Development of siRNA-probes for studying intracellular trafficking of siRNA nanoparticles
Christopher A.
Alabi
ab,
Gaurav
Sahay
a,
Robert
Langer
abc and
Daniel G.
Anderson
*abc
aDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, MA 02142, USA. E-mail: dgander@mit.edu; Tel: +1 617-258-6843
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cHarvard-MIT division of Health Sciences & Technology, Cambridge, MA 02139, USA
First published on 29th August 2012
Abstract
One important barrier facing the delivery of short interfering RNAs (siRNAs) via synthetic nanoparticles is the rate of nanoparticle disassembly. However, our ability to optimize the release kinetics of siRNAs from nanoparticles for maximum efficacy is limited by the lack of methods to track their intracellular disassembly. Towards this end, we describe the design of two different siRNA-based fluorescent probes whose fluorescence emission changes in response to the assembly state of the nanoparticle. The first probe design involves a redox-sensitive fluorescence-quenched probe that fluoresces only when the nanoparticle is disassembled in a reductive environment. The second probe design is based on a FRET-labeled siRNA pair that fluoresces due to the proximity of the siRNA pair when the nanoparticle is intact. In both approaches, the delivery vehicle need not be labeled. The utility of these probes was investigated with a lipidoid nanoparticle (LNP) as proof-of-concept in both extracellular and intracellular environments. Fluorescence kinetic data from both probes were fit to a two-phase release and decay curve and subsequently quantified to give intracellular disassembly rate constants. Quantitative analysis revealed that the rate constant of siRNA release measured via the fluorescence-quenched probe was almost identical to the rate constant for nanoparticle disassembly measured via the FRET-labeled probes. Furthermore, these probes were utilized to determine subcellular localization of LNPs with the use of automated high-resolution microscopy as they undergo dissociation. Interestingly, this work shows that large amounts of siRNA remain inside vesicular compartments. Altogether, we have developed new siRNA probes that can be utilized with multiple nanocarriers for quantitative and qualitative analysis of nanoparticle dissociation that may serve as a design tool for future delivery systems.
Insight, innovation, integrationA key component of siRNA delivery is the interaction of nanocarriers with cells. Integration of nanoparticle disassembly kinetics with dynamic processes of subcellular trafficking is required for the design of nanocarriers that can both contain their payloads, and release them at their final target site. To this end, we have developed siRNA probes for studying the intracellular disassembly kinetics of siRNA nanocarriers. Our study provides insight into the extent and rate of nanoparticle disassembly in a cancer cell line from two different viewpoints; siRNA release and nanoparticle disassembly. Construction of the probes on the siRNA and not the delivery vehicle allows for the possibility of investigating and comparing the intracellular disassembly kinetics of a wide range of nanocarriers without the limitation of nanocarrier chemical structure. Biophysical information gleaned from the application of these probes may aid the design of future nanocarriers for the development of siRNA therapeutics. |
Introduction
Successful delivery of nanoparticles containing siRNAs to the RNA induced silencing complex (RISC) in the cell requires efficient nanoparticle assembly, cellular uptake, subcellular trafficking and timely intracellular release.1–3 During the process of designing nanoparticles for siRNA delivery, structural properties of the delivery vehicle such as charge density, degree of hydrophobicity and extent of siRNA association are optimized. Successful delivery requires that the nanoparticles can stay intact in the presence of serum binding proteins, which are often responsible for extracellular disassembly of nanoparticles.4 However, once siRNA containing nanoparticles reach the extracellular milieu and enter the cell, they will need to disassemble to engage RISC and subsequently down regulate the expression of the target protein. Premature release in the extracellular milieu, as well as lack of intracellular release due to tight nanoparticle–siRNA interactions may lead to inefficient gene silencing.5 Despite its importance, intracellular disassembly has only been investigated in a few siRNA nanoparticles6–8 due to the considerable challenge associated with making this measurement in cells.Current methods employed for measuring intracellular disassembly involve labeling both the delivery vehicle and nucleic acid with different fluorophores or quantum dots and observing co-localization of their emissions qualitatively via microscopy,6,9 or labeling both species with a pair of fluorophores/quantum dots that undergo Förster resonance energy transfer (FRET).7,8,10,11 Both approaches require labeling the delivery agent and are thus limited to delivery agents that have sites available for conjugation and are robust enough such that fluorophore conjugation does not hamper the activity of the delivery agent. Due to these stringent conditions, this approach has not enjoyed widespread use. However, restricting fluorophore labeling to just the nucleic acid will circumvent the afore-mentioned issues and extend utility to all delivery agents. We adopted this strategy in the design of two siRNA-based fluorescent probes that can non-invasively track intracellular nanoparticle disassembly. The first probe design involves the use of a siRNA tethered to a black hole quencher (BHQ3) and AlexaFluor 647 (AF647) that turns on only in reducing environments, such as the intracellular space. Thus, nanoparticles encapsulating this probe will remain dark until they release their siRNA cargo into the intracellular space. This gives rise to an “off–on” system. The second probe design involves the use of the recently discovered pair of FRET-labeled siRNA probes, siRNA-AlexaFluor 594 (siAF594) and siRNA-AlexaFluor 647 (siAF647) conjugates, which fluoresce due to their proximity within the nanoparticle and turn off once the nanoparticle disassembles.12 As a result, this gives rise to an “on–off” system. We demonstrate functionality of both probes with lipidoid nanoparticles (LNPs) in both extracellular and intracellular environments. Although the two probe sets operate differently, we show that information gathered from their data are complimentary.
Results
The “off–on” fluorescence-quenched probe was designed to fluoresce only in the reducing environment of the intracellular space. This was accomplished via the conjugation of a quencher and a fluorophore to each strand of the RNA duplex. Specifically, a BHQ3 dark quencher was conjugated to the 3′ end of the sense strand via a reducible disulfide bond and the fluorophore AF647 was conjugated to the 5′ end of the complimentary DNA strand via an amide linkage. Hybridization of the two strands brings BHQ3 in proximity to AF647 and results in efficient quenching of the AF647 fluorescence via a combination of contact quenching and FRET13,14 (Scheme 1). However, reduction of the disulfide linkage via glutathione (GSH), which occurs in the reducing environment of the intracellular space,15 should result in the release of BHQ3 and subsequent de-quenching of the AF647 fluorescence. Exposure of both 1 and 3 to solutions with and without GSH (Fig. 1a and b) demonstrates that the fluorescence of probe 3 is activated (∼7-fold increase) only in the presence of GSH. No such increase is observed with 1. In fact, a slight decrease (∼1.3-fold) in the fluorescence of 1 was observed the presence of GSH. | ||
| Scheme 1 Assembly of the fluorescence-quenched “off–on” probe 3 from hybridization of DNA-AF647 with RNA-SS-BHQ3. Cleavage of 3 with GSH turns the probe on via de-quenching of AF647 by BHQ3. | ||
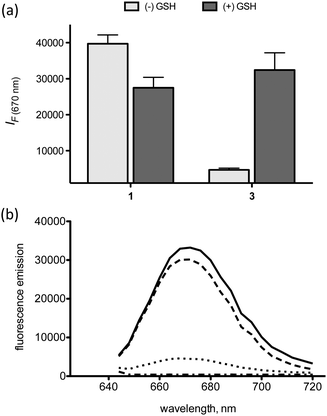 | ||
| Fig. 1 (a) Fluorescence of 1 and 3 (excitation at 640 nm) in the presence and absence of GSH, (b) fluorescence emission curves of 1: (—), 2: (− - −), 3: (···), and 3 + GSH: (– – –) (excitation at 620 nm). Error bars represent s.d., n = 3. | ||
To test if fluorescence-quenching properties of probe 3 can be used to detect nanoparticle disassembly, we encapsulated probe 3 in a LNP and examined its fluorescence emission in the presence and absence of GSH and Triton-X, a non-ionic surfactant that disassembles LNPs via lipid membrane disruption. The fluorescence of a LNP containing probe 3 was activated only when both Triton-X and GSH were present (Fig. 2). This means that nanoparticles encapsulating 3 will fluoresce only after disassembly in a reducing environment such as the cytosol. To contrast these results, we also tested LNPs with encapsulated probe 4 under the same conditions. Probe 4 is a control siRNA probe without the BHQ3 quencher and was synthesized via hybridization of 1 with a non-labeled complimentary sequence. Treatment of LNPs containing 4 with and without GSH in the presence of Triton-X (+/+ and +/−, Fig. 2) resulted in similarly high fluorescence intensities. This further confirms that BHQ3 plays a major role in quenching the fluorescence of AF647 in probe 3. However, we also observe that the fluorescence of probe 4 is about 4-fold lower in the absence of Triton-X, indicating that confinement of the siRNAs in the LNP leads to partial self-quenching of the AF647 fluorescence. Nevertheless, the role of self-quenching is minimal when compared to BHQ mediated quenching (∼40-fold difference) which accounts for ∼90% of the total quenching observed (Fig. 2).
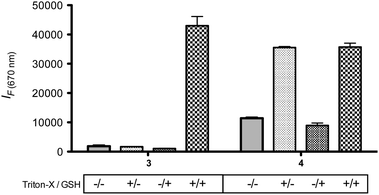 | ||
| Fig. 2 LNPs containing 3 and 4 were incubated with and without 1% Triton-X in the presence and absence of 10 mM GSH at 37 °C. Excitation/emission: 640/670 nm. Error bars represent s.d., n = 2. | ||
The second probe design involves a FRET-labeled pair of siRNAs, which fluoresce only when encapsulated in a LNP (Scheme 2). Proof-of-concept of this probe has recently been described for ligand–siRNA complexes.12 The FRET donor is an AlexaFluor 594 fluorophore conjugated to the 5′ end of the sense strand of a siRNA duplex, while the acceptor is an AlexaFluor 647 fluorophore conjugated to the 5′ end of the sense strand of another siRNA duplex. Both labeled siRNAs have the same sequence. Upon encapsulation in the LNP, the aggregated probes are within the Förster radius of the AlexaFluor 594 and 647 (∼8.5 nm). Under these conditions, excitation of the donor at 540 nm leads to emission from the acceptor at 690 nm (Fig. 3a).
 | ||
| Scheme 2 FRET-labeled siRNA probes in the “on” and “off” state. | ||
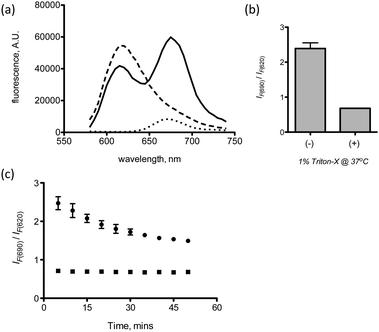 | ||
| Fig. 3 (a) Fluorescence emission (excitation at 540 nm) of LNP-siAF594 (– – –), LNP-siAF647 (···) and LNP-siAF594/647 (—), (b) LNP-si594/647 with (+) and without (−) 1% Triton-X, (c) kinetics of LNP-siAF594/647 disassembly in the absence (●) and presence (■) of 1% Triton-X. Error bars represent s.d., n = 2. | ||
The FRET signal is measured as a ratio of the fluorescence due to FRET at 690 nm to the residual emission from the donor at 620 nm. To test the sensitivity of the probe to disassembly, the LNP containing the FRET-labeled probes was exposed to 1% Triton-X for 30 minutes. Fig. 3b shows a 3-fold decrease in FRET signal in the presence of Trition-X, consistent with LNP disassembly. Furthermore, the kinetics of LNP disassembly in the 1× PBS can also be monitored as shown in Fig. 3c. The kinetic plot shows a fast disassembly phase, which occurs in the first 30 minutes, followed by a slower disassembly phase thereafter.
The above studies on the fluorescence-quenched and FRET-labeled probes demonstrate their ability to detect siRNA release and LNP disassembly outside the cellular space. Next, we focused on demonstrating the utility of these probes in vitro. To show activity of the fluorescence-quenched probe 3in vitro, we contrasted its uptake in HeLa cells to that of the traditional “always on” probe, 4. Thus, formulated LNPs containing probe 3 and 4 were transfected into HeLa cells at different time points and the total fluorescence at each time point was quantified via flow cytometry. An increase in fluorescence intensity with time is observed due to an increase in siRNA release as well as increased LNP accumulation. At earlier time points (between 0–2 hours), the results in Fig. 4 clearly show a larger difference between the fluorescence intensities of probe 3 and 4 than at later time points. The larger difference at earlier time points occurs because a majority of LNPs containing probe 3 are still intact and thus the fluorescence of AF647 in probe 3 is quenched. However, as time progresses, LNPs containing probe 3 disassemble and the fluorescence intensity from the sample increases and slowly approaches the fluorescence of probe 4, which is always “on”. The increase in fluorescence intensity of LNPs containing probe 3 relative to LNPs containing probe 4 should be due to de-quenching of the AF647 fluorophore on the released probe 3via intracellular mediated GSH cleavage of BHQ3. Thus, the ratio of the fluorescence intensity of probe 3 to 4 (Fig. 4b) represents the percentage non-encapsulated intracellular siRNA. This ratio also normalizes for the increase in fluorescence due to LNP unpacking (Fig. 2) prior to GSH cleavage of probe 3.
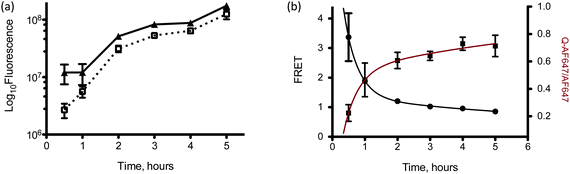 | ||
| Fig. 4 (a) Flow cytometry analysis of LNPs containing 3 (□) and 4 (▲) after transfection in HeLa cells at different time points, (b) flow cytometric analysis of FRET-labeled siRNAs (●) transfected into HeLa cells at different time points. The data was fit to a two-phase decay function (black line). Also plotted is the ratio of the fluoresce data of LNPs containing 3 to that of LNPs containing 4 (■). This data was fit to a two-phase association function (red line). Error bars represent s.d., n = 3. | ||
The utility of the FRET-labeled probes in vitro was also demonstrated in HeLa cells using LNPs with encapsulated siAF594 and siAF647. The FRET output shown in Fig. 4b is the emission at 695 nm (excitation with a yellow-green laser at 561 nm) normalized to the emission from AlexaFluor 647 (excitation/emission 633/660 nm) alone. By doing this, the FRET signal from the LNPs is always normalized to the total amount of LNPs taken up by the cells. The results show that the FRET signal decreases with time, indicative of LNP disassembly. Similar to the extracellular kinetics data in Fig. 3c, we see a two-phase exponential decay of LNPs in the cell; a fast burst occurs in the first two hours followed by a slower decay thereafter. This two-phase decay is in agreement with the kinetics of siRNA release measured by the fluorescence-quenched probe 3. The kinetic profiles of both FRET and fluorescence-quenched data were fit to the two-phase exponential kinetic decay and release models; y = yo+ A1(e−kft) + A2(e−kst) and y = yo + A1(1 – e−kft) + A2(1 − e−kst) respectively, where kf and ks represent the rate constants of the fast and slow phase respectively.
The exponential kinetic functions used to model the experimental kinetic data showed very good fits for both fluorescence-quenched and FRET-labeled probes (R2 > 0.99). Both fast and slow dissociation rate constants obtained for the fluorescence-quenched probe 3 are in good agreement with those obtained for the FRET-labeled probe (Table 1). In both cases, the initial burst release, characterized by kf is about 20 times faster than the rate constant for the slow release phase, ks.
| Probe | k f (h−1) | k s (h−1) | R 2 |
|---|---|---|---|
| Fluorescence-quenched probe, 3 | 2.24 | 0.12 | 0.9908 |
| FRET-labeled probe | 2.35 | 0.09 | 0.9999 |
Finally, we visualized intracellular trafficking of the LNPs with encapsulated fluorescence-quenched and FRET-labeled probes via automated confocal microscopy. LNPs containing FRET probes after incubation with HeLa cells for one hour accumulate in the endosomal (punctate dots) and cytosolic space (diffuse staining) (Fig. 5). Representative images of cells transfected with LNPs containing probe 3 show an increase in fluorescence intensity with time due to increase in siRNA release as well as increased LNP accumulation. Similar results are obtained with LNPs containing probe 4, with the exception that the overall fluorescence intensities at all points are higher due to this probe being “always on”. Upon close inspection, we can also visually observe that the difference in fluorescence intensities between probe 4 and 3 is greater at the earlier time points than at later time points. These results are in agreement with the quantitative data from flow cytometry experiments in Fig. 4a. Cells transfected with LNPs containing FRET-labeled probes showed extensive co-localization (yellow color) between the siAF594 and siAF647 fluorophores (Fig. 5, bottom panel), indicative of siRNA entrapment within the endosome.
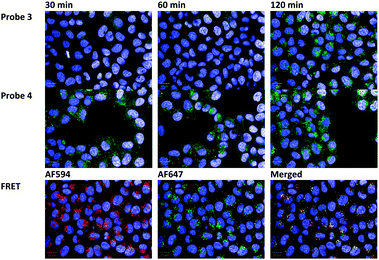 | ||
| Fig. 5 (Top panel) LNPs containing fluorescence-quenched probe 3 after incubation with HeLa cells for 30, 60 and 120 minutes. (Middle panel) LNPs containing probe 4 after incubation with HeLa cells for 30, 60 and 120 minutes. (Bottom panel) LNPs containing FRET-labeled siRNAs after 120 minutes of incubation with HeLa cells. The nucleus of the cell is stained blue with DAPI. | ||
Discussion
The discovery of siRNAs and their gene silencing potential has lead to the synthesis and development of numerous synthetic delivery vehicles. However, progress on the development of molecular analytics that can be used to probe these synthetic vehicles and report their biophysical properties in their active environment is lagging. So far, biophysical properties such as nanoparticle size, surface charge and siRNA entrapment have been well documented on numerous synthetic nanoparticles thanks to light scattering techniques and RNA detection strategies such as the Ribogreen™ assay. By correlating these properties with siRNA delivery efficiency, we now have a better idea on the size, surface charge and loading requirements necessary for efficient delivery.16 However, the relationship between other biophysical properties such as nanoparticle elasticity, surface hydrophobicity and the intracellular rate constant of disassembly, among others are yet to be well understood due to the lack of molecular probes required for their measurement. In this article, we described the design and operation of a fluorescence-quenched probe 3 and FRET-labeled probes for measuring the intracellular rate constant of nanoparticle disassembly.Both designs involve labeling the nucleic acid with fluorophores that respond to environmental cues. The fluorescent-quenched probe 3 was designed to mimic the activity of molecular beacons.17 The latter are hairpin shaped molecules that are quenched in the hairpin mode, due to the alignment of a fluorophore–quencher pair, but fluoresce when bound to a target nucleic acid. In our approach, we use a similar design to achieve quenching, i.e. placing the quencher and fluorophore adjacent to one another at the 5′ and 3′ ends of the nucleic acid. However, de-quenching is triggered through GSH mediated cleavage of the disulfide bond used to tether the quencher molecule to the nucleic acid. GSH was chosen as the environmental cue due to its intracellular abundance (∼1–10 mM) in healthy cells.15,18 As a result, a nanoparticle containing this probe fluoresces only after intracellular disassembly and siRNA release. This is in contrast to traditional probes, such as probe 4, whose fluorescence is largely independent on the state of assembly of the nanoparticle (Fig. 2).
The second probe design involves a pair of FRET-labeled probes composed of two identical siRNAs, each labeled with fluorophores capable of undergoing distance dependent energy transfer, i.e. FRET. Upon nanoparticle formation, the siRNA-bound fluorophores become locally aggregated within the nanoparticle and undergo FRET. However, upon disassembly, these labeled siRNAs drift away from one another resulting in a decrease in FRET signal. Thus, this probe can be used to report on the rate and extent of nanoparticle disassembly as shown in Fig. 3 and 4. It is important to note that neither of these strategies involves labeling the delivery vehicle, a key advantage that allows this technique to be used on a variety of nanoparticles. After establishing proof-of-concept with each probe (Fig. 1–3), we tested the intracellular response of each probe encapsulated in LNPs using HeLa cells. Both molecular probes showed a two-phase kinetic decay and release profile for the LNPs containing siRNAs. Moreover, the rate of nanoparticle disassembly as measured by the FRET-labeled probes was in very good agreement with the rate of siRNA release measured by the fluorescence-quenched probe. The fact that both probes give near identical rate constants validates both measurements and shows that they both can be used interchangeably. It is important to note that the disassembly rates as measured by flow cytometry represent the averaged rate of LNP disassembly from all the cellular compartments, including the endosome, endoplasmic reticulum, Golgi, lysosomes and cytosol. Compartment specific disassembly rates can be achieved using the FRET probe by quantitative microscopy if individual intracellular compartments can be clearly delineated. The utility of the quencher probe in this instance will depend on the reducing potential of the compartment in question.
With regards to implementation in the research workflow, the fluorescence-quenched probe is relatively simple to implement because it involves the use of only one fluorophore (quencher is non-fluorescent). This means that only one filter set is required for imaging and quantitation. The fluorescence-quenched probe being “off” in the extracellular media will also be an asset in live cell microscopy as a result of the potential for low background signal. Finally, because this probe responds to GSH, it may also find use as a reporter for intracellular GSH as well as oxidative stress in cells both in vitro and in vivo.19,20 However, intracellular rate constants of siRNA release measured with this probe in cells that are under severe oxidative stress may be longer than that in normal cells due to lower GSH concentrations. Unlike the fluorescence-quenched probe, the FRET-labeled probe requires the use of two fluorophores and thus two emission filters. However, this probe is independent of GSH and can be used to measure disassembly in both healthy and oxidatively stressed cells. Thus, depending on the available instrumentation and type of cells under investigation, one or both probes might prove useful for quantifying the rate and extent of siRNA nanoparticle disassembly.
Conclusion
We describe the design and synthesis of a fluorescence-quenched probe and a pair of FRET-labeled probes for measuring the rate of disassembly of siRNA bound nanoparticles in both extracellular and intracellular environments. Both probes circumvent the need to label the delivery vehicle, allowing for their use in delivery vehicles that are difficult to label. The fluorescence-quenched and FRET-labeled probes were in evaluated in a cancer cell line with LNPs and each displayed a two-phase exponential release and decay profile respectively. The rate constants for siRNA release and LNP disassembly obtained from the fits to the intracellular kinetic data were in agreement, further validating both experiments. The intracellular disassembly rate constant will be a valuable parameter for use in in vitro and in vivo computational models for nucleic acid delivery as well as understanding and improving nanoparticle design through structure function correlations.Materials and methods
Materials
siRNA duplexes labeled at the 5′ end of the sense strand with either Alexa Fluor594 or Alexa Fluor647 dyes were purchased from Integrated DNA Technologies (HPLC purified and desalted). The sequences are: (sense) 5′-Alex594-GAUUAUGUCCGGUUAUGUAUU-3′, 5′-Alex647-GAUUAUGUCCGGUUAUGUAUU-3′ and (antisense) 5′-UACAUAACCGGACAUAAUCUU-3′. The following sequences were used for the preparation of the fluorescence-quenched probe: (sense) 5′-CUGGCUGAAUUUCAGAGCAdTsdT-(SS-CH2CH2NH2)-3′ (RNA-SS-NH2, bases in bold are 2′OMe modified), obtained from Alnylam pharmaceuticals and (antisense) 5′-(Alexa647) TGC TCT GAA ATT CAG CCA GTT-3′ (Compound 1) obtained from Integrated DNA Technologies. Phenol red-free DMEM, fetal bovine serum (FBS) and 0.25% trypsin-EDTA were obtained from Invitrogen. Black Hole Quencher-3 carboxylic acid succinimidyl ester (BHQ3-NHS) was purchased from BIOSEARCH technologies. Cholesterol and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) were obtained from Sigma Aldrich and mPEG2000-DMG was synthesized by Alnylam Pharmaceuticals.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38.5:10:1.5 in ethanol and siRNA-probe (3, 4 or FRET-labeled probes) in pH 3 citrate buffer (40 ng μL−1). This mixture was subsequently diluted at a 1:1 volume ratio with 1× PBS and dialyzed overnight with a 3500 MWCO dialysis membrane (Thermoscientific PIERCE).
:38.5:10:1.5 in ethanol and siRNA-probe (3, 4 or FRET-labeled probes) in pH 3 citrate buffer (40 ng μL−1). This mixture was subsequently diluted at a 1:1 volume ratio with 1× PBS and dialyzed overnight with a 3500 MWCO dialysis membrane (Thermoscientific PIERCE).
Fluorescence-quenched and FRET assays
LNPs containing probe 3 or 4 were diluted to 1 ng μL−1 in 1× PBS, 1× PBS with 1% Triton-X, and/or 1× PBS with 10 mM GSH in a black 96-well plate. Fluorescence was measured via excitation at 640 nm and emission at 670 nm. Fluorescence emission plots were measured between 640 and 720 nm via excitation at 620 nm. LNPs containing an equimolar mixture of siRNAs labeled with Alexa Fluor594 and Alexa Fluor647 were diluted to 1 ng μL−1 in 1× PBS or 1× PBS with 1% Triton-X in a black 96-well plate. To measure FRET, the samples were excited at 540 nm and the fluorescence intensity was read at 690 and 620 nm using a Tecan Safire 2 Microplate reader. FRET was determined as the ratio of the fluorescence intensities at 690/620 nm. The FRET signals for all samples were normalized to a control FRET signal from the siAF594 and siAF647 pair without LNP. All experiments were conducted at room temperature unless stated otherwise.Cell culture
HeLa cells, stably expressing firefly luciferase and Renilla luciferase, were generously donated by Alnylam pharmaceuticals. The cells were maintained in phenol red-free DMEM media (no antibiotics) with 10% FBS (growth media) at 37 °C in a 5% CO2 humidified atmosphere.Intracellular disassembly kinetic measurements
100 μL of cells at a cell density of 2 × 104 cells per well were seeded in black tissue culture treated 96-well plates and maintained for 24 hours. On the day of transfection, 50 ng of siRNA probes in LNPs were added to each well and the cells were incubated for the indicated time period. All experiments were carried out in triplicates. After transfection, the media was aspirated and cells were washed with calcium and magnesium free 1× PBS and trypsinized with 30 μL of 0.25% trypsin-EDTA. The trypsined cells were then neutralized with 120 μL of quenching media (25% cell culture media in 1× PBS) and transferred to a 96-well v-bottom plate and analyzed via flow cytometry. For each sample, 10000 events were monitored and evaluated by a BD LSR II HTS flow cytometer (BD Bioscience). Samples with probe 3 and 4 were analyzed using settings for the Alexa Fluor647 channel: excitation via a 633 nm (red, HeNe) laser and emission via a 660/20 nm filter. For FRET analysis, samples were excited with a 561 nm excitation laser and their emission was observed with a 695/40 nm emission filter set. The emission signal obtained was normalized to the emission signal obtained from the Alexa Fluor647 channel to normalize for the total LNP uptake at each time point. In addition, each channel was compensated for bleed through using single fluorophore controls.
Confocal microscopy
HeLa cells were seeded at 1.5 × 104 cells per well in black (clear bottom) 96-well plates (Greiner Bio-one) in incubated for 24 hours prior to transfection. Cells were then transfected with LNPs containing 50 ng of fluorescence-quenched probe 3, probe 4, or FRET-labeled probes for the indicated time points. Cells were washed, fixed, and counterstained in PBS containing Hoechst (2 μg mL−1) for nuclei identification. The cells were then imaged using an automated spinning disk confocal microscope (OPERA, Perkin Elmer) with a 40× objective. The same defined pattern of 20 fields from each well was acquired to eliminate bias and provide a statistically significant number of cells for analysis. The exposure rates were maintained constant for individual fluorophores across the 96-well plate. The images were acquired from the Acapella software with a 20 μm scale bar. Data represents images from 20 different fields. All experiments were performed in triplicate.Acknowledgements
The Authors thank the Koch Institute Flow Cytometry Core at MIT for use of their flow cytometer for data collection. The Authors thank Alnylam pharmaceuticals for use of their automated spinning disk confocal microscope (OPERA, Perkin Elmer). The Authors also thank the NIH grants R37-EB000244, R01-CA132091 and R01-CA115527 for funding. C.A.A. thanks the NIH for his Postdoctoral Fellowship.References
- A. Mescalchin, A. Detzer, M. Wecke, M. Overhoff, W. Wünsche and G. Sczakiel, Expert Opin. Biol. Ther., 2007, 7, 1531–1538 CrossRef CAS.
- E. Wagner, Acc. Chem. Res., 2012, 45, 1005–1013 CrossRef CAS.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS.
- K. Buyens, M. Meyer, E. Wagner, J. Demeester, S. C. De Smedt and N. N. Sanders, J. Controlled Release, 2010, 141, 38–41 CrossRef CAS.
- V. S. David, A. F. Nick, D. Nily and A. L. Douglas, Biotechnol. Bioeng., 2000, 67, 598–606 CrossRef.
- B. Lucas, K. Remaut, N. N. Sanders, K. Braeckmans, S. C. De Smedt and J. Demeester, Biochemistry, 2005, 44, 9905–9912 CrossRef CAS.
- H. Lee, I.-K. Kim and T. G. Park, Bioconjugate Chem., 2010, 21, 289–295 CrossRef CAS.
- S. Jiang and Y. Zhang, Langmuir, 2010, 26, 6689–6694 CrossRef CAS.
- C. Leal, N. F. Bouxsein, K. K. Ewert and C. R. Safinya, J. Am. Chem. Soc., 2010, 132, 16841–16847 CrossRef CAS.
- C. Grigsby and Y. Ho, Nanomedicine, 2012, 7, 565–577 CrossRef CAS.
- Y. Wu, Y.-P. Ho, Y. Mao, X. Wang, B. Yu, K. W. Leong and L. J. Lee, Mol. Pharmaceutics, 2011, 8, 1662–1668 CrossRef CAS.
- C. A. Alabi, K. T. Love, G. Sahay, T. Stutzman, W. T. Young, R. Langer and D. G. Anderson, ACS Nano, 2012, 6, 6133–6141 CrossRef CAS.
- M. K. Johansson, H. Fidder, D. Dick and R. M. Cook, J. Am. Chem. Soc., 2002, 124, 6950–6956 CrossRef CAS.
- M. K. Johansson, Methods Mol. Biol., 2006, 335, 17–29 CAS.
- F. Schafer and G. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS.
- A. Akinc, M. Goldberg, J. Qin, J. R. Dorkin, C. Gamba-Vitalo, M. Maier, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, M. Manoharan, V. Koteliansky, I. Rohl, E. S. Leshchiner, R. Langer and D. G. Anderson, Mol. Ther., 2009, 17, 872–879 CrossRef CAS.
- S. Tyagi and F. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- G. Wu, Y. Fang, S. Yang, J. Lupton and N. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- M. H. Lee, J. H. Han, P.-S. Kwon, S. Bhuniya, J. Y. Kim, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 1316–1322 CrossRef CAS.
- N. Shao, J. Jin, H. Wang, J. Zheng, R. Yang, W. Chan and Z. Abliz, J. Am. Chem. Soc., 2010, 132, 725–736 CrossRef CAS.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Y. Sah, A. de Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1864–1869 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |