Centrifugal gas-phase transition magnetophoresis (GTM) – a generic method for automation of magnetic bead based assays on the centrifugal microfluidic platform and application to DNA purification†
Oliver
Strohmeier
*ab,
Alexander
Emperle
a,
Günter
Roth
ab,
Daniel
Mark
b,
Roland
Zengerle
abc and
Felix
von Stetten
ab
aLaboratory for MEMS Applications, Department of Microsystems Engineering – IMTEK, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: vstetten@imtek.uni-freiburg.de; Fax: +49 761 203 73299; Tel: +49 761 203 73213
bHSG-IMIT, Wilhelm-Schickard-Straße 10, 78052 Villingen-Schwenningen, Germany
cBIOSS – Centre for Biological Signalling Studies, University of Freiburg, 79110 Freiburg, Germany
First published on 11th October 2012
Abstract
Transportation of magnetic beads between different reagents plays a crucial role in many biological assays e.g. for purification of biomolecules or cells where the beads act as a mobile solid support. Therefore, usually a complex set-up either for fluidic processing or for manipulation of magnetic beads is required. To circumvent these drawbacks, we present a facile and automated method for the transportation of magnetic beads between multiple microfluidic chambers on a centrifugal microfluidic cartridge “LabDisk”. The method excels by requiring only one stack of stationary permanent magnets, a specific microfluidic layout without actively controlled valves and a predefined frequency protocol for rotation of the LabDisk. Magnetic beads were transported through three fluidically separated chambers with a yield of 82.6% ± 3.6%. Bead based DNA purification from a dilution series of a Listeria innocua lysate and from a lambda phage DNA standard was demonstrated where the three chambers were used for binding, washing and elution of DNA. Recovery of L. innocua DNA was up to 68% ± 24% and for lambda phage DNA 43% ± 10% compared to manual reference purification in test tubes. Complete purification was conducted automatically within 12.5 min. Since all reagents can be preloaded onto the LabDisk prior to purification, no further hands-on steps are required during processing. Due to its modular and generic character, the presented method could also be adapted to other magnetic bead based assays e.g. to immunoassays or protein affinity purification, solely requiring the adjustment of number and volumes of the fluidic chambers.
Introduction
Magnetic beads with micrometre size and specific surface functionalization became prominent materials for a variety of biological assays1–3 mainly due to their broad commercial availability.4 Similar to non-magnetic beads, their high surface-to-volume ratio and the possibility of actively improving mass transport to the (reactive) surface make them superior to stationary solid phases5 especially when it is required to capture minute amounts of target analyte in liquid samples. Thereby, assay sensitivity is improved and reagent volumes can be reduced, which facilitates the integration of beads in microfluidic applications. Various applications including DNA separation6 and extraction,7 mRNA purification,8 immunoassays3,9,10 or cell capture11 and separation12 using magnetic beads in microfluidic structures have been reported. In all these applications, the beads were sequentially brought into contact with a series of different reagents, which was achieved by manipulating the beads by an external magnetic field. The availability of an appropriate bead-handling technique is therefore an essential requirement.In general, methods for handling magnetic beads can be divided into three groups: (1) Beads are (temporarily) immobilized by a magnetic field and sequentially flushed by different reagents.9,13 However, for resuspension and efficient washing of the beads, the magnetic field needs to be removed or switched off and the reagent flow needs to be stopped in order not to drag away any beads. (2) Beads are actively transported between different reagents e.g. by actuated permanent magnets8,14–16 or coil arrays17,18 requiring rather complex and costly set-ups for magnet actuation or fabrication of the coil arrays. (3) Beads are attracted over interfaces of adjacent laminar reagent streams by magnetophoresis19 using permanent magnets3,7,12 thereby facilitating continuous processing. However, successful bead transport is highly sensitive to the interplay of magnetic field strength, orientation and the flow rate of the reagents.
Although automated manipulation of magnetic beads is state of the art in pipetting workstations and various microfluidic platforms3,7,11–13,20,21 smart solutions for bead handling in centrifugal microfluidic platforms are very limited and need to be further explored, especially because centrifugal microfluidic platforms22–25 are expected to have a crucial impact on future automated point-of-care testing.20,26–29
So far, only one centrifugal microfluidic system has been published, which exploited the handling of magnetic beads and sequentially exposed them to different reagents.30 Here, a primary magnet mounted on a linear geared stepping motor underneath the rotating cartridge was used to control the position of a secondary magnet which was integrated in the bottom layer of a double layered cartridge. Depending on the position of the primary magnet, spinning frequency and direction of rotation, the secondary magnet could be moved to four different positions in the bottom layer and thereby manipulate magnetic beads within the microfluidic structure that was integrated into the top layer of the cartridge. During processing, the beads were captured by the magnet in a first chamber and mixed with the sample for immunomagnetic capturing of pathogens. The beads were then flushed with the washing buffer to a second position where they were captured for washing. The washing buffer was transferred to a downstream waste reservoir while the beads remained fixed for subsequent thermal pathogen lysis. The purified cell lysate containing the released DNA could then be used for further analysis. The required fluidic control of the buffer streams was achieved by a complex actuation scheme involving a series of integrated ferrowax valves (LIFM) that could be opened or closed by infra-red laser irradiation.
In this work, we aim at an improved and generic bead handling method on a centrifugal microfluidic LabDisk that excels by requiring solely a stationary permanent magnet, a spinning drive, and a single layered microfluidic LabDisk. The need for costly magnet actuation systems is circumvented by defined positioning of the LabDisk with respect to a stationary permanent magnet. The presented method of bead handling in combination with the modular fluidic layout allows stand-alone processing since all reagents can be loaded onto the LabDisk simultaneously prior to starting the automated frequency protocol. The novel method is demonstrated by magnetic bead based DNA purification, one of the most prevalent applications involving transportation of magnetic beads. Aiming at significant reduction of complexity and costs, the method is meant to facilitate entry of centrifugal microfluidic platforms into the point-of-care market.
Functional principle
Unit operation for transportation of magnetic beads and fluidic processing
In this work, a facile and generic method for transportation of magnetic beads between multiple liquid phases on a centrifugal microfluidic platform is investigated. The generic microfluidic structure required for transportation of beads consists of microfluidic chambers, which are isoradially arranged on a centrifugal microfluidic cartridge (LabDisk). Adjacent chambers are interconnected by a radially inward channel. Depending on the assay requirements, the required number of microfluidic chambers can be concatenated via the defined cross section A–A independently of the chamber volume (Fig. 1a). When the chambers are loaded with liquids, the liquids are separated from each other by an air-gap, i.e. the radially inward channel (Fig. 1b). Transportation of magnetic beads from the liquid in a first chamber (i.e. chamber 1) through the air-gap into the liquid in a second chamber (i.e. chamber 2) is achieved by incremental rotation of the LabDisk with respect to a non-rotating permanent magnet followed by a final centrifugation step (Fig. 1c–f). We designate this method of bead transport between adjacent liquid filled chambers via an air-gap “Centrifugal Gas-Phase Transition Magnetophoresis” (GTM).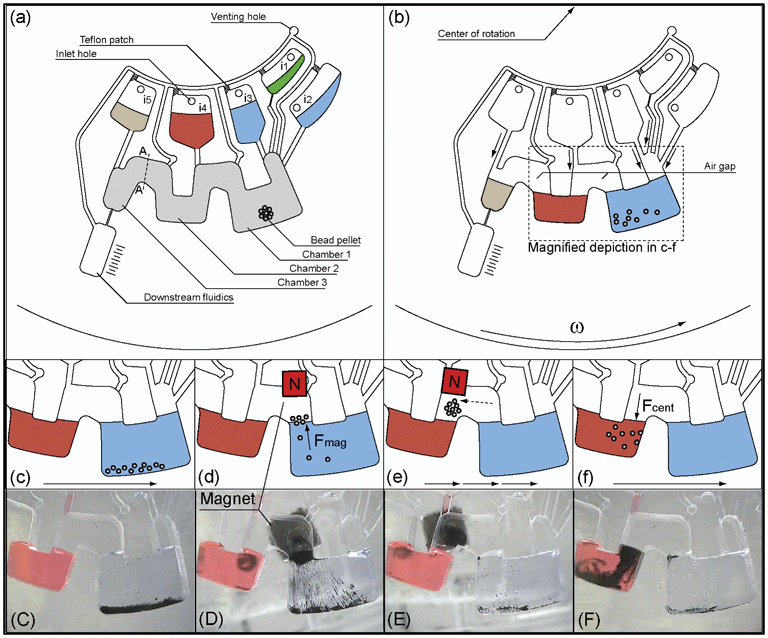 | ||
| Fig. 1 Schematic microfluidic structure for characterisation of GTM. (a) Microfluidic structure with magnetic bead pellet in chamber 1 and reagents in inlet chambers i1–i5 (b) After starting the centrifugal frequency protocol, reagents are transferred from inlet chambers into the associated chambers 1–3. The bead pellet is rehydrated and resuspended. (c) Beads are centrifuged to the rim of chamber 1 by spinning the LabDisk. (d) The LabDisk is stopped at a defined azimuthal position of the liquid phase in chamber 1 (blue) with respect to the external magnet. The magnet attracts the beads, which form clusters and are moved across the liquid-gas interface into the air-gap that connects chamber 1 and chamber 2. (e) The magnet holds the beads fixed while the LabDisk is rotated in 0.5° increments. Thereby, the beads are transported to an azimuthal position radially inwards of chamber 2 (dashed arrow). (f) The LabDisk is reaccelerated and the beads are centrifuged into the liquid phase in chamber 2 (red). (C)–(F) show photographs of the bead transport. Steps (c)–(f) are repeated a second time to transport the beads from chamber 2 into chamber 3 that is connected to a chamber representing downstream fluidics for further assay steps. Grey shaded area represents a hydrophobic Teflon coating (see section: fabrication and preparation of the LabDisk). | ||
The only required components to perform GTM are a LabDisk with one single microfluidic layer comprising the bead-transfer structure and a spinning device equipped with a permanent magnet fixed above the disk. Due to the modular layout of the GTM unit operation, the number of bead-transfer steps can be adjusted to the number of liquids that the beads have to transit. Additionally, the single volume of the chambers can be adjusted to the respective volumes of the liquids. For characterisation of GTM, a LabDisk with a microfluidic structure for two serial bead-transfers was designed and fabricated (Fig. 1). The structure consists of chamber 1 (volume 250 μL), chamber 2 (vol. 130 μL) and chamber 3 (vol. 50 μL), separated by two air-gaps between adjacent chambers. Furthermore, chambers 1–3 are connected to upstream inlet chambers i1–i5. Chamber 1 is connected to three inlet chambers to allow preloading of several liquids that are merged after a first centrifugation step. Magnetic beads are prestored in chamber 1 (described in chapter 1 ESI†).
Initially, the LabDisk is fixed to the rotor of the processing device and a stack of three neodymium iron boron (NdFeB) magnets (#W-07-N and #S 03 01-N, Supermagnete.de, Germany) is positioned above the microfluidic chambers (Fig. S1, ESI†). The magnet stack has a radial position of r = 36 mm, a radial distance of i = 2.5 mm to the liquid–gas interface and was positioned with a distance of d = 0.5 mm above the microfluidic chambers (Fig. 2a–b). The position of the magnet stack remains constant during the entire operation. To initiate bead transfer by GTM, the LabDisk is stopped at a defined azimuthal position of chamber 1 with respect to the magnet. The magnet attracts the beads which form clusters and are moved across the liquid–gas interface into the air-gap that connects chamber 1 and chamber 2. The LabDisk is then rotated in increments of 0.5° while the beads are held by the magnet. Thereby, the beads are transported to the azimuthal position of chamber 2. Subsequent acceleration centrifuges the beads into the liquid phase of chamber 2. The complete sequence of GTM is depicted in Fig. 1 and in a movie file provided in the ESI.† After incubation of the beads in the liquid of processing chamber 2, GTM is conducted a second time to transfer the beads into the liquid phase of chamber 3.
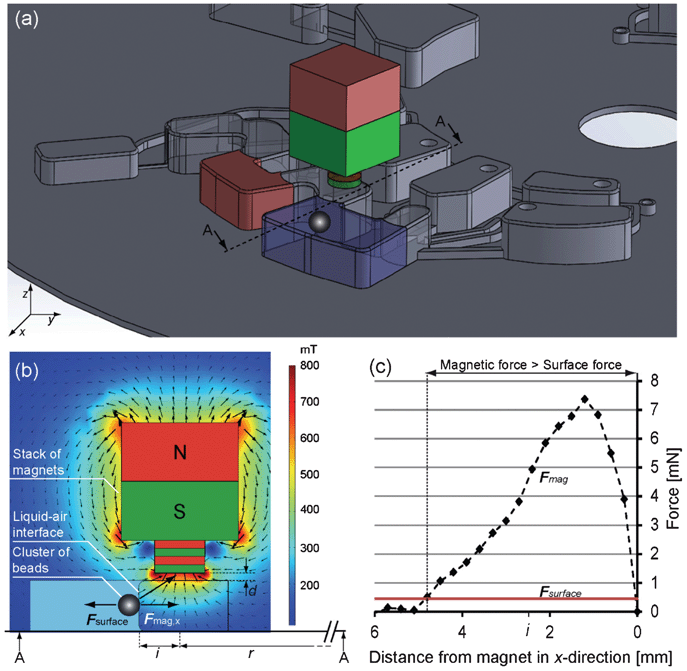 | ||
| Fig. 2 Forces acting on the magnetic beads. (a) Scheme of microfluidic structure with liquid phase in chamber 1 (blue) and a second liquid phase in chamber 2 (red). The cluster of magnetic beads is attracted in the x direction. Cross-section A–A is magnified in (b) showing the opposing forces acting on the beads. The magnetic field was calculated using COMSOL. (c) Magnetic force in the x direction (calculated using eqn (3) and values from Table 1) versus opposing surface force (calculated with eqn (4) and values from Table 1). For radial distances between liquid–gas interface and magnet ≲5 mm, the magnetic force overcomes the surface force and the beads are attracted over the interface. For the experiments, the distance was set to i = 2.5 mm. | ||
Theoretical consideration on bead transfer over liquid gas interfaces
The starting point of the consideration is the state of the resuspended magnetic beads located in the liquid phase of the first chamber (Fig. 1c–d). A stack of three neodymium iron boron (NdFeB) magnets (#W-07-N and #S-03-01-N, Supermagnete.de, Germany) is then used to transport the beads out of the liquid phase, through the air-gap and to the second chamber.The prerequisite for the beads to cross the liquid–gas interface is that the magnetic force Fmag overcomes the counteracting force of the surface tension Fsurface (eqn (1)):
| Fmag > Fsurface | (1) |
The magnetic force can be calculated from the volume of the bead cluster Vcluster, the difference in magnetic susceptibilities of the magnetic beads χmag and surrounding liquid χH2O, the strength and gradient of the magnetic field (grad(B))·B and the magnetic vacuum permeability μ0 (eqn (2)).4
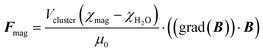 | (2) |
For the calculations, only the x-component of the magnetic force is considered (Fig. 2a–b) (3).1
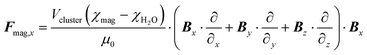 | (3) |
The counteracting force from the surface tension is given in (eqn (4)).
| Fsurface = 61/3 · π2/3 · σliquid · V1/3cluster | (4) |
| a Measured on analytical balance (Ohaus Adventurer ARRV70, China). b Measured with vibrating sample magnetometer. Kindly provided by Dr A. Tschöpe (Universität des Saarlandes, Germany). c Locally dependent magnetic field components in the x, y and z direction were measured by a Koshava 4 teslameter (Wuntronic, Germany). The teslameter was mounted to an automatic positioning system. Measurements were conducted in 0.3 mm increments. From adjacent points of measurement, the local derivatives of the magnetic field components in the x, y and z direction were calculated. | ||||
|---|---|---|---|---|
| Volume of bead cluster | V cluster | ∼4 | mm³ | Measureda |
| Volume susceptibility of mag. beads | χ mag | 0.29 | Measuredb | |
| Magnetic field | B | B x, By, Bz | T | Measuredc |
| Magnetic vacuum permeability | μ 0 | 1.25 × 10−6 | N A-2 | Constant |
| Surface tension of water | σ H2O | 72.5 | mN m−1 | Matter constant |
Microfluidic integration of magnetic bead-based DNA purification
To demonstrate the feasibility of bead transport by GTM, magnetic bead based purification of DNA as one of the most prevalent applications has been selected. A bench-top protocol comprising bind–wash–elute steps33 has been implemented into the microfluidic structure described in Fig. 1. Magnetic beads with silica coating (Cat. No #1026883, MagAttract Suspension G, Qiagen, Germany) are used for unspecific binding of DNA under chaotropic conditions.33 In brief, a bacterial lysate, binding buffer and magnetic beads are brought into contact (chamber 1). The beads bind the DNA and are transported to the washing buffer (chamber 2). After washing, the beads are transferred to the elution buffer (PCR grade water, chamber 3), where DNA is eluted from the beads.For the experiments, a mixture of 200 μL of a Listeria innocua lysate and 5 μL of a DNA standard was used as sample. The sample was split into two aliquots of 102.5 μL that were then loaded into inlets i1 and i3 respectively. Furthermore, 50% (v/v) isopropanol, washing buffer (buffer AW2, Qiagen, Germany) and elution buffer (PCR grade water) were loaded into inlets i2, i4 and i5 as described in Table 2.
| Inlet | Reagent | Volume [μL] |
|---|---|---|
| i1 | Sample lysate | 102.5 |
| i2 | 50% (v/v) isopropanol | 50 |
| i3 | Sample lysate | 102.5 |
| i4 | Washing buffer AW2 | 130 |
| i5 | PCR grade water | 50 |
The magnetic beads were prestored in chamber 1 as dried pellet. Finally, the inlet holes were sealed with adhesive tape and stand-alone DNA purification was started by the frequency protocol described in Table 3. The protocol automatically performs release of reagents into processing chambers 1–3 used for binding, washing and elution. Afterwards the eluted DNA was removed from the LabDisk by pipetting and stored for further analysis.
| Frequency [rpm] | Duration [ms] | Notes | Action |
|---|---|---|---|
| 600 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
All reagents are transferred from inlet chambers to binding, washing or elution chamber | |
| 480 | 2000 | Pellet resuspension | |
| 120 | 10000 |
Alternation between 120 and 480 rpm repeated 30 times | Beads are magnetically attracted radially inwards at 120 rpm while centrifuged radially outwards at 480 rpm. DNA binds to the surface of the beads. |
| 480 | 10000 |
||
| 0 | 20000 |
Defined positioning of LabDisk | Beads are magnetically attracted out of the lysate. |
| 0 | 1000 | Incremental rotation of 0.5°, repeated 30 times | Beads are moved from binding chamber to washing chamber through air-gap |
| 480 | 15000 |
Beads are centrifuged into washing buffer | |
| 0 | 20000 |
Defined positioning of LabDisk | Beads are magnetically attracted out of washing buffer. |
| 0 | 1000 | Incremental rotation of 0.5°, repeated 30 times | Beads are moved from washing chamber to elution chamber through air-gap |
| 480 | 15000 |
Beads are centrifuged into elution buffer |
Materials and methods
Fabrication and preparation of the LabDisk
All microfluidic structures were designed using CAD software (AutoCAD, Autodesk Inc., USA). Fabrication of the LabDisk was performed by the HSG-IMIT Microfluidic Design & Foundry Service (Freiburg, Germany). The structures were replicated from a PDMS master using the μTSL process.34 Therefore, the microfluidic structures were micro milled (Kern Evo, Kern Microtech, Germany) into PMMA blanks with a thickness of 4 mm (Maertin, Germany). The milled PMMA structures were then cast with PDMS (Elastosil RT607, Wacker, Germany) forming a negative mold insert for blow molding. The final LabDisks with a diameter of 130 mm, used for all experiments described, were fabricated from 188 μm thick cyclic olefin polymer foils (COP ZF 14, Zeon Chemicals, USA) by blow molding. After blow molding, inlet and venting holes were drilled manually. Residuals from the manufacturing process were removed by cleaning the LabDisks with isopropanol and DI water. To obtain hydrophobic surfaces, chambers 1–3 for binding, washing and elution and the air-gaps were manually coated by pipetting 2 × 100 μL of a 0.5% (w/w) Teflon AF solution (DuPont, USA) dissolved in Fluorinert FC 77 (3M, Belgium). Connections between inlet chambers and ventilation channels were coated with a 0.2 μL Teflon patch containing 0.5% (w/w) carbon-black (Type 901, Degussa, Germany) to avoid capillary priming.35 Before sealing the LabDisk with pressure sensitive polyolefin foil (#900 320, HJ Bioanalytik, Germany), a pellet of magnetic beads was placed in each binding chamber. A resulting LabDisk is depicted in Fig. 3. | ||
| Fig. 3 LabDisk, replicated by the μTSL process featuring two independent GTM structures, each with three buffer chambers designed for a bind-wash-elute protocol used in magnetic bead based DNA purification. | ||
Processing device
For microfluidic processing, the LabDisk was fixed to the rotor of a centrifugal processing device (Abbis – bio process automation, Germany). The instrument features a motor with a maximum spinning speed of 7800 rpm. The motor encoder enables defined azimuthal positioning of the shaft with a minimum resolution of 0.025°. A predefined frequency protocol controls spinning speed, acceleration, direction of rotation and azimuthal position of the LabDisk.Preparation of the sample for DNA purification
As sample for DNA purification, a lysate of Listeria innocua was used. Therefore, a stock culture of L. innocua (DSM 20649, Leibniz Institute DSMZ – German Collection of Microorganisms and Cell Cultures, Germany) was prepared by overnight incubation in 25 mL of brain heart infusion (BHI) (Carl Roth, Germany) at 37 °C and shaken at 200 rpm. The culture was put on ice. A log10 dilution series of the cell culture was prepared to determine the viable cell count using the drop plate method36,37 on BHI agar plates with 40 individual 10 μL drops per dilution step7 resulting in a viable cell count of 8.5 × 108 cfu mL−1. At the same time, 1.5 mL of the stock solution were mixed with 50 μL Proteinase K (#03115879001, Roche Applied Science, Germany) and 1.5 mL of a lysis-binding buffer containing a chaotropic agent (buffer ML, Qiagen, Germany). After incubation in a thermomixer (56 °C, 1200 rpm for 20 min) a log10 dilution series from the lysate in a 50% (v/v) mixture of BHI and buffer ML was prepared. Hence, each dilution contains the same concentration of chaotropic agents. Since the viable cell count can deviate significantly from the total count determined by quantitative PCR,7 5 μL of lambda phage DNA (#D3654-5UM, Sigma Aldrich, Germany) with a defined concentration of 3500 DNA copies μL−1 were added to each 200 μL aliquot as an internal standard resulting in a total volume of 205 μL per sample.Reference DNA purification
As reference, DNA was purified in 2 mL reaction tubes using a magnetic separation rack (Chemagen, Germany) in the following way. For DNA binding, 50 μL of 50% (v/v) isopropanol were added to each 205 μL sample. To facilitate DNA binding, all samples were incubated in a thermomixer (Eppendorf, Germany) (15 min at 1200 rpm). The beads were then collected at the wall of the reaction tube by using the magnetic separation rack, the lysate was discarded and 130 μL washing buffer AW2 were added. Three times pulse-vortexing ensured proper washing. Again, the beads were collected at the wall of the reaction tube and the washing buffer was removed and discarded. Finally, 50 μL of elution buffer (PCR grade water) were added to elute the DNA from the beads. The tubes were pulse-vortexed again and the eluate was transferred to fresh reaction tubes for subsequent analysis by real time PCR.Quantification of recovered DNA by real-time PCR
After DNA purification, the number of recovered DNA copies was quantified by real-time PCR using a Rotor-Gene 2000 real-time PCR thermocycler (Formerly Corbett Research now Qiagen, Germany). Every PCR was conducted in triplicate to enhance statistical significance. Furthermore, every PCR run included no template controls (NTC) and a set of three external quantification standards with known DNA concentration. Compositions of the PCR reaction mixes and sequences of primers and probes are described in Table S1, ESI†Results and discussion
Efficiency of bead transport
Losses of magnetic beads during GTM between different liquid phases may negatively influence the DNA recovery. Therefore, five bead pellets were prepared and separately weighed on a high precision balance (precision: 0.1 μg; Sartorius SC2, Germany). Each pellet was then placed in chamber 1 of the presented fluidic layout and GTM was conducted as described in Fig. 1. Afterwards, successfully transported beads were recovered from chamber 3 using a pipette, dried and weighed again. The experiment was repeated five times always using new fluidic structures. For this experiment, DNA extraction reagents were replaced by deionized water since buffer constituents like chaotropic salts adhering to the beads would contribute to the weight. To compare the amount of the inserted and the recovered beads, the weight of trehalose acting as the pelleting agent had to be subtracted from the initial weight since it dissolves during bead transport. The amount of trehalose in a single pellet was measured gravimetrically in dried form before and after dissolving in deionized water. From the difference in both measurements, the amount of trehalose per pellet was taken into account with 1.85 mg (n = 3). After transportation from the binding chamber through the washing chamber into the elution chamber, 82.6% ± 3.6% of inserted beads could successfully be recovered as shown in Table 4. The missing beads are mainly trapped in chamber 1 in the interstice between the thermoformed LabDisk and the pressure sensitive sealing as a result of the high initial centrifugation speed. In future, the loss in magnetic beads can be avoided by using a second magnet on a radial outer path that collects all beads within the liquid phase to one bead cluster before the entire cluster is transported across the liquid–gas interface and through the air-gap as described.| Pellet # | Before transport [mg] | After transport [mg] | Yield % |
|---|---|---|---|
| 1 | 1.94 | 1.52 | 78.4 |
| 2 | 1.97 | 1.63 | 82.7 |
| 3 | 1.99 | 1.64 | 82.4 |
| 4 | 2.09 | 1.70 | 81.3 |
| 5 | 2.05 | 1.81 | 88.3 |
| Mean yield | 82.6 | ||
| Std. dev. | 3.6 | ||
Performance of LabDisk based DNA purification
Prior to starting the LabDisk based GTM for automated magnetic bead based bind-wash-elute purification of DNA, the reagents were loaded into the corresponding inlets (Table 2). Subsequently, the frequency protocol was started. After DNA binding, the beads were transported through the washing chamber into the elution chamber where the attached DNA was released. For further analysis, the eluate was then taken from the LabDisk. From each dilution of the L. innocua lysate containing the lambda phage DNA standard, three independent LabDisk based DNA purifications were conducted always using a new LabDisk. Concurrently, three reference purifications were conducted from the same dilution in standard reaction tubes. The amount of the log10 diluted L. innocua used per extraction ranged from 8.5 × 107 down to 8.5 × 102 “lysed CFU” while the amount of inserted lambda phage DNA standard was constantly 17.500 DNA copies. Performance of the DNA purification was subsequently assessed by quantitative real-time PCR in triplicates targeting L. innocua and lambda phage DNA genomic sequences. It should be noted that the amount of successfully recovered DNA was eluted in 50 μL of elution buffer but only 1 μL from the elution buffer was used in a PCR reaction. It was observed that single PCR reactions failed that were conducted from the eluate of the two most diluted samples containing 8.5 × 102 and 8.5 × 103 “lysed CFU” of L. innocua.Due to the small number of DNA copies, real-time PCR results at these dilutions are dominated by statistical effects. Therefore, PROBIT regression analysis using the statistical software SPSS was applied to estimate the concentration of recovered DNA of the two most diluted samples.38,39 By using the PROBIT method, the DNA concentration is determined by the ratio of positive PCR reactions to the overall number of PCR reactions performed per dilution. PROBIT analysis is described in detail in the ESI† (Fig. S2, Table S2). The LabDisk based purification of L. innocua DNA yielded between 29 ± 25% and 68 ± 24% compared to the manual reference purification in tubes (Fig. 4, Table 5). From the total number of 18 LabDisk and 18 reference purifications in tubes (6 dilutions, 3 replicates per dilution) integrated lambda phage DNA standard was measured by real time PCR (Table S3, ESI†). Compared to the theoretical maximum of 17500 inserted lambda phage DNA copies, 31 ± 10% could be recovered after the LabDisk based purification and 72 ± 21% by the manual reference in tube. Thus, the yield of recovered lambda phage DNA on the LabDisk versus manual reference is calculated to be 43 ± 10% (Table 6).
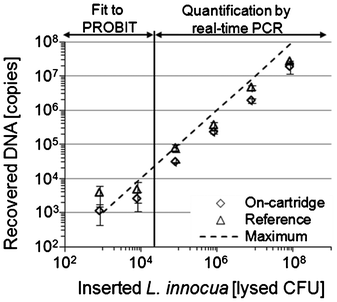 | ||
| Fig. 4 Number of recovered L. innocua copies obtained by LabDisk based (diamond) or reference (triangle) purification from lysed L. innocua cultures. The investigated concentration range comprised six orders of magnitude. Quantification of DNA yield was performed by quantitative real time PCR. For concentrations ≤ 104 “lysed CFU” per sample, PROBIT regression analysis was used for calculation of recovery. | ||
| Inserted number of lysed L. innocua | LabDisk recovered number of DNA copies | Reference recovered number of DNA copies | Ratio LabDisk/Reference (Errors calc. by Gaussian error propagation) |
|---|---|---|---|
| a According to PROBIT analysis, more DNA seems to be recovered at the highest dilution that was actually inserted into the reaction. This might be attributed firstly to low sample size for statistical PROBIT analysis and secondly to the comparison to plate counting which might be defective. | |||
| (8.5 ± 2.5) × 107 | (1.7 ± 0.6) × 107 | (2.5 ± 0.1) × 107 | 68% ± 24% |
| (8.5 ± 2.5) × 106 | (1.8 ± 0.3) × 106 | (4.4 ± 0.6) × 106 | 41% ± 9% |
| (8.5 ± 2.5) × 105 | (2.2 ± 0.2) × 105 | (3.4 ± 0.9) × 105 | 65% ± 18% |
| (8.5 ± 2.5) × 104 | (2.8 ± 0.1) × 104 | (7.5 ± 1.9) × 104 | 37% ± 10% |
| (8.5 ± 2.5) × 103 | (2.5 ± 1.4) × 103 | (4.6 ± 2.8) × 103 | 54% ± 45% |
| (8.5 ± 2.5) × 102 | (1.1 ± 0.6) × 103a | (3.7 ± 2.3) × 103a | 29% ± 25% |
| Inserted number of lambda phage DNA copies | LabDisk recovered number of DNA copies | Reference recovered number of DNA copies | Ratio LabDisk/Reference (%) | Ratio on LabDisk/theor. maximum (%) |
|---|---|---|---|---|
| 17.5 × 103 | (5.5 ± 1.7) × 103 | (12.6 ± 3.7) × 103 | 43 ± 10 | 31 ± 10 |
In summary, DNA recovery of LabDisk based DNA purification versus manual reference was 43 ± 10% for lambda phage DNA and between 29 ± 25% and 68 ± 24% for a log10 dilution series of L. innocua lysate covering a range of six orders of magnitude in DNA concentration. Hence, the recovery should be further optimized for applications that require high sensitivity; for genotyping applications the recovery achieved would be already sufficient. The first and very obvious reason for the reduced DNA recovery of the LabDisk based purification is the loss of almost 20% of magnetic beads during the experiments reported, directly explaining 20% loss in DNA recovery. One second reason for the reduced DNA recovery of the LabDisk based purification might be attributed to a not fully optimized rehydration and resuspension of the dried magnetic bead pellet leading to a reduced silica surface available for DNA binding. Although in tube experiments, similar results for DNA purification using dry magnetic bead pellets or magnetic beads in suspension were obtained (chapter 6 in ESI; Fig. S3†), it has to be considered that for those experiments vortexing was used to resuspend the beads, which very likely might have been more efficient than resuspension of the pellet in the LabDisk. On the LabDisk, mixing was mainly conducted by cyclic variation of rotational speed resulting in a movement of the magnetic beads between a radial inward and a radial outward position (i.e. as a result of dominating magnetic or centrifugal forces). Insufficient resuspension results in less available bead surface for DNA binding. Hence, one way to improve resuspension of magnetic beads might be a more efficient mixing.
A more straightforward approach would replace the bead pelleting agent trehalose which was used in the experiments reported by an agent that better supports rehydration and resuspension of the dried magnetic bead pellet. For example polyvinylpyrrolidone has successfully been used for pelleting and rehydration of beads.40
Conclusion and outlook
In this paper, we introduced a novel and generic method for the automation of magnetic bead based assays on the centrifugal microfluidic platform which was demonstrated by LabDisk based DNA purification using a bind-wash-elute protocol. The key element of the novel method is a mechanism for transportation of magnetic beads between adjacent liquid phases through a separating gas-phase which we named “gas-phase transition magnetophoresis” (GTM). Transportation of the magnetic beads is achieved by a permanent magnet which is mounted at a fixed position of the LabDisk processing device. This magnet first pulls the beads out of a first liquid phase into an air-gap. By incremental rotation of the LabDisk, the beads are transported to the azimuthal position of the second chamber. The beads are then transferred into the liquid phase located in the second chamber by centrifugal forces due to acceleration of the LabDisk.This novel method excels by several features: First, the technical requirements are low: only one fluidic layer and no actively controlled valves integrated into the LabDisk facilitate production. Furthermore, the only requirement for the LabDisk spinning device is to have a permanent magnet installed at a fixed position and to allow azimuthal positioning of the disk. Second, the design of the microfluidic structure for GTM is modular and allows easy adjustment to the requirements of the assay, just by varying number and volume of the buffer chambers. Remaining shortcomings at the time of reporting comprise a limited bead transport efficiency of 82.6 ± 3.6% after two transportation steps.
As an application example for this novel method, DNA purification with commercially available DNA extraction reagents was demonstrated and extensively characterized. Yields of up to 68% compared to manual reference purifications were achieved. The yield may be further enhanced by improving resuspension of the prestored, pelleted beads or using different pelleting agents like water soluble polymers. The process of DNA purification was automated by a predefined frequency protocol and didn't require any human interaction during processing. In the future, lysis will also be included. For this operation mode, instead of the lysate, the original sample (bacteria stock, whole blood, or blood plasma) could be loaded into inlet i2 while the combined lysis and binding buffer could be loaded into inlet i3. The DNA extraction structure is composed in such a way that it may be concatenated with structures for multiplex real-time PCR41 or RPA42 analysis paving the way for automatic nucleic acid sample-to-answer analytics on a disposable foil LabDisk. In future, reagent prestorage in glass ampoules43 or stick-packs44 may be incorporated further enhancing the degree of integration.
Additional applications using magnetic bead based immunoassays or protein affinity purification may also be conducted with the presented method by simply adjusting number and volume of the isoradially arranged chambers.
Acknowledgements
We gratefully acknowledge financial support from the Baden-Württemberg Foundation for funding this project (MST II – 17). Furthermore, we would like to acknowledge Dr Ralf Himmelreich (Qiagen) for providing reagents and magnetic beads for DNA purification. Additionally, we want to thank Dominique Kosse and the Team of HSG-IMIT Microfluidic Design & Foundry Service for fabrication of the LabDisks. We want to thank Dr Andreas Tschöpe (Universität des Saarlandes) for conducting the VSM measurements.References
- M. A. M. Gijs, Microfluidics Nanofluidics, 2004, 1, 22–40 CAS.
- M. A. M. Gijs, F. Lacharme and U. Lehmann, Chem. Rev., 2010, 110, 1518–1563 CrossRef CAS.
- S. A. Peyman, A. Iles and N. Pamme, Lab Chip, 2009, 9, 3110–3117 RSC.
- N. Pamme, Lab Chip, 2006, 6, 24–38 RSC.
- E. Verpoorte, Lab Chip, 2003, 3, 60–68 RSC.
- P. S. Doyle, J. Bibette, A. Bancaud and J. L. Viovy, Science, 2002, 295, 2237–2237 CrossRef CAS.
- M. Karle, J. Miwa, G. Czilwik, V. Auwärter, G. Roth, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 3284–3290 RSC.
- S. M. Berry, E. T. Alarid and D. J. Beebe, Lab Chip, 2011, 11, 1747–1753 RSC.
- S. Bronzeau and N. Pamme, Anal. Chim. Acta, 2008, 609, 105–112 CrossRef CAS.
- J. Do and C. H. Ahn, Lab Chip, 2008, 8, 542–549 RSC.
- V. I. Furdui and D. J. Harrison, Lab Chip, 2004, 4, 614–618 RSC.
- N. Pamme and C. Wilhelm, Lab Chip, 2006, 6, 974–980 RSC.
- M. Slovakova, N. Minc, Z. Bilkova, C. Smadja, W. Faigle, C. Futterer, M. Taverna and J. L. Viovy, Lab Chip, 2005, 5, 935–942 RSC.
- M. Shikida, K. Takayanagi, H. Honda, H. Ito and K. Sato, J. Micromech. Microeng., 2006, 16, 1875–1883 CrossRef.
- J. Pipper, M. Inoue, L. F. P. Ng, P. Neuzil, Y. Zhang and L. Novak, Nat. Med., 2007, 13, 1259–1263 CrossRef CAS.
- J. Pipper, Y. Zhang, P. Neuzil and T. M. Hsieh, Angew. Chem., Int. Ed., 2008, 47, 3900–3904 CrossRef CAS.
- A. Rida, V. Fernandez and M. A. M. Gijs, Appl. Phys. Lett., 2003, 83, 2396–2398 CrossRef CAS.
- U. Lehmann, C. Vandevyver, V. K. Parashar and M. A. M. Gijs, Angew. Chem., Int. Ed., 2006, 45, 3062–3067 CrossRef CAS.
- N. Pamme and A. Manz, Anal. Chem., 2004, 76, 7250–7256 CrossRef CAS.
- D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153–1182 RSC.
- S. Haeberle and R. Zengerle, Lab Chip, 2007, 7, 1094–1110 RSC.
- D. A. Duford, D. D. Peng and E. D. Salin, Anal. Chem., 2009, 81, 4581–4584 CrossRef CAS.
- M. C. R. Kong, M. C. R. A. P. Bouchard and E. D. Salin, Micromachines, 2012, 3, 1–9 CrossRef.
- A. P. Bouchard, D. A. Duford and E. D. Salin, Anal. Chem., 2010, 82, 8386–8389 CrossRef CAS.
- M. Madou, J. Zoval, Guangyao Jia, H. Kido, J. Kim and N. Kim, Lab on a CD, 2006 Search PubMed.
- Abaxis Inc., 2009, http://www.abaxis.com/pdf/Piccolo%20Centrifugation%20and%20Capillarity.pdf (Last Accessed 04.10.2012).
- R. Gorkin, J. Park, J. Siegrist, M. Amasia, B. S. Lee, J. M. Park, J. Kim, H. Kim, M. Madou and Y. K. Cho, Lab Chip, 2010, 10, 1758–1773 RSC.
- J. Ducree, S. Haeberle, S. Lutz, S. Pausch, F. von Stetten and R. Zengerle, J. Micromech. Microeng., 2007, 17, 103–115 CrossRef.
- J. Ducree, M. Grumann, T. Brenner and R. Zengerle, Proc. of BMT, 2005 Search PubMed.
- Y. K. Cho, J. G. Lee, J. M. Park, B. S. Lee, Y. Lee and C. Ko, Lab Chip, 2007, 7, 565–573 RSC.
- G. P. Arrighin, M. Maestro and R. Moccia, J. Chem. Phys., 1968, 49, 882 CrossRef.
- W. M. Spees, D. A. Yablonskiy, M. C. Oswood and J. J. H. Ackerman, Magn. Reson. Med., 2001, 45, 533–542 CrossRef CAS.
- R. Boom, C. J. A. Sol, M. M. M. Salimans, C. L. Jansen, P. M. E. Wertheimvandillen and J. Vandernoordaa, J. Clin. Microbiol., 1990, 28, 495–503 CAS.
- M. Focke, D. Kosse, D. Al-Bamerni, S. Lutz, C. Müller, H. Reinecke, R. Zengerle and F. von Stetten, J. Micromech. Microeng., 2011, 21, 115002 (11pp) CrossRef.
- L. Riegger, M. M. Mielnik, A. Gulliksen, D. Mark, J. Steigert, S. Lutz, M. Clad, R. Zengerle, P. Koltay and J. Hoffmann, J.Micromech.Microeng, 2010, 20, 087003 (5pp) Search PubMed.
- H. J. Hoben and H.J. P.Somasegaran, Appl. Environ. Microbiol., 1982, 44, 1246–1247 CAS.
- B. Herigstad, M. Hamilton and J. Heersink, J. Microbiol. Methods, 2001, 44, 121–129 CrossRef CAS.
- M. Smieja, J. B. Mahony, C. H. Goldsmith, S. Chong, A. Petrich and M. Chernesky, J. Clin. Microbiol., 2001, 39, 1796–1801 CrossRef CAS.
- M. Weidmann, O. Faye, O. Faye, R. Kranaster, A. Marx, M. R. T. Nunes, P. F. C. Vasconcelos, F. T. Hufert and A. A. Sall, J. Clin. Virol., 2010, 48, 187–192 CrossRef CAS.
- J. Siegrist, R. Gorkin, M. Bastien, G. Stewart, R. Peytavi, H. Kido, M. Bergeron and M. Madou, Lab Chip, 2010, 10, 363–371 RSC.
- M. M.Focke, F. Stumpf, B. Faltin, P. Reith, D. Bamarni, S. Wadle, C. Müller, H. Reinecke, J. Schrenzel, P. Francois, D. Mark, G. Roth, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 2519–2526 RSC.
- F. S.Lutz, F. P.Weber, M. Focke, M. B.Faltin, J. Hoffmann, C. Müller, D. Mark, G. Roth, P. Munday, N. Armes, O. Piepenburg, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 887–893 RSC.
- J. Hoffmann, D. Mark, S. Lutz, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 1480–1484 RSC.
- T. van Oordt, Y. Barb, R. Zengerle and F. von Stetten, Proc. μTAS, 2011, 437–439 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2lc40866j |
| This journal is © The Royal Society of Chemistry 2013 |