Imaging of genetically engineered T cells by PET using gold nanoparticles complexed to Copper-64†
Parijat
Bhatnagar
a,
Zheng
Li
b,
Yoonsu
Choi
c,
Jianfeng
Guo
b,
Feng
Li
b,
Daniel Y.
Lee
b,
Matthew
Figliola
c,
Helen
Huls
c,
Dean A.
Lee
cd,
Tomasz
Zal
e,
King C.
Li
b and
Laurence J. N.
Cooper
*cd
aBaylor College of Medicine & Texas Children's Hospital, Department of Obstetrics & Gynecology, Division of Maternal Fetal Medicine, Houston, TX 77030, USA
bDepartment of Radiology, The Methodist Hospital Research Institute, Houston, TX 77030, USA
cDivision of Pediatrics, The University of Texas MD Anderson Cancer Center, Pediatrics-Research, Unit 907, 1515 Holcombe Boulevard, Houston, TX 77030, USA. E-mail: ljncooper@mdanderson.org; Fax: +1 713 792-9832; Tel: +1 713 563-3208
dThe University of Texas Graduate School of Biomedical Sciences, Houston, TX 77030, USA
eDepartment of Immunology, MD Anderson Cancer Center, Houston, TX 77030, USA
First published on 18th September 2012
Abstract
Adoptive transfer of primary T cells genetically modified to have desired specificity can exert an anti-tumor response in some patients. To improve our understanding of their therapeutic potential we have developed a clinically-appealing approach to reveal their in vivo biodistribution using nanoparticles that serve as a radiotracer for imaging by positron emission tomography (PET). T cells electroporated with DNA plasmids from the Sleeping Beauty transposon–transposase system to co-express a chimeric antigen receptor (CAR) specific for CD19 and Firefly luciferase (ffLuc) were propagated on CD19+ K562-derived artificial antigen presenting cells. The approach to generating our clinical-grade CAR+ T cells was adapted for electro-transfer of gold nanoparticles (GNPs) functionalized with 64Cu2+ using the macrocyclic chelator (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTA) and polyethyleneglycol (GNP–64Cu/PEG2000). MicroPET/CT was used to visualize CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells and correlated with bioluminescence imaging. These data demonstrate that GNPs conjugated with 64Cu2+ can be prepared as a radiotracer for PET and used to image T cells using an approach that has translational implications.
Insight, innovation, integrationClinical-grade primary T cells can be used to prevent and treat malignancies. Ex vivo manipulation of T cells improves in vivo effector functions. The link between what can be achieved in the manufacturing suites and what is achieved after infusion is compromised by an inability to assess T-cell spatio-temporal distribution. A clinically-appealing approach is needed to assess the biodistribution of T cells. Therefore, we developed gold nanoparticles (GNPs) for positron emission tomography (PET) by coupling to 64Cu. Since we have adapted electroporation to genetically modify T cells for human application, we electro-transferred GNPs into T cells. These T cells could report their distribution in vivo by PET and represent a step towards developing GNPs as radiolabels for cell-based therapies. |
Introduction
Adoptive cell therapy infusing primary T cells genetically modified to express a chimeric antigen receptor (CAR) specific for a tumor-associated antigen has been shown to be effective against hematologic malignancies and solid tumors.1 The genetic manipulation of primary T cells can improve potency through the engineering of CAR2 to impart a fully-competent activation signal as measured in part by the persistence and homing after infusion. To assess biodistribution of systemically-administered CAR+ T cells, investigators typically undertake quantitative PCR and flow cytometry using CAR-specific probes from serially sampled tissues and peripheral blood.3 However, this is invasive and does not provide real-time whole-body spatio-temporal distribution of infused T cells. Longitudinal non-invasive imaging can be undertaken on T cells genetically modified to enforce expression of reporter genes, such as Firefly luciferase (ffLuc) for bioluminescent imaging (BLI)4 in animal models and thymidine kinase (TK) and associated muteins from herpes simplex virus-1 for positron emission tomography (PET).5 Locally administered human TK+ T cells and intravenously-infused macaque T cells have been imaged by PET.6 Additional studies are needed regarding improving sensitivity and reducing immunogenicity before systemically administered TK+ T cells can be reproducibly imaged by PET in clinical trials. Compounding the difficulties associated with human application of this approach to PET is that 18F-based probes require enzymatic trapping of the radiotracer in the cytoplasm by recombinant TK and the infused non-metabolized 18F creates a background signal from pools within tissues and undermines sensitivity.3,6b,7 The short radioactive half-life (t1/2 = 109.8 min) of 18F also imposes practical limitations and requires an on-site cyclotron or expedited delivery of up to 500 mCi 18F-based probes as starting materials for single infusion of 10 mCi per patient. 64Cu conjugated to lipophilic chelator pyruvaldehyde-bis(N4-methylthiosemicarbazone) for PET tracking has been proposed as an alternative and used to track C6 rat glioma cells in mice for up to 20 hours, although leakage of 64Cu from cells was observed.8 Gold nanoparticles, however, have been investigated for their uptake and have been found appropriate for intracellular retention.9 Therefore, as an alternative to in vivo labeling we developed an ex vivo approach to radiolabel primary T cells with gold nanoparticles (GNPs) conjugated to 64Cu (GNP–64Cu/PEG2000) using electroporation that renders T cells capable of being imaged by PET.Engineering of immunotherapies is a burgeoning field with active contribution from physical technologies10 and can be used to address an important challenge and an unmet clinical need for the in vivo tracking of tumor-targeting T cells.11 Assessments of T-cell trafficking kinetics to tumor locations have been made.12 A recent study by Koya et al.12b using signal correlation from PET and BLI reporter genes showed that T cells home within 2 to 5 days leading to reduction in tumor sizes. We demonstrated that cultured CAR+ T cells pre-labeled with 64Cu before infusion could be tracked in vivo using μPET/CT. Although room for improvement still remain (e.g. towards the impact of an electroporation process on T-cell death and contribution of free GNP–64Cu/PEG2000 released from necrotic cells) our work has translational implications as we use an approach that can be undertaken in compliance with current good manufacturing practice for Phase I/II trials.
Results and discussion
We have developed a strategy to genetically modify primary peripheral blood mononuclear cells (PBMC) and propagate CD19-specific CAR+ T cells that have application in clinical trials for patients with B-cell malignancies (INDs# 14193, 14577, and 14739). Our approach uses the Sleeping Beauty (SB) transposon–transposase system,13 a non-viral gene delivery method based on electro-transfer of DNA plasmids, to introduce the CAR and designer artificial antigen presenting cells (aAPCs) to retrieve and numerically expand genetically modified primary T cells.14 We adapted the electroporation process to co-express a 2nd generation CAR (that signals through CD28 and CD3-ζ) and ffLuc. This was achieved using a process we dubbed “double transposition” (Fig. 1A).13,14 Thawed PBMC isolated from the peripheral blood of healthy volunteer donors were electroporated with three SB DNA supercoiled plasmids expressing three codon optimized15 (CoOp) genes: (i) CAR transposon (Fig. 1B), (ii) fusion of enhanced Green Fluorescent Protein (EGFP), ffLuc, hygromycin phosphotransferase (Hy), thymidine kinase (TK) to create the EGFPffLucHyTK transposon (Fig. 1C), and (iii) SB11 transposase (Fig. 1D). The genetically modified T cells (designated CAR+EGFPffLucHyTK+) were selectively propagated on γ-irradiated designer aAPC in the presence of recombinant human cytokines IL-2 and IL-21 in the presence of cytocidal concentration of hygromycin B. The aAPCs (clone #4)16 are derived from K562 and genetically modified to express truncated CD19 along with desired co-stimulatory molecules (CD64, CD86, CD137L, and membrane bound IL-15 (mIL-15) co-expressed with EGFP). The electroporated and propagated T cells homogeneously expressed CD3 with 12% co-expressing CD4 and 84% co-expressing CD8. Flow cytometry also revealed 68% expression of CAR and 78% expression of EGFP (Fig. S1, ESI†). To track the T cells in vivo we modified the electroporation procedure to load the genetically modified T cells with GNPs modified to function as a reporter for PET. 64Cu2+ was chosen as a radioisotope for PET based on longer half-life of 64Cu (t1/2 = 12.7 h) compared to 18F, and the absence of high-energy γ-emission that could otherwise lead to DNA damage in the cells.1764Cu2+ was conjugated to 7 nm GNPs using the macrocyclic chelator, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)18 and coated with long chain polyethylene glycol to make it biocompatible (GNP–64Cu/PEG2000) for the final size of 35 nm (Fig. 1E).19 Although, endocytosis has been investigated as a usual mode for internalization of GNP, it requires extended periods of time,20 and is a practical limitation with radioactive material which has short half life. Therefore, we developed electro-transfer as a method to label T cells with GNP–64Cu/PEG2000 due to its ability to instantly label the cells.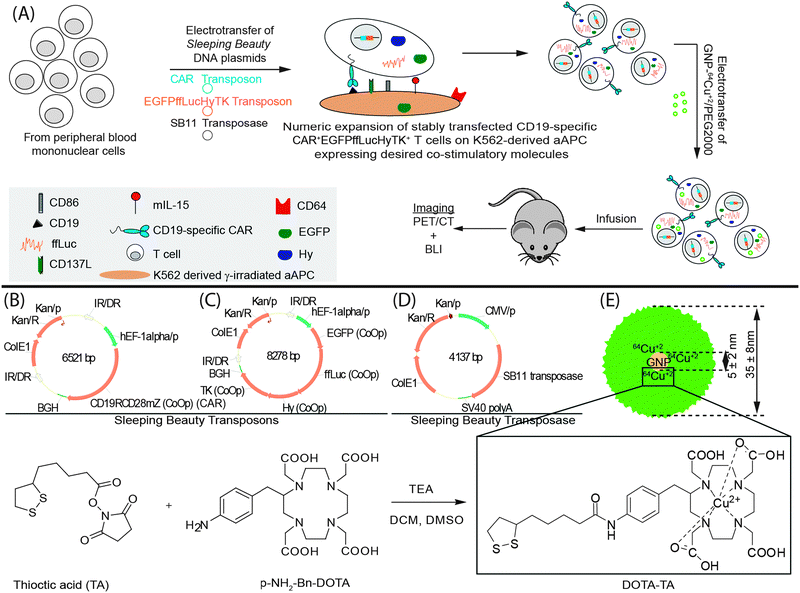 | ||
| Fig. 1 (A) Schematic of the processes for the generation of CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells. Components used include (B) CAR transposon (CD19RCD28mZ(CoOp)/pSBSO). (C) SB11 transposon (pKan-CMV-SB11) (D) ffLuc transposon (EGFPffLucHyTK) (E) GNP–64Cu/PEG2000; synthesis of DOTA-TA). 7 nm GNP (shown) were compared with 15 nm GNP. All other parameters stayed the same during this comparison. TEA, triethylamine; DCM, dichloromethane. | ||
In vitro testing of electroporated and propagated CAR+EGFPffLucHyTK+ primary T cells revealed ffLuc activity at 2.17 ± 0.08 CPM per cell upon administration of D-luciferin, compared with 0.011 ± 0.001 CPM per cell when no D-luciferin was administered (data not shown). The ffLuc activity is a measure of T-cell viability as this enzyme requires ATP as a co-factor which is present only in live cells. Electroporation parameters were adjusted to improve uptake of GNPs. Initially, acute T-cell leukemia cell lines (Jurkat cells) were used to assess the efficiency of electro-transfer by EGFP expression using control DNA plasmid (designated as pmaxGFP). The highest transfection efficiency, as measured by transient GFP expression 24 hours after electroporation was observed at the field strength of 1 kV cm−1 (200 V across 2 mm electrode spacing) applied for 5 ms using a square-wave pulse generator (Fig. 2A) (this finding was supported by repeating the experiment (Fig. S2, ESI†)). Although, conditions for electroporation may differ for cell types, 1 kV cm−1 has been previously reported in the literature as an appropriate value for transfection in mammalian cells and was therefore adopted for future experiments.21 GNP/PEG2000 (synthesized in a similar fashion as GNP–64Cu/PEG2000 but without 64Cu2+ addition) was used to determine the concentration of GNP–64Cu/PEG2000 to be used in the electroporation reaction. Using the conditions determined for electro-transfer of DNA plasmid into T-cell lines (Jurkat cells), we first investigated the effect of GNP/PEG2000 concentration and its electro-transfer into 5 × 106 GFP+ffLuc+ T-cell lines (Jurkat cells) (Fig. 2B). Since primary cell response differ from donor to donor, we repeated this investigation on 20 × 106 CAR+EGFPffLucHyTK+ T cells using the cells from the donor that was used in vivo (Fig. 2C). A GNP/PEG2000 concentration of 1.5 × 1012 particles in 100 μL of electroporation reaction was considered as appropriate for electro-transfer into 20 × 106 CAR+EGFPffLucHyTK+ primary T cells in context of cell recovery, ffLuc expression and cell viability determined by a trypan blue exclusion method. To assess the impact of electro-transferred GNPs on BLI, we electroporated CAR+EGFPffLucHyTK+ T cells with 1.5 × 1012 GNP/PEG2000 using 1 kV cm−1 for 5 ms and intravenously injected 20 × 106 T cells in a mouse. There was no significant difference in average radiance from the mouse infused with T cells that did (1.33 × 104 p s−1 cm−2 per sr) and did not (1.12× 104 p s−1 cm−2 per sr) carry the GNPs (Fig. 2D) which supports the premise that although the process of electroporation may have the potential to affect cell viability, the presence of GNP/PEG2000 in the cells did not detract from their ability to affect ffLuc activity.
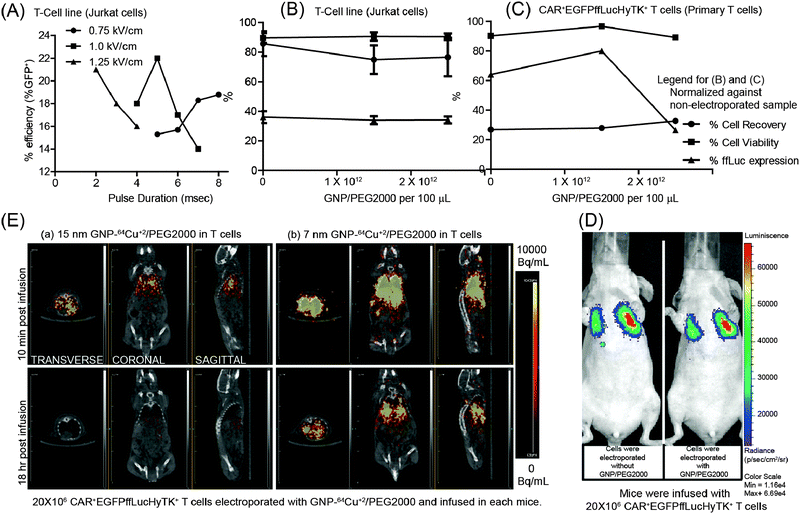 | ||
| Fig. 2 (A) Effect of electric field intensity and pulse duration on efficiency of electro-transfer of DNA plasmid (pmaxGFP) into T-cell lines (Jurkat cells). (B and C) Cell recovery, cell viability and ffLuc expression after electroporation of (B) GFP+ffLuc+ T-cell lines (Jurkat cells) and (C) CAR+EGFPffLucHyTK+ T cells with GNP/PEG2000 (normalized against a non-electroporated sample) as a function of GNP/PEG2000 concentration. (D) Effect of intracellular GNP/PEG2000 on an in vivo BLI signal from ffLuc. (E) Transverse, coronal, and sagittal planes of mice infused with T cells labeled with 15 nm and 7 nm GNP–64Cu/PEG2000, respectively, imaged after 10 min and 18 hours by μPET/CT. | ||
To assess the impact of size of GNPs to cross pores in the T-cell membrane introduced by electroporation, we electro-transferred 15 nm and 7 nm GNPs conjugated with 64Cu2+ and PEG2000 into 20 × 106 CAR+EGFPffLucHyTK+ primary T cells which resulted in 1 μCi and 5.8 μCi of radioactivity for the two sets of cells. T cells were injected into two mice and μPET imaging of transverse, coronal, and sagittal planes was undertaken beginning at 10 minutes post infusion (Fig. 2E). Although an initial PET signal was observed from both mice, a longer term in vivo PET signal was observed only from mice that received T cells bearing 7 nm GNP–64Cu/PEG2000. We hypothesize that 15 nm GNP–64Cu/PEG2000 were less efficiently electro-transferred into T cells due to the increased size of the GNP.
The 7 nm GNP–64Cu/PEG2000 were then used to label genetically modified CAR+EGFPffLucHyTK+ T cells using the established electroporation conditions (20 × 106 T cells, 1.5 × 1012 GNP–64Cu/PEG2000, 100 μL, 1 kV cm−1, 5 ms) to deliver 3 μCi. 20 × 106 radiolabeled primary T cells were intravenously injected and imaged using μPET/CT and BLI. The PET signals from transverse, coronal, and sagittal planes of the mouse co-localized with the BLI signal (Fig. 3A). As the BLI signal indicates the presence of live cells, the co-localized PET/CT and BLI data support the hypothesis that viable (metabolically active) genetically modified T cells can be tracked in vivo using a positron emitter, 64Cu, and imaged by a μPET/CT scanner.
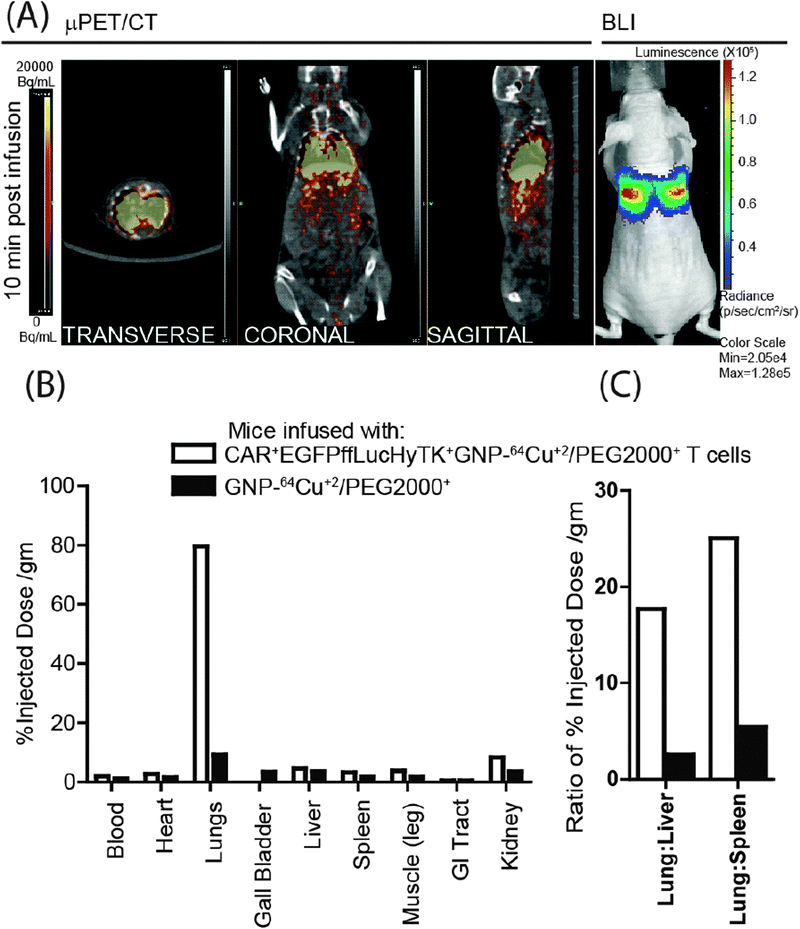 | ||
| Fig. 3 (A) PET images (transverse, coronal and sagittal planes) from CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells infused in a mouse correlated with a BLI signal. (B) Comparison of post-mortem biodistribution of the GNP–64Cu/PEG2000 when enveloped inside the T cells compared to when infused directly, 14 hours after intravenous infusion. (C) Lung-to-liver and lung-to-spleen ratios of GNP–64Cu/PEG2000 when enveloped inside the T cells compared to direct injection, 14 hours after intravenous injection. | ||
GNPs loaded within primary T cells are expected to bypass clearance by the liver and spleen which are part of the reticuloendothelial system (RES). This was assessed by comparing the biodistribution of 20 × 106 T cells radiolabeled with GNP–64Cu/PEG2000 compared with free GNP–64Cu/PEG2000 (90 μCi) also administered via tail vein. The post-mortem biodistribution of the radioactivity, 14 hours after infusion, revealed that the T cells preferentially carried the GNP–64Cu/PEG2000 to the lungs. Whereas, when free GNP–64Cu/PEG2000 were administered, comparatively larger fraction was found outside the pulmonary system (Fig. 3B) and into major RES organs (liver and spleen).22 Kennedy et al.23 also reported four-fold more efficient delivery of GNP to the tumor site when transported within T cells to the tumor locations, compared to accumulation via enhanced permeability and retention effect. The loading of GNP–64Cu/PEG2000 in T cells resulted in less uptake by RES and desired delivery to the lungs. Cell-free GNP–64Cu/PEG2000 were taken up 17.5-fold more in liver and 25-fold more in spleen compared to when packaged in T cells (Fig. 3C). This supports the premise that T cells can act as delivery vehicles for nanostructures without being sequestered by the RES.23,24
In this report, as another example of contribution of engineering approaches to immunotherapy,10 we show that 64Cu conjugated to GNP and coated with PEG could be used to image primary T cells in vivo. A barrier to successful implementation of cell-based therapies is determination of optimal dosage and schedule for administering the biologic and ascertaining that these cells reach their desired target locations.25 As a result, non-invasive real-time monitoring of infused cells has been an area of active research as it can inform on the dosage needed to achieve therapeutically effective numbers of infused cells within desired tissues. Various methods under consideration for tracking include ex vivo and in vivo labeling of cells for imaging and each has pros and cons.11 An advantage of our approach using GNPs is that the conjugation to 64Cu as well as the electro-transfer into T cells can be undertaken using methods in compliance with current good manufacturing practice for Phase I/II trials.
Materials and methods
DNA plasmids
Three DNA plasmids expressing kanamycin resistance gene were constructed coding for three codon optimized (CoOp) transgenes flanked by IR/DR sequences and used to generate CAR+EGFPffLucHyTK+ T cells. (1) CD19-specific CAR expressed under the human elongation factor 1-α (hEF-1α) promoter (CD19RCD28mZ(CoOp)/pSBSO) (Fig. 1B),26 (2) multi-function protein fusing ffLuc downstream of EGFP and upstream of hygromycin phosphotransferase (Hy) and thymidine kinase (TK, HyTK) expressed under the hEF-1α promoter (EGFPffLucHyTK) (Fig. 1C), (3) SB11 transposase expressed under the cytomegalovirus (CMV) promoter (pKan-CMV-SB11) (Fig. 1D).26 Supercoiled DNA plasmid pmaxGFP, coding for EGFP, was used to assess the efficiency of electro-transfer by measuring fluorescence.GNP–64Cu/PEG2000
GNPs with an average diameter of 7 nm were purchased from Nanopartz, Inc, CO. 64CuCl2 was produced on a CS-15 biomedical cyclotron at Washington University School of Medicine. DOTA-TA was synthesized by linking p-NH2–Bn–1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (Macrocyclics Inc., TX, USA) and thioctic acid (TA) (Acros, NJ, USA) with a di-amine linker.27 GNPs were functionalized by forming gold–thiol bond with DOTA-TA (GNP–DOTA).27 GNP–DOTA were incubated with 64Cu2+ at pH 6.5 at 50 °C followed by PEGylation using thiolated PEG (Item# MPEG-SH-2000; Laysan Bio, Inc, AL, USA) (GNP–64Cu/PEG2000)28 (Fig. 1E). Radiolabeled GNP–64Cu/PEG2000 were measured 35 nm in diameter by dynamic light scattering and the labeling efficiency was >95% determined by radio thin layer chromatography.Artificial antigen presenting cells (aAPCs)
K562-derived aAPCs (referred to as clone #4) were transduced with lentivirus to co-express CD19, CD64, CD86, CD137L (CD137L), and membrane-bound (m) interleukin (IL)-15 (mIL-15, co-expressed with EGFP, Fig. 1A) and used to support the ex vivo numeric expansion of CD19-specific CAR+EGFPffLucHyTK+ T cells.16Peripheral Blood Mononuclear Cells (PBMC)
PBMC were isolated from blood by Ficoll-Paque density gradient centrifugation (Cat # 17-1440-02, GE Healthcare Bio-sciences AB, Sweden), re-suspended in freezing medium (40% RPMI-1640, 50% fetal bovine serum (FBS), 10% DMSO) at the density of 20 × 106 cells per mL per vial, and stored in liquid nitrogen.14aGeneration of EGFP+ acute T-cell leukemia cell lines (Jurkat cells)
On the day of electroporation (Day 0), 106 Jurkat cells were re-suspended in 100 μL of phosphate buffered saline (PBS) (Cat# D8537, Sigma-Aldrich, Inc., MO, USA) along with pmaxGFP (2 μg supercoiled DNA, Lonza Cologne GmbH, Germany), transferred to a single cuvette, and electroporated with specific pulse duration and amplitude using an ECM 830 BTX electroporation apparatus (BTX Instrument Division, Harvard Apparatus, Inc., MA, USA). The cells were cultured for 24 hours at 37 °C in phenol-free RPMI-1640 containing 10% FBS and 2 mM L-alanyl-L-glutamine (GlutaMAX™, Cat # 35050, Invitrogen Corp., CA, USA) before analyzing for EGFP expression.Generation of GFP+ffLuc+ acute T-cell leukemia cell lines (Jurkat cells)
Jurkat cells were cultured in RPMI-1640 containing 10% FBS and 2 mM L-alanyl-L-glutamine (GlutaMAX™, Cat # 35050, Invitrogen Corp., CA, USA) and transduced with lentiviral particles carrying ffLuc-F2A-GFP reporter gene expressed under blasticidin resistance gene (Cat# LVP323, GenTarget Inc., San Diego, CA, USA) using manufacturer's protocol. Briefly, lentiviral particles were thawed at room temperature and 50 μL of virus was added to 0.5 mL cells (diluted to 106 cells per mL). Blasticidin S (Cat # ant-bl-5, InvivoGen, CA, USA) was added to the culture beginning 4 days after transduction and maintained at a concentration of 0.1 mg mL−1 until cryopreservation or use.Generation and analysis of CAR+EGFPffLucHyTK+ T cells
CD19-specific CAR+EGFPffLucHyTK+ T cells were propagated from PBMC after SB transposition as previously described14a and depicted in Fig. 1A. On the day of electroporation (Day 0), 20 × 106 PBMC were thawed at 37 °C in a water bath and re-suspended in 100 μL of Amaxa Nucleofector solution (Human T cell Kit, Cat # VPA-1002) along with CAR transposon (CD19RCD28mZ(CoOp)/pSBSO, 7.5 μg supercoiled DNA, Fig. 1B), ffLuc transposon (EGFPffLucHyTK, 7.5 μg supercoiled DNA, Fig. 1C), and SB transposase (pKan-CMV-SB11, 5 μg supercoiled DNA, Fig. 1D) and transferred to a single cuvette and electroporated (Program U-14) using a Nucleofector II (Lonza Cologne GmbH, Germany). The cells were rested for 2 to 3 hours at 37 °C in incomplete phenol-free RPMI-1640 and subsequently cultured overnight in phenol-free RPMI-1640 containing 10% FBS and 2 mM L-alanyl-L-glutamine (GlutaMAX™, Cat # 35050, Invitrogen Corp., CA, USA). The following day γ-irradiated (100 Gy) K562-aAPCs (clone #4) were added at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 T cell/aAPC ratio. Hygromycin B (HygroGold™, Cat # ant-hg-1, InvivoGen, CA, USA) was added to the co-culture beginning 5 days after electroporation and maintained at a concentration of 0.2 mg mL−1, as suggested by the manufacturer, until cryopreservation of T cells. Additional γ-irradiated aAPCs were added at 1:2 T cell/aAPC on days 7, 14, 21, 28, and 35 after electroporation. Soluble recombinant human IL-21 (Cat # 34-8219-85, eBioscience, Inc., CA, USA) was added at a concentration of 30 ng mL−1 beginning the day after electroporation, and soluble recombinant human IL-2 (Chiron Corp (Novartis V&D), CA, USA) was added to the cultures at 50 U mL−1 beginning 7 days after electroporation. These exogenous cytokines (IL-2, IL-21) continued to be supplemented on a Monday–Wednesday–Friday schedule. The cultures were monitored by flow cytometry for the unwanted presence of a CD3negCD56+ cell population and if the percentage exceeded 10% of the total population (which usually occurred between days 10 and 14) a depletion for CD3negCD56+ NK cells was accomplished using CD56 beads (Cat # 130-050-401, Miltenyi Biotech Inc., CA, USA) on a LS column (Cat # 130-042-401, Miltenyi Biotech Inc., CA, USA) according to the manufacturer's instructions. T cells were enumerated every 7 days and viable cells counted based on trypan blue exclusion using a Cellometer automated cell counter (Auto T4 Cell Counter, Nexcelom Bioscience, MA, USA). 35 days after electro-transfer of SB plasmids the CAR+EGFPffLucHyTK+ cells were re-suspended in freezing medium at 20 × 106 cells per mL per vial and stored in liquid nitrogen.
:2 T cell/aAPC ratio. Hygromycin B (HygroGold™, Cat # ant-hg-1, InvivoGen, CA, USA) was added to the co-culture beginning 5 days after electroporation and maintained at a concentration of 0.2 mg mL−1, as suggested by the manufacturer, until cryopreservation of T cells. Additional γ-irradiated aAPCs were added at 1:2 T cell/aAPC on days 7, 14, 21, 28, and 35 after electroporation. Soluble recombinant human IL-21 (Cat # 34-8219-85, eBioscience, Inc., CA, USA) was added at a concentration of 30 ng mL−1 beginning the day after electroporation, and soluble recombinant human IL-2 (Chiron Corp (Novartis V&D), CA, USA) was added to the cultures at 50 U mL−1 beginning 7 days after electroporation. These exogenous cytokines (IL-2, IL-21) continued to be supplemented on a Monday–Wednesday–Friday schedule. The cultures were monitored by flow cytometry for the unwanted presence of a CD3negCD56+ cell population and if the percentage exceeded 10% of the total population (which usually occurred between days 10 and 14) a depletion for CD3negCD56+ NK cells was accomplished using CD56 beads (Cat # 130-050-401, Miltenyi Biotech Inc., CA, USA) on a LS column (Cat # 130-042-401, Miltenyi Biotech Inc., CA, USA) according to the manufacturer's instructions. T cells were enumerated every 7 days and viable cells counted based on trypan blue exclusion using a Cellometer automated cell counter (Auto T4 Cell Counter, Nexcelom Bioscience, MA, USA). 35 days after electro-transfer of SB plasmids the CAR+EGFPffLucHyTK+ cells were re-suspended in freezing medium at 20 × 106 cells per mL per vial and stored in liquid nitrogen.
Generation of CAR+EGFPffLucHyTK+ T cells loaded with GNP–64Cu/PEG2000
20 × 106 CD19-specific CAR+EGFPffLucHyTK+ T cells were thawed and stimulated with γ-irradiated aAPCs (clone #4) in the presence of IL-2/21 and hygromycin B as described above.14a 37 days later the cells were centrifuged and 20 × 106 T cells were re-suspended in 100 μL in the presence of 1 × 1012 GNP–64Cu/PEG2000 (Fig. 1E) in PBS (Cat# D8537, Sigma-Aldrich, Inc., MO, USA) and transferred to a single cuvette and electroporated at 200 V with a 5 ms pulse using an ECM 830 BTX electroporation apparatus (BTX Instrument Division, Harvard Apparatus, Inc., MA, USA).In vivo measurement of the PET signal from CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells
Micro-PET/CT imaging was acquired by the Inveon Preclinical Multimodel SPECT/PET/CT System (Siemens AG, USA). Immunocompromised (nu/nu) mice were ordered from Charles River Laboratories International Inc., MA. CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells were intravenously injected (20 × 106 cells per mouse). All mice imaging was performed at The Methodist Hospital in accordance with guidelines from Animal Care and Use Committee. Mice were anesthetized, placed on an imaging bed attached with heating pad, laser-aligned to the center of the scanner field-of-view and serially imaged by μPET/CT scan. Images were acquired at 10 min after injection of T cells. CT image reconstruction was achieved using a Common Cone-Beam Reconstruction (COBRA) method (Siemens) and PET images were reconstructed by a 2D Ordered Subset Expectation Maximization (OSEM2D) algorithm. The PET and CT images were co-registered and viewed using Inveon Research Workplace software (Siemens).In vivo measurement of the BLI signal from CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells
After PET imaging, mice were injected with 150 mg kg−1D-luciferin (XenoLight™, Cat # 122796, Caliper Life Sciences, MA, USA) and BLI was obtained using an IVIS-200 optical imaging system (Caliper Life Sciences, MA, USA). BLI was acquired using 1 minute exposure time and displayed in the same scale of intensity.Biodistribution studies
Two mice were injected intravenously with CAR+EGFPffLucHyTK+GNP–64Cu/PEG2000+ T cells and GNP–64Cu/PEG2000. Mice were euthanized at 14 hour post injection. Blood, heart, liver, spleen, kidney, lung, stomach, intestine and muscle were harvested and weighed. Radioactivity of different organs was measured with the gamma counter (Perkin Elmer, MA). Uptake of radiolabeled T cells in tissues was calculated as the percentage of the injected dose per gram of tissue (%ID per g).Disclosure of potential conflicts of interest
None.Grant support
This work was supported by funding from: Cancer Center Core Grant (CA16672); RO1 (CA124782, CA120956, CA141303); R33 (CA116127); P01 (CA148600); Albert J. Ward Foundation; Alliance for NanoHealth (W81XWH-09-02-0139, W81XWH-10-02-0125); Cancer Prevention and Research Institute of Texas; Center for Transport Oncophysics – Physical Science Oncology Center at The Methodist Hospital Research Institute (U54CA 143837); Department of Defense; Burroughs Wellcome Fund; CLL Global Research Foundation; Gillson Longenbaugh Foundation; Harry T. Mangurian, Jr., Fund for Leukemia Immunotherapy; Institute of Personalized Cancer Therapy; Leukemia and Lymphoma Society; Lymphoma Research Foundation; Miller Foundation; Mr and Mrs Joe H. Scales; Mr Thomas Scott; National Foundation for Cancer Research; Pediatric Cancer Research Foundation; Production Assistance for Cellular Therapies; Sister Institution Network Fund; William Lawrence and Blanche Hughes Children's Foundation.Acknowledgements
We thank Dr Carl June at the University of Pennsylvania for help generating and providing the aAPC (clone #4), Dr Perry Hackett at the University of Minnesota for help with the SB system, and the flow cytometry core at MDACC.References
- (a) N. P. Restifo, M. E. Dudley and S. A. Rosenberg, Adoptive immunotherapy for cancer: harnessing the T cell response, Nat. Rev. Immunol., 2012, 12, 269–281 CrossRef CAS; (b) S. A. Rosenberg, Cell transfer immunotherapy for metastatic solid cancer-what clinicians need to know, Nat. Rev. Clin. Oncol., 2011, 8, 577–585 CrossRef CAS; (c) S. A. Grupp and C. H. June, in Cancer Immunology and Immunotherapy, ed. G. Dranoff, 2011, vol. 344, pp. 149–172 Search PubMed; (d) C. Bonini, M. K. Brenner, H. E. Heslop and R. A. Morgan, Genetic Modification of T Cells, Biol. Blood Marrow Transplant., 2011, 17, S15–S20 CrossRef CAS; (e) B. Jena, G. Dotti and L. J. N. Cooper, Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor, Blood, 2010, 116, 1035–1044 CrossRef CAS; (f) L. Gattinoni, D. J. Powell, S. A. Rosenberg and N. P. Restifo, Adoptive immunotherapy for cancer: building on success, Nat. Rev. Immunol., 2006, 6, 383–393 CrossRef CAS.
- D. B. Kohn, G. Dotti, R. Brentjens, B. Savoldo, M. Jensen, L. J. N. Cooper, C. H. June, S. Rosenberg, M. Sadelain and H. E. Heslop, CARs on track in the clinic, Mol. Ther., 2011, 19, 432–438 CrossRef CAS.
- E. D. Nair-Gill, C. J. Shu, C. G. Radu and O. N. Witte, Non-invasive imaging of adaptive immunity using positron emission tomography, Immunol. Rev., 2008, 221, 214–228 CrossRef CAS.
- B. A. Rabinovich, Y. Ye, T. Etto, J. Q. Chen, H. I. Levitsky, W. W. Overwijk, L. J. N. Cooper, J. Gelovani and P. Hwu, Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14342–14346 CrossRef CAS.
- H. Singh, A. M. Najjar, S. Olivares, R. Nishii, U. Mukhopadhyay, M. Alauddin, P. R. Manuri, H. Huls, D. A. Lee, G. Dotti, C. Bollard, P. J. Simmons, E. J. Shpall, R. E. Champlin, J. G. Gelovani and L. J. N. Cooper, PET imaging of T cells derived from umbilical cord blood, Leukemia, 2009, 23, 620–622 CrossRef CAS.
- (a) G. Dotti, M. Tian, B. Savoldo, A. Najjar, L. J. N. Cooper, J. Jackson, A. Smith, O. Mawlawi, R. Uthamanthil, A. Borne, D. Brammer, V. Paolillo, M. Alauddin, C. Gonzalez, D. Steiner, W. K. Decker, F. Marini, S. Kornblau, C. M. Bollard, E. J. Shpall and J. G. Gelovani, Repetitive Noninvasive Monitoring of HSV1-tk-Expressing T Cells Intravenously Infused into Nonhuman Primates Using Positron Emission Tomography and Computed Tomography with F-18-FEAU, Mol. Imaging, 2009, 8, 230–237 Search PubMed; (b) S. S. Yaghoubi, M. C. Jensen, N. Satyamurthy, S. Budhiraja, D. Paik, J. Czernin and S. S. Gambhir, Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma, Nat. Clin. Pract. Oncol., 2009, 6, 53–58 CrossRef CAS.
- (a) K. Dobrenkov, M. Olszewska, Y. Likar, L. Shenker, G. Gunset, S. Cai, N. Pillarsetty, H. Hricak, M. Sadelain and V. Ponomarev, Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging, J. Nucl. Med., 2008, 49, 1162–1170 CrossRef; (b) S. A. Collins, K. Hiraoka, A. Inagaki, N. Kasahara and M. Tangney, PET imaging for gene & cell therapy, Curr. Gene Ther., 2012, 12, 20–32 CrossRef CAS.
- N. Adonai, K. N. Nguyen, J. Walsh, M. Iyer, T. Toyokuni, M. E. Phelps, T. McCarthy, D. W. McCarthy and S. S. Gambhir, Ex vivo cell labeling with Cu-64-pyruvaldehyde-bis(N-4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3030–3035 CrossRef CAS.
- (a) E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, Cellular Uptake and Fate of PEGylated Gold Nanoparticles Is Dependent on Both Cell-Penetration Peptides and Particle Size, ACS Nano, 2011, 5, 6434–6448 CrossRef CAS; (b) D. B. Chithrani, Intracellular uptake, transport, and processing of gold nanostructures, Mol. Membr. Biol., 2010, 27, 299–311 CrossRef CAS; (c) P. Nativo, I. A. Prior and M. Brust, Uptake and intracellular fate of surface-modified gold nanoparticles, ACS Nano, 2008, 2, 1639–1644 CrossRef CAS.
- M. A. Swartz, S. Hirosue and J. A. Hubbell, Engineering approaches to immunotherapy, Sci. Transl. Med., 2012, 4, 148 CrossRef.
- M. F. Kircher, S. S. Gambhir and J. Grimm, Noninvasive cell-tracking methods, Nat. Rev. Clin. Oncol., 2011, 8, 677–688 CrossRef CAS.
- (a) A. C. Parente-Pereira, J. Burnet, D. Ellison, J. Foster, D. M. Davies, S. van der Stegen, S. Burbridge, L. Chiapero-Stanke, S. Wilkie, S. Mather and J. Maher, Trafficking of CAR-Engineered Human T Cells Following Regional or Systemic Adoptive Transfer in SCID Beige Mice, J. Clin. Immunol., 2011, 31, 710–718 CrossRef CAS; (b) R. C. Koya, S. Mok, B. Comin-Anduix, T. Chodon, C. G. Radu, M. I. Nishimura, O. N. Witte and A. Ribas, Kinetic phases of distribution and tumor targeting by T cell receptor engineered lymphocytes inducing robust antitumor responses, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14286–14291 CrossRef CAS; (c) K. Matsui, Z. Wang, T. J. McCarthy, P. M. Allen and D. E. Reichert, Quantitation and visualization of tumor-specific T cells in the secondary lymphoid organs during and after tumor elimination by PET, Nucl. Med. Biol., 2004, 31, 1021–1031 CrossRef CAS.
- P. B. Hackett, D. A. Largaespada and L. J. N. Cooper, A Transposon and Transposase System for Human Application, Mol. Ther., 2010, 18, 674–683 CrossRef CAS.
- (a) H. Singh, M. J. Figliola, M. J. Dawson, H. Huls, S. Olivares, K. Switzer, T. J. Mi, S. Maiti, P. Kebriaei, D. A. Lee, R. E. Champlin and L. J. N. Cooper, Reprogramming CD19-Specific T Cells with IL-21 Signaling Can Improve Adoptive Immunotherapy of B-Lineage Malignancies, Cancer Res., 2011, 71, 3516–3527 CrossRef CAS; (b) H. Singh, P. R. Manuri, S. Olivares, N. Dara, M. J. Dawson, H. Huls, P. B. Hackett, D. B. Kohn, E. J. Shpall, R. E. Champlin and L. J. N. Cooper, Redirecting specificity of T-cell populations for CD19 using the sleeping beauty system, Cancer Res., 2008, 68, 2961–2971 CrossRef CAS.
- (a) A. Cid-Arregui, V. Juarez and H. zur Hausen, A synthetic E7 gene of human papillomavirus type 16 that yields enhanced expression of the protein in mammalian cells and is useful for DNA immunization studies, J. Virol., 2003, 77, 4928–4937 CrossRef CAS; (b) S. S. Patterson, H. M. Dionisi, R. K. Gupta and G. S. Sayler, Codon optimization of bacterial luciferase (lux) for expression in mammalian cells, J. Ind. Microbiol. Biotechnol., 2005, 32, 115–123 CrossRef CAS.
- P. V. R. Manuri, M. H. Wilson, S. N. Maiti, T. J. Mi, H. Singh, S. Olivares, M. J. Dawson, H. Huls, D. A. Lee, P. H. Rao, J. M. Kaminski, Y. Nakazawa, S. Gottschalk, P. Kebriaei, E. J. Shpall, R. E. Champlin and L. J. N. Cooper, piggyBac Transposon/Transposase System to Generate CD19-Specific T Cells for the Treatment of B-Lineage Malignancies, Hum. Gene Ther., 2010, 21, 427–437 CrossRef CAS.
- T. K. Nayak and M. W. Brechbiel, Radioimmunoimaging with Longer-Lived Positron-Emitting Radionuclides: Potentials and Challenges, Bioconjugate Chem., 2009, 20, 825–841 CrossRef CAS.
- M. Shokeen and C. J. Anderson, Molecular imaging of cancer with copper-64 radiopharmaceuticals and positron emission tomography (PET), Acc. Chem. Res., 2009, 42, 832–841 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Renal clearance of quantum dots, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells, Nano Lett., 2006, 6, 662–668 CrossRef CAS.
- (a) C. Chen, S. W. Smye, M. P. Robinson and J. A. Evans, Membrane electroporation theories: a review, Med. Biol. Eng. Comput., 2006, 44, 5–14 CrossRef CAS; (b) C. Favard, D. S. Dean and M. P. Rols, Electrotransfer as a non-viral method of gene delivery, Curr. Gene Ther., 2007, 7, 67–77 CrossRef CAS.
- (a) W. D. James, L. R. Hirsch, J. L. West, P. D. O'Neal and J. D. Payne, Application of INAA to the build-up and clearance of gold nanoshells in clinical studies in mice, J. Radioanal. Nucl. Chem., 2007, 271, 455–459 CrossRef CAS; (b) T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, PEG-modified gold nanorods with a stealth character for in vivo applications, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS.
- L. C. Kennedy, A. S. Bear, J. K. Young, N. A. Lewinski, J. Kim, A. E. Foster and R. A. Drezek, T cells enhance gold nanoparticle delivery to tumors in vivo, Nanoscale Res. Lett., 2011, 6, 283 CrossRef CAS.
- (a) M. R. Choi, K. J. Stanton-Maxey, J. K. Stanley, C. S. Levin, R. Bardhan, D. Akin, S. Badve, J. Sturgis, J. P. Robinson, R. Bashir, N. J. Halas and S. E. Clare, A cellular Trojan horse for delivery of therapeutic nanoparticles into tumors, Nano Lett., 2007, 7, 3759–3765 CrossRef CAS; (b) K. Harrington, L. Alvarez-Vallina, M. Crittenden, M. Gough, H. Chong, R. M. Diaz, G. Vassaux, N. Lemoine and R. Vile, Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery?, Hum. Gene Ther., 2002, 13, 1263–1280 CrossRef CAS.
- W. J. Lesterhuis, J. Haanen and C. J. A. Punt, Cancer immunotherapy - revisited, Nat. Rev. Drug Discovery, 2011, 10, 591–600 CrossRef CAS.
- J. K. Davies, H. Singh, H. Huls, D. Yuk, D. A. Lee, P. Kebriaei, R. E. Champlin, L. M. Nadler, E. C. Guinan and L. J. N. Cooper, Combining CD19 Redirection and Alloanergization to Generate Tumor-Specific Human T Cells for Allogeneic Cell Therapy of B-Cell Malignancies, Cancer Res., 2010, 70, 3915–3924 CrossRef CAS.
- G. D. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS.
- Z. B. Li, W. B. Cai, Q. Z. Cao, K. Chen, Z. H. Wu, L. N. He and X. Y. Chen, 64Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor alpha(v)beta(3) integrin expression, J. Nucl. Med., 2007, 48, 1162–1171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ib20093g |
| This journal is © The Royal Society of Chemistry 2013 |