Low-blank chemistry for Zn stable isotope ratio determination using extraction chromatographic resin and double spike-multiple collector-ICP-MS
Akio
Makishima
* and
Eizo
Nakamura
The Pheasant Memorial Laboratory for Geochemistry and Cosmochemistry, Institute for Study of the Earth's Interior, Okayama University at Misasa, Misasa, Tottori-ken 682-0193, Japan. E-mail: max@misasa.okayama-u.ac.jp; Fax: +81-858-43-2184; Tel: +81-858-43-1215
First published on 15th November 2012
Abstract
A new low-blank Zn separation method employing an extraction chromatographic resin using Aliquat 336 (commercially available as TEVA resin) has been developed for Zn isotope composition determination by double spike-multiple collector-ICP-MS. The silicate sample solution containing 0.1 μg Zn in 0.5 mol L−1 HBr containing 0.15% ascorbic acid (AA) is passed through a 1 mL column with 0.33 mL TEVA resin. While Zn is absorbed, major elements are eluted with 0.5 mol L−1 HBr with AA. Afterwards, Zn was recovered with 2 mol L−1 HNO3. Recovery yield using silicate samples was 96.7 ± 6.8% (n = 10) and total blank was 0.05 ng (n = 6). The blank level is from 1/300 to 1/20 of previous studies, meaning 0.1 μg of Zn is sufficient for analysis. The 67Zn–70Zn double spike method was used in MC-ICP-MS. δ66Zn of three USGS standard silicate reference materials (BHVO-1, AGV-1 and PCC-1) and seven GSJ silicate reference materials (JB-1, -2, -3, JA-1, -2, -3 and JP-1) were determined. Reproducibility of δ66Zn of the total silicates was 0.19‰ (2SD). δ66Zn of three carbonaceous chondrites (Orgueil, Murchison and Allende) were also determined and compared with those of references.
Introduction
Zinc has five stable isotopes, 64Zn, 66Zn, 67Zn, 68Zn and 70Zn. Since isotopic fractionation of Zn was observed, Zn stable isotopes have been used for studies of the solar system.1–4The most popular method for purification of Zn is anion exchange chromatography employing Zn(II) chloro-complex and anion exchange resin, AG MP-1,5 Chelex-100,6 or a combination.7,8
In this method, Zn is absorbed by a high concentration of HCl (e.g., 7 mol L−1), and recovered with diluted HNO3 (e.g., 0.5 mol L−1). The merit of this method is that simultaneous separation of Cu and Fe is available. However, the total eluent volume became 50 mL and extremely large even though 1.6 mL of the AG MP-1 resin is used.5 The currently described procedures in literature, however, suffer from a high Zn blank as low as 1–15 ng.5,8 Thus an aim of this study was to develop a method with low Zn blank levels. A further aim of the method was to correct mass fractionation by employing the Zn double spike technique.
In order to reduce the blank, extraction chromatographic resin employing trioctylmethylammonium chloride (Aliquat 336), which is sold as TEVA resin from Eichrom Technologies, Inc., USA,9–11 with eluent of 0.5 mol L−1 HBr containing ascorbic acid (AA) was used.
In a double spike technique,12–14 a double spike enriched in two isotopes with known isotope ratios is mixed with sample isotopes, and the isotope ratios of the mixtures are measured by multiple collector-inductively coupled plasma source-mass spectrometry (MC-ICP-MS). Then the stable isotope composition is calculated. The largest merit of the double spike technique is that mass discrimination occuring in chemical separation and/or mass spectrometry can be corrected.
In order to determine reproducibility of the method developed in this study, δ66Zn of three US Geological Survey (USGS) silicate reference materials (BHVO-1, AGV-1 and PCC-1) and seven GSJ (Geological Survey of Japan) silicate reference materials (JB-1, -2, -3, JA-1, -2, -3 and JP-1) were determined. δ66Zn in three carbonaceous chondrites (Orgueil, Murchison and Allende) was also determined and compared with those in the references.
Reagents and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg mL−1, Specpure, Product no. 14404, Alfa Aesar, USA) was used in the mixing experiment. Multi-element standard solutions (Specpure, Product no. 42885 and 44270, Alfa Aesar, USA) were used in the column calibration. IRMM-3702 Zn standard solution (3.09 μg mL−1, Lot no. 000069, Belgium) was diluted to be 0.1 μg mL−1 Zn solution with 0.5 mol L−1 HNO3. This Zn solution was used as Zn isotopic standard solution for MC-ICP-MS. Atomic absorption spectrometry standard solutions of Zn and Te (1000 μg mL−1, Kanto Chemical Co. Inc., Japan) were used in a mass interference test.
000 μg mL−1, Specpure, Product no. 14404, Alfa Aesar, USA) was used in the mixing experiment. Multi-element standard solutions (Specpure, Product no. 42885 and 44270, Alfa Aesar, USA) were used in the column calibration. IRMM-3702 Zn standard solution (3.09 μg mL−1, Lot no. 000069, Belgium) was diluted to be 0.1 μg mL−1 Zn solution with 0.5 mol L−1 HNO3. This Zn solution was used as Zn isotopic standard solution for MC-ICP-MS. Atomic absorption spectrometry standard solutions of Zn and Te (1000 μg mL−1, Kanto Chemical Co. Inc., Japan) were used in a mass interference test.
A theory of a double spike is briefly explained here. As Zn has five stable isotopes, 64Zn, 66Zn, 67Zn, 68Zn and 70Zn, there are a lot of combinations for the double spike. We chose a 67Zn–70Zn double spike, which is recommended in Rudge et al.1364Zn was used as an denominator isotope. Each Zn ratio is written as follows using an exponential law:
| α = [ln(Rc-66/64/Rmeas-66/64)]/[ln(m66/m64)]/m64 | (1) |
| Rc-67/64/Rmeas-67/64 = (m67/m64) ^ [αm64] | (2) |
| Rc-70/64/Rmeas-70/64 = (m70/m64) ^ [αm64] | (3) |
In order to determine Rsample-i/64 (i = 66, 67 and 70) of the unknown sample with an unknown degree of fractionation, the double spike method has been developed.12,13,16 To a sample the enriched 67Zn and 70Zn are added. The spike isotope ratios of Rspike-i/64 (i = 66, 67 and 70) are determined to hold eqn (1)–(3) in advance (the spike calibration method is related later). For the mixture, the following equation holds:
| Rmix-i/64 = [bi (Rsample-i/64) + (Rspike-i/64)spike]/(1 + bi) (i = 66, 67 and 70) | (4) |
The spike calibration method is simple. First, only the double spike with higher concentration is measured. Thus, one set of spike solution, Rspike-i/64 (i = 66, 67 and 70) are obtained. In this set, when one of Rspike-j/64, for example Rspike-66/64, is determined, the other two Rspike-j/64 (j = 67 and 70) are determined. Secondly, lots of spike-standard (the IRMM-3702 solution) mixtures are made, then the mixture isotope ratios are measured. More than ten mixtures are made and measured. Finally, by changing the spike isotope ratio of Rspike-66/64, the best-fit spike isotope ratio is searched to minimize the differences of IRMM-3702 between the measured and ideal values. Thus the Zn double spike was calibrated to be 66Zn/64Zn = 1.8425, 67Zn/64Zn = 88.293 and 70Zn/64Zn = 88.877 by this method, however, this is off the ideal value of the double spike.
Ideal values of the double spike were 42.65 and 57.35% for 67Zn and 70Zn, respectively.13 Such pure spike cannot be obtained and 64Zn and 66Zn are contained, therefore, the spike values in this study were 1.8425, 88.293, 88.877 for 66Zn/64Zn, 67Zn/64Zn and 70Zn/64Zn, respectively. The spike: sample mole ratio is required to be 37:63,13 therefore, 0.05 μg spike was added with 0.1 μg of sample. In order to achieve this ratio, a Zn amount of the sample was determined before isotope analysis.16
Carbonaceous chondrite powders of Orgueil (CI1), Murchison (CM2) and Allende (CV3) were digested in a similar manner to the silicate reference materials.
The column chemistry procedure developed in this study is summarized in Table 1. All experiments were done in a clean room.17 The column was pre-washed with 6.4 mL of 2 mol L−1 HNO3 and 1.6 mL of water, followed by 3.2 mL of 0.5 mol L−1 HBr. The resin bed was then conditioned with 0.5 mol L−1 HBr containing 0.15% (w/v) AA. The dried sample was dissolved with 0.1 mL of 0.5 mol L−1 HBr containing 0.15% (w/v) AA and loaded on the resin bed. The major elements (Na, Mg, Al, P, Ca, Cr, Mn and Fe, etc.) were eluted by addition of 3.2 mL of 0.5 mol L−1 HBr containing 0.15% (w/v) AA. Subsequently, the resin bed was washed with 0.8 mL of 0.5 mol L−1 HBr, and Zn was collected by addition of 0.8 mL of water and 5.6 mL of 2 mol L−1 HNO3. The purpose of 0.8 mL of 0.5 mol L−1 HBr and water before Zn collection are to prevent AA from forming complex organic materials and to prevent HBr from generating gaseous Br2 or NOx by reaction with HBr and HNO3, respectively.
| Washing | ||
| 2 mol L−1 HNO3 | 6.4 mL | |
| Water | 1.6 mL | |
| 0.5 mol L−1 HBr | 3.2 mL | |
| Conditioning | ||
| 0.5 mol L−1 HBr with 0.15% ascorbic acid | 3.2 mL | |
| Loading the sample | ||
| 0.5 mol L−1 HBr with 0.15% ascorbic acid | 0.1 mL | |
| Removing major elements | ||
| 0.5 mol L−1 HBr with 0.15% ascorbic acid | 3.2 mL | |
| 0.5 mol L−1 HBr | 0.8 mL | |
| Collecting Zn | ||
| Water | 0.8 mL | |
| 2 mol L−1 HNO3 | 5.6 mL | |
Then the Zn fraction was dried on a hot plate at 120 °C in a clean fume hood with the addition of 1 drop of HClO4 to decompose organic materials and small resin particles. To remove HClO4 completely from the sample, the dried sample was further heated at 195 °C for 6 hours, and then dissolved with 1 mL of 0.5 M HNO3.
| δ66Zn = [(66Zn/64Zn)sample/(66Zn/64Zn)IRMM-3702 − 1] × 1000 | (5) |
| a The amplifier with a resistor of 1 TΩ. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1. Sample introduction and ICP conditions | |||||||||
| Nebulizer | Micro-flow PFA nebulizer, PFA-50 (ESI, USA), self-aspiration (flow rate: ∼50 μL min−1) | ||||||||
| Desolvator | Aridus II | ||||||||
| Ar gas flow rate | 8–9 L min−1 | ||||||||
| Plasma power | 1.2 kW (27.12 MHz) | ||||||||
| Torch | Quartz glass torch with a sapphire injector | ||||||||
| Plasma Ar gas flow rate | 15 L min−1 | ||||||||
| Auxiliary Ar gas flow rate | 0.80 L min−1 | ||||||||
| Nebulizer Ar gas flow rate | 0.90 L min−1 | ||||||||
| 2. Interface | |||||||||
| Sampling cone | Jet cone, made of Ni, with additional rotary pump | ||||||||
| Skimmer cone | X-skimmer, made of Ni | ||||||||
| 3. Data acquisition conditions | |||||||||
| Resolution | ∼500 | ||||||||
| Washing time | 480 s after measurement | ||||||||
| Uptake time | 90 s | ||||||||
| Background data integration | 4 s for 1 scan | ||||||||
| 20 scans in one run | |||||||||
| On-top zeroes | |||||||||
| Sample data integration | 4 s for 1 scan | ||||||||
| 20 scans in one run | |||||||||
| 4.Cup configuration | |||||||||
| L4 | L3 | L2 | L1 | C | H1 | H2 | H3 | H4 | |
| 62Nia | 63Cu | 64Zn | 65Cu | 66Zn | 67Zn | 68Zn | 70Zn | ||
One run consists of 20 scans, and the average and standard error (1SE) were obtained. One measurement takes almost 15 min including washing time.
Results and discussion
TEVA column chemistry
In the early stage of this study, 2 mol L−1 HCl, 0.05, 0.25 and 0.5 mol L−1 HBr mixed with 0.15% ascorbic acid (AA) were tested as eluent. Elution curves of Fe, Cu and Zn in these tests are shown in Fig. 1(a)–(d), respectively. The diluted multi-element standard solutions of Specpure no. 42885 and 44270 were used as the sample solution.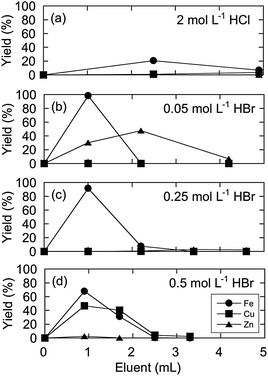 | ||
| Fig. 1 Elution curves of Fe, Cu and Zn obtained by (a) 2 mol L−1 HCl with 0.15% ascorbic acid (AA), (b) 0.05 mol L−1 HBr with AA, (c) 0.25 mol L−1 HBr with AA and (d) 0.5 mol L−1 HBr with AA. The horizontal axis indicates the eluent volume (mL). The first eluent includes that of sample loading (0.1–0.2 mL). The vertical axis shows the yield (%) of each element relative to the loaded amount of the sample. | ||
Fig. 1(a) shows the elution curves of Fe, Cu and Zn when 2 mol L−1 HCl with AA was used. Fe, Cu and Zn adsorb on the TEVA resin forming chloro-complex but do not elute. This elution condition cannot be used because there is no separation of Fe from Zn. In the next step, the sample solution and eluents were changed into HBr. Using 0.05 mol L−1 HBr, Fe elutes together with Zn, but Cu does not appear (Fig. 1(b)), resulting in no separation between Fe and Zn. In a case of 0.25 mol L−1 HBr with AA, Fe and Zn can be separated, however, Cu is not separated from Zn (Fig. 1(c)). Thus, this condition also does not fit to the Zn purification. However, when 0.5 mol L−1 HBr with AA is applied, Cu as well as Fe elute, while Zn is still on the resin. Therefore, Zn can be purified from Cu and Fe. Likewise, alkaline, alkaline earth and rare earth elements are separated. Luck et al.3 used AG 1 × 4 and HBr, but, the details of the method are not described in their publication.
Performance of TEVA resin chemistry
Alkaline, alkaline earth and rare earth elements of >99% are removed in the first 1.6 mL of 0.5 mol L−1 HBr with AA. V, Cr, Mn, Fe, Co, Ni, Ga, Ge, Zr, La, Ce and Hf show similar behavior to Fig. 1(d). Coexistent elements in the Zn fraction are ∼20% of Nb, Ta and W, and >90% of Cd, In, Sn, Tl, Pb and Bi, which are obtained by using the mixture of multi-element standard solutions of Specpure no. 42885 and 44270. Total amounts of Nb, Ta and W in the sample solution should be lower than these values, because coprecipitation with Ti oxides is expected to occur in natural silicate analysis.19 Generally, abundances of these coexistent elements are low compared to Zn, thus, our method suits most silicate samples.The recovery yield of Zn in the various actual silicate composition containing 0.1 μg Zn was 96.7 ± 6.8% (n = 10). Small loss in the column chemistry should cause mass fractionation in column chemistry,20 however, as the double spike method is employed in this study, the fractionation in the column chemistry should be corrected.
The flow rate of the eluent was 0.3 mL min−1, therefore, the column chemistry can be finished within two hours including the resin wash and column conditioning procedure. This is another advantage of the TEVA resin column chemistry developed in this study compared to the conventional methods, for example, the flow rate of the eluent using 1.6 mL of AG MP-1 is ∼0.1 mL min−1, and total elution volume is 50 mL.5 Thus it takes ∼8 hours for the Zn column purification. If the washing time of the resin is included, total separation time is much longer.
The total blank including sample digestion was 0.05 ng (n = 6), which was determined by a calibration curve method. This blank was determined by measuring solution obtained after all sample digestion and column chemistry procedures. As 0.1 μg of Zn is separated, and isotope composition between the sample and blank is assumed <±10‰, this blank levels affect <±0.01‰ to the sample isotope composition, which can be neglected. This blank level is 1/300 of Marechal et al. (15 ng)5 or 1/20 of Archer and Vance (1 ng).8 In other words, 300 or 20 times Zn are required in the conventional methods to make blank effects negligible.5,8 Such low blank is one of advantages of this new column chemistry, and 0.1 μg of Zn is a sufficient amount to determine Zn isotope composition.
Isobaric interferences
In Fig. 2, interferences on Zn are summarized.21 Argides such as ArMg+, ArAl+ and ArSi+ should not exist, because Mg, Al and Si are removed in the TEVA column chemistry. Barium, La and Ce form interfering ions of 138Ba2+, 138La2+ and 140Ce2+, however, they are also removed by the TEVA column, therefore, there is no need to consider them. Ge has an isobaric isotope with Zn, 70Ge. In our sample digestion method using HF, Ge is removed as gaseous GeF4.17,22 Tellurium and Xe, form interfering doubly charged ions, 128Te2+ and 132Xe2+ in the plasma. Tellurium could remain in the Zn fraction. Even if 100% of Te remains in the Zn fraction, its effect should be <0.03‰ and negligible, because Te/Zn of general silicates is <1000,22 Te2+/Te+ is <0.1% and sensitivity of Te/Zn = ∼0.4. The double charge forming ratio and sensitivity ratio between Te and Zn were determined by the measurement condition in Table 2. | ||
| Fig. 2 Typical background levels of Neptune with a Jet cone, an X-skimmer cone, and a large rotary pump. The horizontal axis indicates mass (m) over charge (z) plotted in linear scale. The vertical axis shows the background levels in A in log scale. The background levels were obtained by NEPTUNE introducing 0.5 mol L−1 HNO3 through the desolvator at low (LR), middle (MR) and high (HR) mass resolutions (M/ΔM = ∼500, ∼3000 and ∼7500), respectively. The error bars indicate 1SE (standard error) of 20 measurements. The possible major interferences in the Zn measurement are shown at the top of the figure. | ||
In order to estimate the effects of 40Ar12C12C+, 132Xe2+, 40Ar12C16O+, 40Ar14N14N+, and 40Ar14N16O+ on m/z = 64, 66, 67, 68 and 70, respectively, backgrounds were measured at low (LR; M/ΔM = ∼500), middle (MR; M/ΔM = ∼3000) and high (HR; M/ΔM = ∼7500) resolutions (Fig. 2). As the resolution increases, the transmission of the mass spectrometer decreases so the background level decreases. At the highest resolution, the pattern of peaks becomes almost flat, indicating peculiar molecular ions are eliminated. However, at the low resolution, backgrounds show a zigzag pattern with higher backgrounds at m/z = 64, 66 and 68. As only 0.5 mol L−1 HNO3 through the desolvator is introduced into the plasma, these higher backgrounds are considered to be caused by 40Ar12C12C+, 132Xe2+, 40Ar14N14N+ and 40Ar12C16O+. The intensities of these backgrounds are 3–5 × 10−14 A. After the double spike addition, the smallest peak is 66Zn+ of ∼6 × 10−11 A, therefore, the largest effect is on the background of 66Zn+ and 0.5‰. The standard and sample solutions are basically the same Zn solution, it can be assumed that the intensity of 132Xe2+ should not change larger than 10%. If so, the accuracy of 66Zn+ is <0.05‰.
Performance of double spike-MC-ICP-MS
The average and standard deviation of double spike-MC-ICP-MS for the Zn standard of IRMM-3702 were δ66Zn = −0.00 ± 0.04 (2SD; n = 58) in this study. This error of 0.04 is considered to be the minimum error in the double spike technique including errors in the spike isotope ratio estimation.If the silicate sample is already digested and diluted with HNO3, the total analysis time is 3 days. The first day is for the column preparation and column separation; the second day is for drying HClO4 and final dissolution into 0.5 mol L−1 HNO3; and the third day is for measurement by double spike-MC-ICP-MS.
Mixing experiment of double spike-MC-ICP-MS
It is difficult to evaluate the accuracy for unconventional stable isotope systems such as Zn, because there are a few standard solutions for Zn23, which are difficult to obtain. One of the available standard solutions was IRMM-3702, which is already used for sample-standard bracketing measurements and also for calculating the isotope ratio as δ-notation. If there are two standards, evaluation of analytical methods or data becomes far easier. In this study, in order to estimate the accuracy, the mixed Zn solutions are made by mixing of two samples, and δ66Zn was compared with the calculated one. Such mixing experiments were done in Tl isotope determination to evaluate the accuracy of the method.24 The two samples were chosen to have different isotope and matrix compositions, therefore, matrix effects on isotopes were also examined in this test.For the two samples, the JB-2-digested solution (sample #1; 0.33 μg mL−1) of δ66Zn = −0.13 ± 0.08 (2SD; n = 5) and the diluted Alfa Aesar Specpure Zn standard solution (sample #2; 0.10 μg mL−1) of δ66Zn = −10.01 ± 0.24 (2SD; n = 29) were used. δ66Zn of sample #2 was determined by a bracketing method, because both sample #2 and IRMM-3702 were the same concentration Zn solutions with similar high purity. Sample #1 and sample #2 solutions were taken to be 0.1 μg with three different compositions. Then the mixed solutions were added with the Zn double spike, 0.4 mL 8 mol L−1 HBr and dried, re-dissolved, and passed through the TEVA column. Then δ66Zn of the mixtures was determined by the double spike-MC-ICP-MS. The calculated δ66Zn was obtained from the mixing ratio and δ66Zn of the samples #1 and #2. The error of the calculated δ66Zn of the mixture was obtained from errors of concentration of ∼5% of the two solutions and pipetting error of ∼1%.
Measurement results of the δ66Zn values of the three mixtures are shown in Table 3. The calculated value is also shown in Table 3. The results are also plotted in Fig. 3. The observed isotope fractionation is consistent with the calculated δ66Zn within error. Although the three samples have different isotope ratios and major element compositions, the observed δ66Zn in mixture is consistent with the calculated δ66Zn. Thus, it is concluded that the double spike-MC-ICP-MS of this study gives the analytical results independent of concentration of major element concentrations and δ66Zn, resulting in high reliability and accuracy.
| Calculated | 2SD (‰) | Measured | 2SD (‰) | n | |
|---|---|---|---|---|---|
| δ 66Zn | δ 66Zn | ||||
| Mixture #1 | −1.67 | −0.10 | −1.54 | 0.14 | 4 |
| Mixture #2 | −3.45 | −0.20 | −3.73 | 0.18 | 7 |
| Mixture #3 | −6.94 | −0.40 | −7.27 | 0.10 | 4 |
 | ||
| Fig. 3 Analytical results of the mixing experiments. The samples #1 and #2 are mixed, and their observed and calculated values of δ66Zn are plotted. The diagonal line shows slope 1. The dotted lines indicate errors of isotope ratios combined by the samples #1 and #2 (0.26‰, 2SD). Error bars are 2SD. | ||
Reproducibility of Zn isotope ratios in silicate reference materials
Reproducibility is obtained by 2SD of the same sample starting from different sample digestion. In Table 4, the δ66Zn values with 2SD for the USGS standard reference materials, BHVO-1, AGV-1 and PCC-1 are shown. δ66Zn of GSJ reference materials, JB-1, 2, 3, JA-1, 2, 3 and JP-1 and reproducibility (2SD) are also in Table 4. There are no δ66Zn values to compare to those of this study, so future extensive studies are required. The reproducibility of all measurements of the silicate samples in Table 4 was 0.08–0.32‰ (the average was 0.19‰ in 2SD), so this average (0.19‰) can be considered as the reproducibility in actual silicate sample analysis of this study.| Sample | δ 66Zn | 2SD | n |
|---|---|---|---|
| Average | (‰) | ||
| BHVO-1 | −0.42 | 0.26 | 5 |
| AGV-1 | −1.90 | 0.30 | 3 |
| PCC-1 | −0.09 | 0.10 | 5 |
| JB-1 | −0.04 | 0.32 | 3 |
| JB-2 | −0.13 | 0.08 | 5 |
| JB-3 | −0.29 | 0.20 | 3 |
| JA-1 | −0.03 | 0.08 | 3 |
| JA-2 | −0.33 | 0.14 | 4 |
| JA-3 | −0.11 | 0.18 | 3 |
| JP-1 | −0.15 | 0.20 | 4 |
Comparison of δ66Zn values of chondrites with those in other laboratories
The δ66Zn values of carbonaceous chondrites, Orgueil, Murchison and Allende are shown in Table 5. It is difficult to compare our data with those of other laboratories, because each laboratory uses different standards. For example, Moynier et al.2 and Luck et al.3 use Zn from Johnson Matthey Company (JMC) 400882B, called “Lyon solution” as the standard. Cloquet et al.23 reported that δ66Zn of Lyon solution is −0.32 ± 0.16 (2SD) compared to IRMM-3702. This correction factor makes δ66Zn of carbonaceous meteorite data of Moynier et al.2 and Luck et al.3 possible to be compared with our data. The error of correction factor of 0.16 (ref. 23) is considered as 2SD error of ref. 2 and 3. Ghidan and Loss4 use IRMM-3702, therefore, their data can be directly used here.| Sample | δ 66Zn | 2SD | n | References |
|---|---|---|---|---|
| Average | (Error is 2SD) | |||
| Orgueil | 0.59 | 0.26 | 3 | 0.14 ± 0.163, 0.20 ± 0.163, 0.60 ± 0.304 |
| Murchison | 0.16 | 0.04 | 4 | 0.12 ± 0.162, 0.01 ± 0.163, 0.13 ± 0.163 |
| Allende | −0.06 | 0.12 | 3 | 0.06 ± 0.163, −0.32 ± 0.163, 0.76 ± 0.324 |
In Table 5, δ66Zn of recalculated reference values are shown. In Fig. 4, the data of this study are compared with those of the reference values. From Fig. 4, δ66Zn of the carbonaceous chondrites measured in this study seem to be consistent with the reported values, indicating that our method can yield accurate results. However, variations of Allende in references are too large. This possibly indicates heterogeneity of the Allende meteorite.
 | ||
| Fig. 4 δ 66Zn of carbonaceous chondrites in this study vs. δ66Zn of those in ref. 2–4 The error bars are 2SD. A diagonal line indicates slope = 1. The solid circle, square and triangle indicate δ66Zn of Orgueil, Murchison and Allende, respectively. | ||
Conclusions
A new low-blank column chemistry for separating Zn has been developed employing HBr-ascorbic acid and TEVA resin. Recovery yield using silicate samples was 96.7 ± 6.8% (n = 10) and blank was 0.05 ng (n = 6). The blank level is from 1/300 to 1/20 of previous studies, meaning 0.1 μg of Zn is sufficient for analysis. Therefore, 0.1 μg of Zn was used in this study. We found that the total separation time of Zn including resin washing time can be reduced to < two hours.The δ66Zn value was determined using double spike MC-ICP-MS. The IRMM-3702 was used as the standard. The δ66Zn values of 0.04‰ (2SD; n = 58) was obtained as reproducibility of the standard solution by the double spike-MC-ICP-MS.
In order to show the applicability of this method, δ66Zn of three USGS and seven GSJ standard silicate materials were determined. The reproducibility of δ66Zn was estimated from the average of total silicate samples to be 0.19‰ (2SD). δ66Zn of carbonaceous chondrites of Orgueil, Murchison and Allende are measured and compared with the results of previous studies to evaluate the accuracy of double spike MC-ICP-MS.
Acknowledgements
The authors thank Kayo Tanaka for performing the column chemistry; T. Moriguti, C. Sakaguchi and all members of PML for maintaining our clean laboratory. The authors also thank the two anonymous journal referees for improving the manuscript.References
- A. D. Anbar and O. Rouxel, Annu. Rev. Earth Planet. Sci., 2007, 35, 717–746 CrossRef CAS.
- F. Moynier, J. Blichert-Toft, P. Telouk, J.-M. Luck and F. Albarede, Geochim. Cosmochim. Acta, 2007, 71, 4365–4379 CrossRef CAS.
- J.-M. Luck, D. Ben Othman and F. Albarede, Geochim. Cosmochim. Acta, 2005, 69, 5351–5363 CrossRef CAS.
- O. Y. Ghidan and R. D. Loss, Meteorit. Planet. Sci., 2011, 46, 830–842 CrossRef CAS.
- C. M. Marechal, P. Telouk and F. Albarede, Chem. Geol., 1999, 156, 251–273 CrossRef CAS.
- A. E. Shiel, J. Barling, K. J. Orians and D. Weis, Anal. Chim. Acta, 2009, 633, 29–37 CrossRef CAS.
- J.-B. Chen, P. Louvat, J. Gaillardet and J.-L. Birck, Chem. Geol., 2009, 259, 120–130 CrossRef CAS.
- C. Archar and D. Vance, J. Anal. At. Spectrom., 2004, 19, 656–665 RSC.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, S. L. Maxwell III and M. R. Nelson, Anal. Chim. Acta, 1995, 310, 63–78 CrossRef CAS.
- A. Makishima, M. Nakanishi and E. Nakamura, Anal. Chem., 2001, 73, 5240–5248 CrossRef CAS.
- Z. Grahek and M. R. Macefat, Anal. Chim. Acta, 2004, 511, 339–348 CrossRef CAS.
- M. H. Dodson, J. Sci. Instrum., 1963, 40, 289–295 CrossRef.
- J. F. Rudge, B. C. Reynolds and B. Bourdon, Chem. Geol., 2009, 265, 420–431 CrossRef CAS.
- T. Arnold, M. Schönbächler, M. Rehkämper, S. Dong, F.-J. Zhao, G. J. D. Kirk, B. J. Coles and D. J. Weiss, Anal. Bioanal. Chem., 2010, 398, 3115–3125 CrossRef CAS.
- E. Nakamura, A. Makishima, T. Moriguti, K. Kobayashi, C. Sakaguchi, T. Yokoyama, R. Tanaka, T. Kuritani and H. Takei, Inst. Space Astronaut. Sci. Rep., 2003, 16, 49–101 Search PubMed.
- C. M. Johnson and B. L. Beard, Int. J. Mass Spectrom., 1999, 193, 87–99 CrossRef CAS.
- A. Makishima and E. Nakamura, Geostand. Geoanal. Res., 2006, 30, 245–271 CrossRef CAS.
- A. Makishima and E. Nakamura, J. Anal. At. Spectrom., 2010, 25, 1712–1716 RSC.
- A. Makishima and E. Nakamura, Geochem. J., 2008, 42, 199–206 CrossRef CAS.
- C. M. Marechal and F. Albarede, Geochim. Cosmochim. Acta, 2002, 66, 1499–1509 CrossRef CAS.
- T. F. D. Mason, D. J. Weiss, M. Horstwood, R. R. Parrish, S. S. Russell, E. Mullane and B. J. Coles, J. Anal. At. Spectrom., 2004, 19, 209–217 RSC.
- A. Makishima and E. Nakamura, Geostand. Geoanal. Res., 2009, 33, 369–384 CrossRef CAS.
- C. Cloquet, J. Carignan and G. Libourel, Environ. Sci. Technol., 2006, 40, 451–463 Search PubMed.
- S. G. Nielsen, M. Rehkämper, J. Baker and A. N. Halliday, Chem. Geol., 2004, 204, 109–124 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |