguyen, D. Teng, K.-C. Wang, L. Flores, B. Alonso-Latorre, J. C. Lasheras and S. Chien, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11110–5 CrossRef CAS.
- Z. Liu, J. L. Tan, D. M. Cohen, M. T. Yang, N. J. Sniadecki, S. A. Ruiz, C. M. Nelson and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9944–9 CrossRef CAS.
- O. G. Shcherbakova, W. I. Weis, B. L. Pruitt, W. James, A. R. Dunn, N. Borghi, M. Sorokina, W. J. Nelson and R. D. Alexander, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19034 Search PubMed.
- C. S. Chen, J. Tan and J. Tien, Annu. Rev. Biomed. Eng., 2004, 6, 275–302 CrossRef CAS.
- W. J. Polacheck, R. Li, S. G. M. Uzel and R. D. Kamm, Lab Chip, 2013, 13, 2252–67 RSC.
- J. Wang, D. Tham, W. Wei, Y. S. Shin, C. Ma, H. Ahmad, Q. Shi, J. Yu, R. D. Levine and J. R. Heath, Nano Lett., 2012, 12, 6101–6 CrossRef CAS.
- G. Camussi, M. C. Deregibus, S. Bruno, V. Cantaluppi and L. Biancone, Kidney Int., 2010, 78, 838–48 CrossRef CAS.
- C. Théry, L. Zitvogel and S. Amigorena, Nature reviews. Immunology, 2002, 2, 569–79 Search PubMed.
- M. A. Antonyak, B. Li, K. Lindsey, J. L. Johnson, J. E. Druso, K. L. Bryant, D. A. Holowka, R. A. Cerione and L. K. Boroughs, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17569 CrossRef CAS.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–40 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |