Rational design of an immunoconjugate for selective knock-down of leukemia-specific E2A–PBX1 fusion gene expression in human Pre-B leukemia
Fatih M.
Uckun
*abc,
Sanjive
Qazi
d,
Ilker
Dibirdik
ac and
Dorothea E.
Myers
ac
aDevelopmental Therapeutics Program, Children's Hospital Los Angeles, Children's Center for Cancer and Blood Diseases, Los Angeles, CA 90027, USA
bDepartment of Pediatrics and Norris Comprehensive Cancer Center, Keck School of Medicine and Keck Medical Center of University of Southern California, Los Angeles, CA 90027, USA
cBiotherapy Program, Parker Hughes Institute, St. Paul, MN 55455, USA
dDepartment of Biology and Bioinformatics Program, Gustavus Adolphus College, St. Peter, MN 56082, USA
First published on 18th September 2012
Abstract
The t(1;19)(q23;p13) is one of the most common chromosomal translocations in acute lymphoblastic leukemia (ALL) and results in production of the transforming oncoprotein E2A–PBX1. Here we first report a novel, biomarker-guided biotherapy strategy for personalized treatment of t(1;19)+ ALL. A supervised interrogation of the gene expression profiles of primary leukemic cells from a cohort of 207 children with high risk B-lineage ALL identified up-regulated CD19 gene expression as a biomarker for t(1;19)+ ALL. A disulfide-linked immunoconjugate of a 5-amino-modified 24 mer phosphorothioate anti-sense E2A–PBX1 oligonucleotide (AON) with a mAb specific for a CD19 receptor (αCD19–AON) was prepared as a CD19-directed and leukemia-specific biotherapeutic agent against E2A–PBX1+ B-lineage ALL. Treatment of E2A–PBX1+ leukemia cells with low nanomolar concentrations of αCD19–AON resulted in selective depletion of E2A–PBX1 transcripts and caused apoptotic destruction and abrogation of clonogenic growth. Subcutaneously administered αCD19–AON at a total dose level of 93 nmol kg−1 delivered over 14 days using a micro-osmotic pump more than doubled the leukemia-free survival time of SCID mice in a xenograft model of E2A–PBX1+ human B-lineage ALL (82.0 ± 1.9 days vs. 37.0 ± 0.1 days, P < 0.0001). Both the AON moiety and the targeting CD19-specific mAb moiety were required for the in vitro as well as in vivo anti-leukemic activity of αCD19–AON. The observed in vitro and in vivo anti-leukemic potency of the αCD19–AON immunoconjugate provides the first preclinical proof-of-principle that t(1;19)+ high risk B-lineage ALL can be treated with leukemia-specific biotherapeutic agents that knock-down E2A–PBX1 expression.
Insight, innovation, integrationThe t(1;19)(q23;p13) is one of the most common chromosomal translocations in acute lymphoblastic leukemia (ALL), the most common form of childhood and adolescent cancer, and results in production of the transforming oncoprotein E2A–PBX1. Here we first report a novel, biomarker-guided biotherapy strategy for personalized treatment of t(1;19) ALL. The observed in vitro and in vivo anti-leukemic potency of the anti(α)-CD19–AON immunoconjugate provides preclinical proof-of-principle that t(1;19)+ high risk B-lineage ALL can be treated with leukemia-specific biotherapeutic agents that knock down E2A–PBX1 expression. Our study also exemplifies how the discovery of a novel biomarker through bioinformatics research can be leveraged to rationally design innovative personalized medicines for cancer patients. |
Introduction
The transcription factor 3 gene (TCF3/E2A) located on the short arm of chromosome 19 at band 19p13.3 encodes two transcription factors, E12 and E47, both of which belong to the class I family of basic helix-loop-helix (bHLH) proteins.1 In B-cell ontogeny, E2A homodimeric complexes exert pivotal regulatory functions, including activation of B-cell specific gene expression and immunoglobulin gene rearrangement.2,3 Targeted disruption of the E2A locus in mice results in a maturational arrest of B-lineage lymphoid progenitor cells at the early pro-B cell stage prior to the onset of Ig heavy chain rearrangement.2,3 The chromosomal translocation t(1;19) (q23;p13.3), the second most common translocation in acute lymphoblastic leukemia (ALL) and the most common TCF3/E2A gene rearrangement,4–7 results in the expression of a nuclear oncoprotein, which is comprised of the N-terminal transactivation domains of E2A fused with the C-terminal DNA binding homeodomain of PBX1 (Pre-B cell leukemic homeobox1) encoded by PBX1 at 1q23.8–10 This chimeric E2A–PBX1 fusion protein acts as a transcriptional activator and is capable of cellular transformation.8–10 A number of genes specifically expressed in t(1;19)+ ALL cell lines were identified, including Wnt16 that encodes a potentially leukemogenic growth factor of the Wnt signaling pathway capable of stimulating proliferation of B-cell precursors.11 t(1;19)+ ALL is an immunophenotypically and prognostically distinct subset of B-lineage ALL that is characterized by expression of cytoplasmic immunoglobulin in leukemia cells (pre-B cell stage of differentiation), specific clinical and biological features and a signature transcriptome.12–16 Although the overall prognostic significance of t(1;19) has been obviated by contemporary risk-adjusted protocols, the balanced t(1;19) translocation remains an adverse prognostic factor.12,14 The purpose of the present study was to evaluate the clinical potential of a new biotherapeutic strategy against t(1;19)+ ALL that employs a rationally designed immunoconjugate for selective knockdown of the E2A–PBX1 fusion transcript expression in pre-B ALL cells.Materials and methods
Bioinformatics and statistical analysis of gene expression profilesGene pattern (http://www.broadinstitute.org/cancer/software/genepattern)17 was used to extract expression values for 11 genes; CD19, CD2, CD22, CD34, CD40, CD5, CD72, IGF1R, IL2RA, IL3RA, IL7R, obtained from matched pair bone marrow specimens of ALL patients at the time of initial diagnosis (1st specimen) and then at first relapse. Matched pair expression values were taken from 59 B-lineage ALL patients at diagnosis and then at relapse combined from GSE3912 (N = 32)18 and GSE18497 (N = 27).19 To determine the differential expression of each gene, paired t-tests were performed for the combined mean centered values from GSE3912 and GSE18497 datasets (unequal variance correction, P < 0.05 deemed significant). Comparison of early (N = 40; <36 months) versus late (N = 19; ≥36 months) relapse subsets for newly diagnosed patients was performed to identify potential biomarkers for early relapse (2-sample t-test, unequal variances). The expression of this gene set was also calculated in samples obtained from 23 E2A–PBX1 positive patients in a cohort of 207 patients with high risk B-lineage ALL (COG P9906, GSE11877, MAS5.0 normalized expression values,20) (t-test, unequal variances, P < 0.05 deemed significant). We used a two-way agglomerative hierarchical clustering technique to organize expression patterns using the average distance linkage method such that gene transcripts (rows) and samples (columns) having similar expression values were grouped together (calculated using the average distance metric). Dendrograms were drawn to illustrate similar gene-expression profiles from joining pairs of closely related gene expression profiles, whereby genes and samples joined by short branch lengths showed most similarity in expression profiles across patient samples and genes (JMP Software, SAS, Cary, NC). The heat map represents the color-coded expression value reported as a mean centered expression level relative to log10 transformed diagnostic samples.
Preparation of an αCD19–AON immunoconjugate
The two-step method that was used for immunoconjugation involved modification of the mAb and AON moieties prior to their incubation to prepare a disulfide-linked mAb–AON conjugate. 24 mer oligonucleotide (ON) phosphorothioates were obtained from the Midland Certified Reagent Company (Midland, Texas). Each ON contained a 6-carbon amino modification at its 5-terminus. The antisense ON (AON) had a sequence complementary to the sequence of the E2A–PBX1 fusion transcript: 5′-GAT ACT CAA AAC ACT GTA GGA GTC-3′. The scrambled ON (SON) consisted of the same number of all 4 bases that were arranged randomly: 5′-TGA GCA GAT AGT ACG TAA CCC AAT-3′. Approximately 2.8 μmol (22 mg) of each amino-modified ON were reacted for 3 h at room temperature with the thiolating agent 2-iminothiolane (2-IT) (30 mM solution in 50 mM dibasic sodium phosphate, pH 8.6) (Pierce Chemical Co., Rockford, IL) at a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (2-IT:ON) to generate an amidine compound that has a free sulfhydryl group. The mixture was concentrated by centrifugation through a Centricon-3 device (Amicon, Beverly, MA) to remove excess 2-IT prior to passage through a prepacked PD-10 column (Pharmacia Biotech, Piscataway, NJ). Purified αCD19–mAb21 (8 mg ml−1 in PBS, pH 7.5) was modified with N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP) (Pharmacia) at a 25:1 molar ratio of SPDP:mAb, as previously reported.22 The amine-reactive portion of SPDP is the N-hydroxysuccinimide (NHS) ester. The sulfhydryl-reactive portion of SPDP is the 2-pyridyldithio group, which reacts optimally with sulfhydryl groups between pH 7 and 8.1. SPDP was dissolved in DMSO at a concentration of 190 mM prior to use. After a 3 h reaction at room temperature, excess SPDP was removed by passage through a PD-10 column equilibrated in PBS. Fractions containing the majority of the SPDP-modified/pyridyldithiol-activated αCD19–mAb protein were concentrated to 8 mg ml−1 using Centricon-30 centrifugal concentrating devices (Amicon) and mixed with an equal volume of the derivatized AON at a final molar ratio of 15:1 (AON:mAb) to prepare a disulfide-linked mAb–AON conjugate. The mixture was gently rotated overnight at room temperature and concentrated 2-fold prior to being filtered (0.2 μm) and injected into a Beckman System Gold HPLC TSKG3000SW 21.5 × 600 mm size exclusion column (TosoHaas, Montgomeryville, PA) to separate unreacted AON from mAb–AON conjugates. The column was equilibrated in 100 mM sodium phosphate buffer, pH 6.8 at a flow rate of 3 ml min−1. The mAb–AON immunoconjugate eluting at 30–40 min post-injection was collected and concentrated.
:1 molar ratio (2-IT:ON) to generate an amidine compound that has a free sulfhydryl group. The mixture was concentrated by centrifugation through a Centricon-3 device (Amicon, Beverly, MA) to remove excess 2-IT prior to passage through a prepacked PD-10 column (Pharmacia Biotech, Piscataway, NJ). Purified αCD19–mAb21 (8 mg ml−1 in PBS, pH 7.5) was modified with N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP) (Pharmacia) at a 25:1 molar ratio of SPDP:mAb, as previously reported.22 The amine-reactive portion of SPDP is the N-hydroxysuccinimide (NHS) ester. The sulfhydryl-reactive portion of SPDP is the 2-pyridyldithio group, which reacts optimally with sulfhydryl groups between pH 7 and 8.1. SPDP was dissolved in DMSO at a concentration of 190 mM prior to use. After a 3 h reaction at room temperature, excess SPDP was removed by passage through a PD-10 column equilibrated in PBS. Fractions containing the majority of the SPDP-modified/pyridyldithiol-activated αCD19–mAb protein were concentrated to 8 mg ml−1 using Centricon-30 centrifugal concentrating devices (Amicon) and mixed with an equal volume of the derivatized AON at a final molar ratio of 15:1 (AON:mAb) to prepare a disulfide-linked mAb–AON conjugate. The mixture was gently rotated overnight at room temperature and concentrated 2-fold prior to being filtered (0.2 μm) and injected into a Beckman System Gold HPLC TSKG3000SW 21.5 × 600 mm size exclusion column (TosoHaas, Montgomeryville, PA) to separate unreacted AON from mAb–AON conjugates. The column was equilibrated in 100 mM sodium phosphate buffer, pH 6.8 at a flow rate of 3 ml min−1. The mAb–AON immunoconjugate eluting at 30–40 min post-injection was collected and concentrated.
Binding and internalization studies
8 mg (∼1 μmol) of the AON were tritiated at the C-8 thymidine position by ChemSyn Laboratories (Lenexa, Kansas). 680 μCi of 3H-AON were obtained and had a specific activity of 0.085 μCi μg−1 (6.6 × 105 cpm nmol−1). 5 mg of this radiolabeled AON were linked to αCD19–mAb as described above for non-radioactive AON. Sepharose CL-6B size exclusion chromatography was used to separate unreacted AON from the mAb–AON conjugate. The conjugate had a specific activity of 6000 cpm μg−1 (9.9 × 105 cpm nmol−1) and contained an average of 1.5 molecules of AON linked to each molecule of mAb. Binding experiments were carried out by incubating 5 × 106 CD19+ B-lineage ALL cells in 200 μl PBS + 2.5% fetal bovine serum (FBS) for 30 min at 4 °C with 9 μg (60 pmol) αCD19–mAb–3H-AON in the presence and absence of a 50-fold molar excess of unconjugated αCD19–mAb or control αCD7 mAb that were added to the cells 1 h prior to addition of the radiolabeled immunoconjugate. At the end of the incubation, cells were washed twice in PBS containing 2.5% FBS to remove any unbound immunoconjugate and cell-associated radioactivity was measured using a Beckman liquid scintillation counter. Internalization studies were carried out using our previously published procedures21 to determine the fate of surface-bound αCD19–3H-AON molecules in B-lineage ALL cells. 135 × 106 NALM-6 cells were incubated for 18 h at 4 °C in RPMI 1640 medium containing 5% FBS and 115 μg (700 pmol) αCD19–3H-AON. The cells were then warmed to 18–25 °C for 5 h before being washed twice with PBS + 5% FBS to remove the unbound radioactive material and cell pellets were resuspended in 2.5 ml of 10 mM Hepes, 1 mM EDTA, 0.25 M sucrose (pH 7.5) containing the protease inhibitors aprotinin (10 μg ml−1) and leupeptin (10 μg ml−1). Homogenization was done using a tight-fitting glass homogenizer, and the mixture was centrifuged at 600 × g for 10 min to remove nuclei. The supernatant was layered over a 20% Percoll solution (Pharmacia Biotech) in 0.25 M sucrose to fractionate cellular organelles by density gradient centrifugation. An in situ gradient was formed by centrifugation at 22700 × g for 40 min in a JA-17 rotor (Beckman Instruments). One milliliter fractions were isolated from the top of the gradient, counted, and analyzed as previously reported.21 Balance tubes contained a mixture of density marker beads (Pharmacia Biotech), layered on top of 20% Percoll in 0.25 M sucrose for calibration of the generated gradients according to the manufacturer's instructions. Free αCD19–3H-AON was also layered over Percoll to indicate the sample loading zone and the position of soluble proteins.
Leukemic cell lines
We evaluated the selective anti-leukemic activity of αCD19–AON using several cell lines, including the (a) Burkitt's/B-ALL leukemia/lymphoma cell lines RAJI (ATCC No. CCL-86) and DAUDI (ATCC No. CCL-213), (b) E2A–PBX1 negative B-lineage ALL cell lines REH (Pre-Pre-B, ATCC No. CRL-8286), NALM-6 (Pre-B, DSMZ No. ACC-128), RS4;11 (MLL-AF4+) (ATCC No. CRL-1873),22–24 and ALL-1 (BCR-ABL+),22–24 (c) E2A–PBX1 positive B-lineage ALL cell line LC1;1916, and (d) T-lineage ALL cell line MOLT-3 (ATCC No. CRL-1552).RT-PCR
Total cellular RNA was extracted from LC1;19 cells and examined for the presence of t(1;19)-specific E2A–PBX1 fusion transcripts using a reverse transcriptase polymerase chain reaction (RT-PCR) assay, as previously described.7 PCR products were separated by electrophoresis in a 1.2% agarose gel, transferred to nylon membranes and subjected to Southern blot analysis with previously reported oligonucleotide probes which were 5′ end-labeled with γ32P-ATP and T4 polynucleotide kinase, as described.7 RNA integrity was analyzed using an ABL RT-PCR as reported.7Confocal laser scanning microscopy
Confocal microscopy was performed using previously published procedures.25 Leukemia cells treated with αCD19–AON or control reagents αCD7–AON, αCD19–SON, and unconjugated αCD19–mAb for 96 h at 37 °C/5% CO2 were attached to poly-L-lysine-coated coverslips and fixed in ice-cold (–20 °C) methanol for 15 min. After fixation, the coverslips were washed for 15 min in phosphate-buffered saline (PBS) + 0.1% Triton X-100. Cells were stained with a rabbit polyclonal anti-tubulin antibody according to the manufacturer's recommendations (Sigma, St. Louis, MO, USA) to visualize their cytoplasms. The secondary antibody was a goat anti-rabbit fluorescein-conjugated antibody. DNA was labeled for 15 min with 1 μM Toto-3, a DNA specific dye (Molecular Probes, Eugene, OR) to visualize the apoptotic changes in the nuclei. Coverslips were inverted and mounted onto slides in Vectashield (Vector Labs, Burlingame, CA) to prevent photobleaching and were sealed with nail varnish. Slides were examined using a Bio-Rad MRC-1024 laser scanning confocal microscope mounted on a Nikon Eclipse E-800 upright microscope equipped for epifluorescence with high numerical aperture objectives.25Clonogenic assays and statistical analysis using planned linear contrasts
Leukemic cell lines were treated with various controls and test agents and then assayed for colony formation in a semi-solid methylcellulose culture system as previously described.22 Controls included samples exposed to 4 Gy γ-rays or 25 μg ml−1 vincristine (VCR), a standard chemotherapy drug used in ALL therapy. In brief, treated cells (105 cells ml−1) were washed twice, and then suspended in alpha-MEM supplemented with 0.9% methylcellulose, 30% fetal calf serum, 2 mM L-glutamine, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. Controls included untreated cells. Duplicate 1 ml samples containing 104 cells per sample were cultured in 35 mm Petri dishes for 7 days at 37 °C in a humidified 5% CO2 atmosphere. On day 7, colonies containing ≥30 cells were counted using an inverted phase microscope with high optical resolution. We constructed 4 General Linear Models for each cell type (LC1;19, RS4;11, ALL-1, MOLT3) with two factors (“treatment” and “experiment (2 replicates per experiment)”) for the analysis of the clonogenic death assay data expressed as colony count (mean no. colonies per 104 cells plated after test treatment)/(mean no. colonies per 104 cells plated after vehicle control treatment). One factor for the dependent variable was the “treatment” (9 levels for LC1;19 and 4 levels for RS4;11, ALL-1, MOLT3 cell lines) that included control, 3 concentrations of αCD19–AON, αCD19–SON, αCD7–AON, 4 Gy γ-rays and VCR treatments. Since 2 replicates were performed for each of the 2 experiments, we included the “experiment” factor (2 levels) to consider 2 sources of variation for the treatment effect arising from replicates within an experiment and variation between experiments. Consistent treatment effects for 4 replicates partitioned across 2 experiments were accounted for by normalizing differences between experiments utilizing the variance component in the “experiment” factor. In this design, differences in treatment effects were discerned in cases where there were significant differences in the overall mean of colony counts for all treatments and replicates between the 2 experiments. The least squares method was used to fit the parameters for the General Linear Models (model for LC1;19 included an intercept, 8 parameters for the treatment factor and 1 parameter for the experiment factor, and the model for the other 3 cell lines included an intercept, 3 parameters for the treatment factor and 1 parameter for the experiment factor). Model parameters were utilized to generate prediction equations and best fit lines visualized by plotting Leverage graphs using standard coding procedures. We examined the distribution of the residuals of the model for equal dispersion around the line of best fit that compared predicted and observed cell death values. The root mean square error term calculated from the deviation of data points from the model was utilized to determine significant treatment effects in the planned linear contrasts (JMP Software, SAS, Cary, NC). These contrasts compared the effect sizes such that the combination of linear parameters to be jointly tested sum to zero for each level of the contrast. Least square mean values were used to construct contrasts between each of the treatment groups (set to −1) and the control group (set to 1) for each cell type. Two-tailed tests for differences between the least square means with P-values less than 0.05 were deemed statistically significant. Post-hoc statistical power analysis for the General Linear Models was also performed for cell death assays at the 5% significance level (JMP Software, SAS, Cary, NC).SCID mouse xenograft model of t(1;19) ALL
Mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC), and all animal care procedures conformed to the Guide for the Care and Use of Laboratory Animals of the National Research Council (National Academy Press, Washington DC 1996). LC1;19 is an E2A–PBX1+ B-lineage ALL cell line established from leukemic SCID mouse xenografts of primary leukemic cells from a t(1;19)+ ALL patient who relapsed at 8.8 months.16 It causes rapidly progressive, disseminated and invariably fatal leukemia in SCID mice.16 Female CB.17 SCID mice (6–8 weeks of age; Taconic/Germantown, NY) were inoculated intravenously with 0.2 ml of a cell suspension containing 1 × 106 LC1;19 cells, as previously reported. On the next day, osmotic minipumps were implanted subcutaneously between the scapulae for continuous delivery of different treatments at a rate of 0.25 μl h−1 over a time period of 14 days (ALZET microosmotic pump, Model 1002). Mice were anesthesized with ketamine (100 mg kg−1) and xylazine (15 mg kg−1) for the procedure. Treatments included unconjugated AON (20 μg per day × 14 days = 1.8 μmol kg−1), αCD19–SON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1), control AON immunoconjugate αCD7–AON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1) and αCD19–AON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1). All SCID mice were electively killed at 120 day unless they died or became moribund earlier due to their disseminated leukemia. At the time of their death or killing, mice were necropsied to confirm leukemia-associated marked hepatomegaly and/or splenomegaly.16 For histopathologic studies, tissues were fixed in 10% neutral buffered formalin, dehydrated, and embedded in paraffin by routine methods. Glass slides with affixed 6-μm tissue sections were prepared and stained with hemotoxylin–eosin. For the analysis of the SCID mouse xenograft data, event-free survival times were measured from the day of inoculation of leukaemia cells to the day of death or killing. The probability of survival was determined and the event-free interval curves were generated using the Kaplan–Meier product limit method, as previously reported.14,16,21 We used the Mantel–Cox log-rank test to assess the effects of various treatments on the probability of survival of SCID mice inoculated with LC1;19 cells, as reported.14,16,21 Statistical power and sample size analysis were performed at the 5% significance level and 95% power using a one-tailed, two-sample test for differences in proportions assuming normal approximations. For these experiments, 12 animals per group for pairwise comparisons provided sufficient power to detect significant differences at the 95% power level. With a total sample size of 75 (60 controls versus 15 CD19–AON mice), we can detect 23% increase in survival at 95% power.Results
Up-regulated CD19 expression as a biomarker for t(1;19)+ B-lineage ALL
CD19 is a B-lineage-specific surface receptor that is expressed on the surface of leukemia cells from 85% of patients with ALL.26 It is absent from parenchymal cells of nonhematopoietic organs, circulating blood myeloid and erythroid cells, T lymphocytes, and bone marrow stem cells.26 Gene expression profiling of primary leukemic cells from matched pair relapse vs. diagnosis bone marrow specimens of 59 patients with B-lineage ALL who relapsed showed similar expression levels for CD19 (Fig. 1, Table 1) (fold-difference relapse vs. diagnosis = 0.96, P = 0.68). Therefore, CD19 is uniquely suited to serve as a target for biotherapy against newly diagnosed as well as relapsed B-lineage ALL. Expression profiles for CD19 and CD22 transcripts were highly correlated forming a subcluster in the hierarchical cluster representation. Intriguingly, comparison of CD19 expression levels in primary leukemic cells in diagnostic specimens from patients who experienced an early (N = 40; time to relapse < 36 months) versus late relapse (N = 19; time to relapse ≥ 36 months) revealed a trend toward higher expression levels for early relapse cases (fold difference early vs. late relapse: 1.44, P = 0.06, Table 2), suggesting that CD19 may be a biomarker for subpopulations of patients who are at high risk for treatment failure and early relapse, such as those with t(1;19)/E2A–PBX1+ ALL. A similar borderline significant increase was detected for expression of the IL7R gene (Table 2).| Gene symbol | Fold (relapse/diagnosis) | Paired t-test P-value | Cluster order |
|---|---|---|---|
| Paired t-tests were performed for the combined log10 mean centered values from GSE3912 and GSE18497 matched pair datasets (N = 59 pairs). Fold increase in relapse and p-values are depicted. | |||
| CD19 | 0.96 | 0.68 | 1 |
| CD22 | 0.93 | 0.47 | 2 |
| CD22 | 0.91 | 0.34 | 3 |
| CD2 | 0.99 | 0.90 | 4 |
| CD40 | 1.02 | 0.69 | 5 |
| CD5 | 1.04 | 0.56 | 6 |
| CD40 | 1.16 | 0.09 | 7 |
| IGF1R | 1.12 | 0.11 | 8 |
| CD40 | 1.08 | 0.47 | 9 |
| IGF1R | 0.93 | 0.50 | 10 |
| CD34 | 1.22 | 0.09 | 11 |
| IL2RA | 1.19 | 0.12 | 12 |
| CD40 | 1.04 | 0.78 | 13 |
| IL3RA | 1.00 | 0.97 | 14 |
| CD22 | 0.93 | 0.49 | 15 |
| CD22 | 0.89 | 0.40 | 16 |
| CD72 | 0.75 | 0.07 | 17 |
| IL2RA | 0.83 | 0.23 | 18 |
| IL7R | 0.79 | 0.14 | 19 |
| IGF1R | 1.15 | 0.36 | 20 |
| Name | Gene symbol | Fold (early/late at diag.) | t-Test P-value |
|---|---|---|---|
| Gene expression profiles based on combined mean centered values of primary leukemia cells in diagnostic specimens of ALL patients who subsequently experienced an early (N = 40) vs. late (N = 19) relapse. Fold increase for early relapse samples and the t-test P-values (unequal variances) are reported. | |||
| 206398_s_at | CD19 | 1.44 | 0.06 |
| 205831_at | CD2 | 1.31 | 0.11 |
| 204581_at | CD22 | 1.06 | 0.71 |
| 217422_s_at | CD22 | 0.97 | 0.90 |
| 220674_at | CD22 | 1.06 | 0.78 |
| 38521_at | CD22 | 1.09 | 0.57 |
| 209543_s_at | CD34 | 1.32 | 0.27 |
| 205153_s_at | CD40 | 1.21 | 0.18 |
| 215346_at | CD40 | 1.01 | 0.98 |
| 222292_at | CD40 | 0.89 | 0.58 |
| 35150_at | CD40 | 1.14 | 0.13 |
| 206485_at | CD5 | 1.21 | 0.18 |
| 215925_s_at | CD72 | 1.26 | 0.34 |
| 203627_at | IGF1R | 0.97 | 0.85 |
| 203628_at | IGF1R | 1.42 | 0.22 |
| 208441_at | IGF1R | 1.12 | 0.29 |
| 206341_at | IL2RA | 1.21 | 0.28 |
| 211269_s_at | IL2RA | 1.39 | 0.23 |
| 206148_at | IL3RA | 0.99 | 0.95 |
| 205798_at | IL7R | 1.61 | 0.07 |
 | ||
| Fig. 1 Surface receptor gene expression profiles of primary leukemic cells from matched pair relapse vs. diagnostic bone marrow specimens of ALL patients. Gene expression values for leukemic cells in matched pair specimens taken from 59 B-lineage ALL patients at diagnosis and then at relapse (combined from GSE3912, N = 32 and GSE18497, N = 27). RMA-normalized values for the GSE18497 dataset and the MAS5-signal intensity values for the GSE3912 dataset were log10 transformed and mean centered to the average value for the diagnosis samples for each gene transcript in each study. A two-way agglomerative hierarchical clustering technique was used to organize expression patterns using the average distance linkage method such that genes (rows) having similar expression across patients and patients with similar gene expression profiles were grouped together (average distance metric). Dendrograms were drawn to illustrate similar gene-expression profiles from joining pairs of closely related gene expression profiles, whereby genes joined by short branch lengths showed most similarity in expression profiles across patients and genes. The heat map represents the color-coded expression value for 59 matched pair diagnostic and relapse samples reported as mean centered expression value relative log10 transformed diagnosis samples. | ||
We next set out to compare the CD19 expression levels of primary leukemic cells from 23 patients with t(1;19)/E2A–PBX1+ ALL vs. 184 patients with t(1;19)/E2A–PBX1− ALL in a cohort of 207 patients with high risk B-lineage ALL (GSE11877). Notably, the gene for CD19 was expressed at significantly higher levels in the t(1;19)/E2A–PBX1+ subset (fold difference: 1.79, P = 0.00013; Fig. 2). Of 9 receptor genes examined, IL7R was the only other gene besides CD19 that was expressed at higher levels in t(1;19)/E2A–PBX1+ cells, but it is not a B-lineage specific receptor like CD19 and is strongly expressed in T-cells and T-cell precursors.27 In agreement with previous reports,13 the lymphoid stem cell marker CD34 was expressed at very low levels in t(1;19)/E2A–PBX1+ cells (fold difference: 0.20, P = 9.9 × 10−20). Transcripts for CD40, IL2RA, and IL3RA were also significantly down-regulated in E2A–PBX1+ patients (Fig. 2).
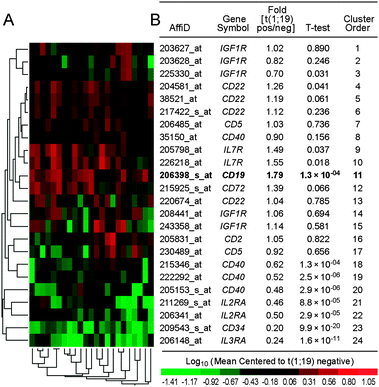 | ||
| Fig. 2 CD19 upregulation in t(1;19)+ ALL. (A) The surface receptor gene expression profiles of primary cells from t(1;19)+ ALL patients (N = 23) were compared to those of primary cells from t(1;19)− patients (N = 184) with high risk B-lineage ALL (COG P9906, GSE11877). The heat map depicts log10 expression values mean centered to E2A–PBX1 negative patients. (B) A two-way agglomerative hierarchical clustering technique was utilized to organize expression patterns such that gene transcripts (rows) and E2A–PBX1 positive samples (columns) having similar expression values were grouped together (calculated using the average distance metric shown by the dendrograms). Differential expression of each gene transcript is reported as fold increase in t(1;19) positive patients and P-values using the t-test corrected for unequal variances. Abbreviation: t-test, P-value obtained in the t-test. | ||
Construction of an anti-CD19 immunoconjugate for targeting t(1;19)/E2A–PBX1+ ALL cells with an anti-sense oligonucleotide
A disulfide-linked immunoconjugate of a 5′-amino-modified 24 mer phosphorothioate anti-sense E2A–PBX1 oligonucleotide with a mAb specific for a CD19 receptor (αCD19–AON) was prepared as a CD19-directed and leukemia-specific anti-sense oligonucleotide (AON) targeting E2A–PBX1 fusion transcript positive B-lineage ALL cells with a t(1;19) chromosomal translocation (Fig. 3A–C). The CD19-specific immunoreactivity of this immunoconjugate with B-lineage ALL cells was confirmed by using standard ligand-binding assays21 using a 3H-labeled radioactive preparation (specific activity: 0.085 μCi μg−1 ∼ 6.6 × 105 cpm nmol−1). At a 300 nM concentration of the αCD19–AON immunoconjugate, B-lineage ALL cells specifically bound 0.94 ± 0.38 × 105 molecules per cell. This specific binding was abolished by a 50-fold molar excess of unconjugated αCD19–mAb, but not by a 50-fold molar excess of unconjugated control αCD7 mAb. | ||
| Fig. 3 Anti-CD19–AON immunoconjugate against E2A–PBX1+ B-lineage ALL. (A) The thiolating agent 2-iminothiolane (2-IT) was used to generate an amidine compound with the amino-modified AON in order to introduce a free sulfhydryl group. The derivatized AON was reacted with the SPDP-modified anti-CD19–mAb to prepare a disulfide-linked mAb-AON conjugate specific for CD19+ cells carrying the leukemia-specific target fusion transcript E2A–PBX1. The conjugate contained an average of 1.5 molecules of AON linked to each molecule of mAb. (B) An HPLC size exclusion column was used to separate unreacted AON from mAb–AON conjugates. The large peak of protein (fraction II) eluting between 30–40 min was collected and concentrated. Non-reducing 5% SDS-PAGE gels of this fraction were stained with Coomassie Blue and revealed a more diffusely stained and slightly larger band than unmodified mAb, as shown in the inset. (C) Fate of a cell-bound αCD19–3H-AON immunoconjugate. NALM-6 cells were incubated with αCD19–3H-AON, washed to remove an unbound immunoconjugate, and homogenized, as described in Materials and methods. Various cellular components were fractionated on density gradients of colloidal silica (Percoll) using our previously published procedure.21 | ||
The destination of surface-bound αCD19–3H-AON molecules was traced in B-lineage ALL cells (Fig. 3C). After a total of 23 h treatment with the immunoconjugate, leukemia cells were washed to remove the unbound immunoconjugate, homogenized, and nuclei were separated by centrifugation at 600 × g. Of 86 pmol of 3H-AON molecules associated with 135 × 106 cells (∼3.8 × 105 molecules per cell), 82.6% (∼3.1 × 105 molecules per cell) were found in the nuclear pellets. The various subcellular components contained in the supernatants were fractionated on Percoll density gradients. A substantial portion of αCD19–3H-AON representing 36.9% of the total cpm recovered from the in situ generated Percoll gradient (6.4% of the total cell associated cpm or 0.2 × 105 molecules per cell) was localized in the plasma membrane (Percoll gradient fractions 3–6, density range: 1.05–1.06) (Fig. 3C). 24.2% of the cpm (4.2% of the total cpm or 0.16 × 105 molecules per cell) representing internalized αCD19–3H-AON was associated with the soluble cytoplasmic fraction (Percoll gradient fractions 1 and 2, density gradient <1.05). In contrast, only 2.3% of the cpm (0.4% of the total cpm or 0.15 × 104 molecules per cell) banded in the region near the bottom of the gradient where the lysosomes are located (Percoll gradient fractions 20–25; density range 1.063–1.08; Fig. 3C).
The ability of αCD19–AON to knock down the expression levels of E2A–PBX1 fusion transcripts in t(1;19)+ B-lineage ALL cells was examined using RT-PCR assays. Treatment of LC1;19 E2A–PBX1+ B-lineage ALL cells with 1 μg ml−1 (∼6.7 nM) αCD19–AON for 96 h resulted in selective depletion of E2A–PBX1 fusion transcripts (Fig. 4A). In contrast, E2A–PBX1 expression levels were not affected by (a) SPDP-modified unconjugated αCD19–mAb (100 μg ml−1 = 670 nM), (b) control αCD19 immunoconjugate prepared with a scrambled oligonucleotide (αCD19–SON) (10 μg ml−1 = 67 nM), or (c) control αCD7 immunoconjugate prepared with the E2A–PBX-1 AON (αCD7–AON) (10 μg ml−1 = 67 nM), indicating that both the AON moiety and the targeting CD19-specific mAb moiety are required for the biological activity of αCD19–AON. Furthermore, preincubation of LC1;19 cells with a 3–30-fold excess of unconjugated αCD19–mAb effectively blocked the αCD19–AON-induced knock-down of E2A–PBX1 expression in a concentration-dependent fashion (Fig. 4A). Unlike the leukemia-specific E2A–PBX1 transcript levels, the ABL transcript levels were not affected by αCD19–AON after 96 h treatment at 67 nM concentration demonstrating the selective nature of the αCD19–AON-mediated knock-down of E2A–PBX1 transcript levels in LC1;19 cells (Fig. 4B).
![Selective depletion of E2A–PBX1 fusion transcripts and induction of apoptosis death in t(1;19)+ ALL cells by αCD19–AON. (A and B) After treatment with αCD19–AON in the presence or absence of excess unconjugated αCD19–mAb, αCD7–AON, or αCD19–SON, LC1;19 cells were lysed, total RNA isolated, and E2A–PBX1 fusion transcript expression levels were examined by using RT-PCR, as previously reported. Normal ABL transcripts were also amplified as a control for RNA integrity. Controls included PCR reaction products of RNA-free reaction mixture 1 plus reaction mixture 2 (NEG CON1) and RNA-free reaction mixture 2 (NEG CON2). (C) LC1;19 cells were treated for 96 h with αCD19–AON, αCD7–AON, or αCD19–SON, stained with a rabbit polyclonal anti-tubulin antibody (green fluorescence) and the DNA-specific dye Toto-3 (blue fluorescence), and then examined by laser scanning confocal microscopy for apoptotic changes, as described in Materials and methods. Cells treated with αCD19–AON (depicted in C1) showed classic signs of advanced apoptosis, including total loss of the tubulin fiber network, destruction of cytoplasm with irregular contours and vacuolation along with nuclear fragmentation and micronuclei formation. Apoptotic bodies containing remnants of nuclear and cytoplasmic material were detected. In contrast, control cells treated with αCD19–SON (depicted in C2) or αCD7–AON (depicted in C3) showed normal tubular architecture and normal round blue nuclei with no signs of damage. (D) The effects of the indicated treatments on the clonogenic survival of human ALL cell lines were examined using in vitro colony assays as described in Materials and methods. % Clonogenic death was calculated using the formula: % clonogenic death = 100 − [100 × (mean no. colonies per 104 cells plated after test treatment)/(mean no. colonies per 104 cells plated after vehicle control treatment)]. Significant effect sizes (p < 0.0001 for all contrasts) were observed for t(1;19)+ LC1;19 cells comparing αCD19–AON treatment and controls.](/image/article/2013/IB/c2ib20114c/c2ib20114c-f4.gif) | ||
| Fig. 4 Selective depletion of E2A–PBX1 fusion transcripts and induction of apoptosis death in t(1;19)+ ALL cells by αCD19–AON. (A and B) After treatment with αCD19–AON in the presence or absence of excess unconjugated αCD19–mAb, αCD7–AON, or αCD19–SON, LC1;19 cells were lysed, total RNA isolated, and E2A–PBX1 fusion transcript expression levels were examined by using RT-PCR, as previously reported. Normal ABL transcripts were also amplified as a control for RNA integrity. Controls included PCR reaction products of RNA-free reaction mixture 1 plus reaction mixture 2 (NEG CON1) and RNA-free reaction mixture 2 (NEG CON2). (C) LC1;19 cells were treated for 96 h with αCD19–AON, αCD7–AON, or αCD19–SON, stained with a rabbit polyclonal anti-tubulin antibody (green fluorescence) and the DNA-specific dye Toto-3 (blue fluorescence), and then examined by laser scanning confocal microscopy for apoptotic changes, as described in Materials and methods. Cells treated with αCD19–AON (depicted in C1) showed classic signs of advanced apoptosis, including total loss of the tubulin fiber network, destruction of cytoplasm with irregular contours and vacuolation along with nuclear fragmentation and micronuclei formation. Apoptotic bodies containing remnants of nuclear and cytoplasmic material were detected. In contrast, control cells treated with αCD19–SON (depicted in C2) or αCD7–AON (depicted in C3) showed normal tubular architecture and normal round blue nuclei with no signs of damage. (D) The effects of the indicated treatments on the clonogenic survival of human ALL cell lines were examined using in vitro colony assays as described in Materials and methods. % Clonogenic death was calculated using the formula: % clonogenic death = 100 − [100 × (mean no. colonies per 104 cells plated after test treatment)/(mean no. colonies per 104 cells plated after vehicle control treatment)]. Significant effect sizes (p < 0.0001 for all contrasts) were observed for t(1;19)+ LC1;19 cells comparing αCD19–AON treatment and controls. | ||
In vitro and in vivo anti-leukemic activity of αCD19–AON against t(1;19)/E2A–PBX1− ALL cells
We next used confocal microscopy to examine the cytotoxicity of αCD19–AON against E2A–PBX1+ B-lineage ALL cells. Treatment with 6.7 nM αCD19–AON caused apoptotic destruction of E2A–PBX1+ LC1;19 cells within 96 h, whereas SPDP-modified unconjugated αCD19–mAb (670 nM, αCD19–SON) (67 nM), or αCD7–AON (67 nM) did not affect their viability (Fig. 4C).Likewise, αCD19–AON at picomolar to low nanomolar concentrations (but not αCD19–mAb, αCD19–SON, or αCD7–AON) killed clonogenic LC1;19 cells with >99% clonogenic death achieved at concentrations ≥6.7 nM (linear contrast, R2 = 0.95, t-values > 12, P-value < 0.00001 for all control versus αCD19–AON treatments) (Table 3, Fig. 4D). Neither γ-rays nor VCR were effective in killing clonogenic LC1;19 cells. Clonogenic fractions of the E2A–PBX1− B-lineage leukemia cell lines ALL1 (BCR-ABL+) or RS4;11 (MLL-AF4+) expressing the target surface receptor CD19 were not killed by αCD19–AON (linear contrast, P-value = 0.42 and 0.56 for ALL1 and RS4;11, respectively). Similarly, αCD19–AON did not inhibit the clonogenic growth of the CD19− T-lineage ALL cell line MOLT3 even at 67 nM (linear contrast, P = 0.44) (Fig. 4D, Table 3). MOLT-3 was moderately sensitive to both γ-rays and VCR (Table 3). These results demonstrate that this rationally designed immunoconjugate is selectively cytotoxic to E2A–PBX1+ B-lineage ALL cells expressing both CD19 which is recognized by its mAb moiety and E2A–PBX1 transcripts, which represent the leukemia-specific molecular target of its AON moiety.
| Treatment | LC1;19 CD19+ B-lineage ALL E2A–PBX1+ | RS4;11 CD19+ B-lineage ALL MLL-AF4+ | ALL-1 CD19+ B-lineage ALL BCR-ABL+ | MOLT3 CD19– T-lineage ALL | ||||
|---|---|---|---|---|---|---|---|---|
| Experiment #1 | # Colonies | % Kill | # Colonies | % Kill | # Colonies | % Kill | # Colonies | % Kill |
| The effects of the indicated treatments on the clonogenic survival of human ALL cell lines were examined using in vitro colony assays as described in Materials and methods. % Clonogenic death was calculated using the formula: % Clonogenic death = 100 − [100 × (mean no. colonies per 104 cells plated after test treatment)/(mean no. colonies per 104 cells plated after vehicle control treatment)]. The results are shown as the mean number of colonies. The actual number of colonies in the individual replicate samples are shown in brackets. Experiments 1 and 2 are two independent experiments. | ||||||||
| Control | 298.5 (283, 314) | — | 122 (109, 135) | — | 193 (187, 199) | — | 139.5 (134, 145) | — |
| αCD19–AON 670 pM | 3 (2, 4) | 99.0 | — | — | — | |||
| αCD19–AON 6.7 nM | 0 (0, 0) | >99.6 | — | — | — | |||
| αCD19–AON 67 nM | 0 (0, 0) | >99.6 | 125 (117, 133) | 0 | 232 (219, 245) | 0 | 140.5 (124, 157) | 0 |
| αCD19–SON 67 nM | 267.5 (254, 281) | 10.4 | — | — | — | |||
| αCD19–mAb 670 nM | 321 (305, 337) | 0 | — | — | — | |||
| αCD7–AON 67 nM | 312 (298, 326) | 0 | — | — | — | |||
| 4 Gy γ-rays | 246.5 (242, 251) | 17.4 | 180 (177, 183) | 0 | 197.5 (196, 199) | 0 | 65 (54, 76) | 53.4 |
| VCR 25 μg ml−1 | 299.5 (279, 320) | 0 | 158 (156, 160) | 0 | 190 (183, 197) | 1.6 | 27 (23, 31) | 80.6 |
| Experiment #2 | ||||||||
| Control | 209 (197, 221) | — | 177.5 (175, 180) | — | 281.5 (273, 290) | — | 121 (97, 145) | — |
| αCD19–AON 670 pM | 8 (7, 9) | 96.2 | — | — | — | |||
| αCD19–AON 6.7 nM | 0 (0, 0) | >99.5 | — | — | — | |||
| αCD19–AON 67 nM | 0 (0, 0) | >99.5 | 190 (188, 192) | 0 | 268 (259, 277) | 0 | 136 (133, 139) | 0 |
| αCD19–SON 67 nM | 191 (163, 219) | 8.6 | — | — | — | |||
| αCD19–mAb 670 nM | 238.5 (225, 252) | 0 | — | — | — | |||
| αCD7–AON 67 nM | 239 (237, 241) | 0 | — | — | — | |||
| 4 Gy γ-rays | 245.5 (218, 273) | 0 | 172 (169, 175) | 0 | 315.5 (303, 328) | 0 | 54 (49, 59) | 55.4 |
| VCR 25 μg ml−1 | 269.5 (251, 288) | 0 | 201.5 (195, 208) | 0 | 256.5 (242, 271) | 8.9 | 15 (13, 17) | 87.6 |
We next evaluated the anti-leukemic efficacy of αCD19–AON against E2A–PBX1+ human B-lineage leukemia in an established SCID mouse xenograft model of human t(1;19)+ ALL in which the E2A–PBX1+ B-lineage ALL cell line LC1;19 causes disseminated and invariably fatal leukemia in SCID mice (Fig. 5). All untreated control mice (N = 65) challenged with an intravenous inoculum of 1 × 106 LC1;19 cells died of overt leukemia with a median event-free survival (EFS) of 37.0 ± 0.1 days. At the time of death, all mice had massive hepatosplenomegaly due to leukemic infiltration. Histopathologic studies showed leukemic infiltration of multiple organs, including bone marrow, brain, liver, spleen, ovaries, and lungs. Involvement of bone marrow ranged from moderate, multifocal to diffuse infiltration with replacement of normal tissue elements by sheets of closely packed leukemic blasts resulting in total or near total effacement of the normal tissue architecture. Involvement of the CNS consisted of infiltration by leukemic blasts in the leptomeninges, and mild multifocal parenchymal infiltration. Likewise, all control mice treated with (a) unconjugated AON (20 μg per day × 14 days = 1.8 μmol kg−1) or αCD19–SON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1; N = 25; median EFS: 49.0 ± 0.4 days) or (b) control AON immunoconjugate αCD7–AON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1; N = 10; median EFS: 43.0 ± 0.5 (d) rapidly died of disseminated leukemia with <10% survival at 60 days (Fig. 5). In contrast, αCD19–AON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1) delivering 74 μg kg−1 per day of the AON (total amount of AON delivered: 1.48 μg per day × 14 days = 1 mg kg−1) more than doubled the median EFS to 82.0 ± 1.9 days. While only 3% of control mice were alive at 60 days and none were alive at 90 days, 87% and 47% of αCD19–AON-treated mice remained alive at 60 days and 90 days, respectively (P < 0.0001) (Fig. 5).
 | ||
| Fig. 5 In vivo anti-leukemic efficacy of αCD19–AON in a SCID mouse xenograft model of human t(1;19) ALL. Female CB.17 SCID mice were inoculated intravenously with 0.2 ml of a cell suspension containing 1 × 106 LC1;19 cells, as previously reported. Unconjugated AON (20 μg per day × 14 days = 1.8 μmol kg−1), αCD19–SON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1), control AON immunoconjugate αCD7–AON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1) and αCD19–AON (20 μg per day × 14 days = 14 mg kg−1 or 93.3 nmol kg−1) were delivered as a continuous 14 day infusion using an ALZET microosmotic pump. All mice were electively killed at 120 day unless they died or became moribund earlier due to their disseminated leukemia. At the time of their death or killing, mice were necropsied to confirm leukemia-associated marked hepatomegaly and/or splenomegaly.16 For the analysis of the SCID mouse xenograft data, event-free survival times were measured from the day of inoculation of leukemia cells to the day of death or killing. Cumulative proportions of mice surviving event-free are shown according to the number of days after inoculation of LC1;19 leukemia cells (A1). A2 and A3 depict the massive hepatosplenomegaly of the original xenografted SCID mice with overt leukemia from which the LC1;19 cell line was derived.16 In the preparation of the two mouse diagrams, the artist used color photographs taken at the time of necropsy. Scans of the photos were used to accurately trace the contours of organ structures and mouse outlines. The color images were converted to grayscale, and image contrast was decreased. The peripheral traced shapes (background and mouse body) were filled with opaque shades of white and grey, while highlighted organ shapes (liver and spleen) were given a percentage of opacity to allow some of the tissue's texture to show through. L, liver; S, spleen; T, tumor. B depicts the life table analysis of the survival data shown in A1. Control mice in the CON2 group that were treated with αCD19–SON or AON had a prolongation of their EFS from 37 days to 49 days. Likewise, control mice treated with αCD7–AON had a prolongation of their EFS from 37 days to 43 days. By contrast, treatment with αCD19–AON prolonged the EFS to 82 days. **P < 0.0001 compared to all other treatment groups and an untreated control group (CON/CON1). | ||
Discussion
Here we first report a novel, biomarker-guided biotherapy strategy for personalized treatment of t(1;19) ALL. Inspired by the results of our bioinformatics studies that provided the first evidence that the gene for the B-lineage specific surface receptor CD19 is constitutively overexpressed in t(1;19) B-lineage ALL, we prepared a rationally-designed, disulfide-linked immunoconjugate of a 5-amino-modified 24 mer phosphorothioate anti-sense E2A–PBX1 oligonucleotide (AON) with a mAb specific for a CD19 receptor (αCD19–AON) as a CD19-directed and leukemia-specific biotherapeutic agent against E2A–PBX1+ B-lineage ALL. Treatment of E2A–PBX1+ leukemia cells with low nanomolar concentrations of αCD19–AON resulted in selective depletion of E2A–PBX1 transcripts and caused apoptotic destruction and abrogation of clonogenic growth. At a dose of 93 nmol kg−1, αCD19–AON more than doubled the leukemia-free survival time of SCID mice challenged with radiochemotherapy-resistant highly aggressive human E2A–PBX1+ B-lineage leukemia cells. Both the AON moiety and the targeting CD19-specific mAb moiety were required for the in vitro as well as in vivo anti-leukemic activity of αCD19–AON. The observed in vitro and in vivo anti-leukemic potency of the αCD19–AON immunoconjugate provides the first preclinical proof-of-principle that t(1;19)+ high risk B-lineage ALL can be treated with leukemia-specific biotherapeutic agents that knock-down E2A–PBX1 expression.Our data indicate that the E2A–PBX1 transcripts are important for the survival of t(1;19)+ B-lineage ALL cells and their in vitro as well as in vivo clonogenic subpopulations. AON acts by targeting specific mRNAs through heteroduplex formation inside the cell, thereby inducing RNase H activation, translational arrest, or alternative splicing.28 The observed in vitro and in vivo anti-leukemic potency of the αCD19–AON immunoconjugate provides preclinical proof-of-principle that t(1;19)+ high risk B-lineage ALL can be treated with leukemia-specific biotherapeutic agents that knock down E2A–PBX1 expression.
CD19 is a 95 kDa B-lineage restricted receptor molecule that functions as a key regulator of transmembrane signals in both B-cells and B-cell precursors.26,29,30 CD19 antigen is acquired at a very early stage of B-cell ontogeny, prior to rearrangement of immunoglobulin genes and expression of other B-precursor antigens such as CD10 and CD22.26,29,30 CD19 antigen is abundantly expressed on the malignant cells from B-lineage leukemia and lymphoma patients, but it is absent on the parenchymal cells of life-maintaining non-hematopoietic organs, circulating blood myeloid and erythroid cells, T-cells as well as bone marrow stem cells.26,29,30 In B-lineage ALL, CD19 antigen is expressed on candidate leukemic stem cell populations with in vivo clonogenic, leukemia initiating and propagating properties in xenograft models using immunocompromised mice. The very favorable B-lineage leukemia/lymphoma vs. normal tissue expression profile of CD19 and its association with stemness properties of leukemic B-cell precursor populations make it an attractive molecular target for biotherapy in relapsed ALL.26,29,30
Several hundred thousand CD19 molecules located on the surface of each B-lineage leukemia/lymphoma cell are rapidly internalized upon ligation with anti-CD19–mAb or immunoconjugates.26,31 CD19 has tyrosine-based internalization motifs in its cytoplasmic domain that were predicted to bind the clathrin adaptor AP-2.26,31 Ingle et al. reported that CD19-directed mAb and mAb-drug conjugates are internalized by dynamin-dependent, clathrin-mediated endocytosis and their cellular uptake as well as intracellular trafficking mimic the endocytic pathway of transferrin.32 Their studies demonstrated that anti-CD19–mAb are transported to late endosomes and lysosomes within 3 h.32 Clathrin inhibitor chlorpromazine has been shown to inhibit the uptake of both transferrin and anti-CD19–mAb.33 Detailed immunoelectron microscopy studies by Pulczynski et al. showed that upon binding of anti-CD19–mAb, CD19 antigen on the surface of pre-B ALL cells is internalized within 30 min via plasmalemmal pits, transferred through the endosomal compartment within 2 h, delivered to multivesicular bodies/late endosomes and lysosomes after 2 h, and recycled.33 Notably, the internalized anti-CD19–mAb-CD19 complexes showed close physical association with/attachment to the endosomal membrane.33 This close association of internalized anti-CD19–mAb with the endosomal membrane likely facilitates the endosomal leakage and/or escape of chemical or biological substances attached to them into the cytosol and provides a cogent explanation for the documented potency of toxin conjugates22 as well as drug conjugates21 of anti-CD19–mAb against CD19+ leukemia/lymphoma cells. The pharmacological effectiveness of oligonucleotide-based therapeutics depends on their cellular uptake, intracellular trafficking, endosomal release, and productive delivery to their target subcellular compartments.34,35 The observed potency of the anti-CD19–mAb-AON immunoconjugate against t(1;19) ALL cells demonstrates that this biotherapeutic agent is inherently capable of effective delivery of AON molecules to relevant subcellular compartments where they can target specific mRNAs through heteroduplex formation.
While our proof-of-concept study employed AON to knock down E2A–PBX1 transcript expression in leukemia cells, there are other technology platforms that could be used for the same purpose, including RNA interference (RNAi) using small interfering RNAs (siRNA) that can be delivered using nanoparticles.34–40 In recent years, several recombinant fusion proteins that consist of a cell surface targeting moiety (e.g. an antibody fragment or ligand) and an oligonucleotide complexation moiety (e.g. truncated protamine) have been designed for targeted delivery of siRNA.34,41–45 Alternatively, mAb mediated delivery of AON to target cancer cells may provide an effective means for selective knock down of cancer-specific oncogenic genes, such as the E2A–PBX1 fusion gene of t(1;19)+ ALL cells. The described αCD19–AON immunoconjugate and its derivatives may offer an effective treatment for t(1;19)+ B-lineage ALL. Whether AON immunoconjugates will prove superior to immunoconjugates containing cytotoxic drugs or immunomodulatory bifunctional αCD19 biotherapeutic agents should be examined in appropriate preclinical and clinical settings.
Conflicts of interest statement
The authors have no conflicts to disclose.Acknowledgements
F.M.U. is supported in part by DHHS grants U01-CA-151837, R21-CA-164098, and R01CA-154471 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. F.M.U. is also supported in part by Children's Hospital Los Angeles Institutional Endowment and Special Funds, 2011 V-Foundation Translational Research Award, Ronald McDonald House Charities of Southern California, Couples Against Leukemia Foundation and Nautica Triathalon and its producer Michael Epstein.References
- K. E. Barber, C. J. Harrison, Z. J. Broadfield, A. R. Stewart, S. L. Wright, M. Martineau, J. C. Strefford and A. V. Moorman, Molecular cytogenetic characterization of TCF3 (E2A)/19p13.3 rearrangements in B-cell precursor acute lymphoblastic leukemia, Genes, Chromosomes Cancer, 2007, 46, 478–486 CrossRef CAS
.
- G. Bain, E. C. Maandag, D. J. Izon, D. Amsen, A. M. Kruisbeek and B. C. Weintraub,
et al., E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements, Cell, 1994, 79, 885–892 CrossRef CAS
- Y. Zhuang, P. Soriano and H. Weintraub, The helix-loop-helix gene E2A is required for B cell formation, Cell, 1994, 79, 875–884 CrossRef CAS
- J. L. Wiemels, B. C. Leonard, Y. Wang, M. R. Segal, S. P. Hunger and M. T. Smith,
et al., Site-specific translocation and evidence of postnatal origin of the t(1;19) E2A-PBX1 fusion in childhood acute lymphoblastic leukemia, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15101–15106 CrossRef CAS
- W. M. Crist, A. J. Carroll, J. J. Shuster, F. G. Behm, M. Whitehead and T. J. Vietti,
et al., Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study, Blood, 1990, 76, 117–122 CAS
- L. M. Secker-Walker, R. Berger, P. Fenaux, J. L. Lai, B. Nelken and M. Garson,
et al., Prognostic significance of the balanced t(1;19) and unbalanced der(19)t(1;19) translocations in acute lymphoblastic leukemia, Leukemia, 1992, 6, 363–369 CAS
- P. S. Gaynon, M. L. Crotty, H. N. Sather, B. C. Bostrom, J. B. Nachman and P. G. Steinherz, Expression of BCR-ABL, E2A-PBX1, and MLL-AF4 fusion transcripts in newly diagnosed children with acute lymphoblastic leukemia: a Children's Cancer Group initiative, Leuk. Lymphoma, 1997, 26, 57–65 CAS
- M. P. Kamps, C. Murre, X. H. Sun and D. Baltimore, A new homeobox gene contributes the DNA binding domain of the t( 1;19) translocation protein in pre-B ALL, Cell, 1990, 60, 547 CrossRef CAS
- J. D. Mellentin, C. Murre, T. A. Donlon, P. S. McCaw, S. D. Smith and A. J. Carroll,
et al., The gene for enhancer binding proteins E12/E47 lies at the t(1;19) breakpoint in acute leukemias, Science, 1989, 246, 379 CrossRef CAS
- J. Nourse, J. D. Mellentin, N. Galili, J. Wilkinson, E. Stanbridge, S. D. Smith and M. L. Cleary, Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mRNA that codes for a potential chimeric transcription factor, Cell, 1990, 60, 535–545 CrossRef CAS
- J. Mazieres, L. You, B. He, Z. Hu, A. Y. Lee and I. Mikami,
et al., Inhibition of Wnt16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis, Oncogene, 2005, 24, 5396–5400 CrossRef CAS
- S. Jeha, D. Pei, S. C. Raimondi, M. Onciu, D. Campana and C. Cheng,
et al., Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1, Leukemia, 2009, 23, 1406–1409 CrossRef CAS
- M. J. Borowitz, S. P. Hunger, A. J. Carroll, J. J. Shuster, D. J. Pullen, C. P. Steuber and M. L. Cleary, Predictability of the t(1;19)(q23;p13) from surface antigen phenotype: implications for screening cases of childhood acute lymphoblastic leukemia for molecular analysis: a Pediatric Oncology Group study, Blood, 1993, 82, 1086–1091 CAS
- F. M. Uckun, M. G. Sensel, H. N. Sather, P. S. Gaynon, D. C. Arthur and B. J. Lange,
et al., Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group, J. Clin. Oncol., 1998, 16, 527–535 CAS
- B. J. Waurzyniak, N. Heerema, M. G. Sensel, P. S. Gaynon, P. Kraft and H. N. Sather,
et al., Distinct in vivo engraftment and growth patterns of t(1;19)+/E2A-PBX1+ and t(9;22)+/BCR-ABL+ human leukemia cells in SCID mice, Leuk. Lymphoma, 1998, 32, 77–787 CAS
- F. M. Uckun, J. R. Downing, R. Gunther, L. M. Chelstrom, D. Finnegan and V. J. Land,
et al., Human t(1;19)(q23;p13) pre-B acute lymphoblastic leukemia in mice with severe combined immunodeficiency, Blood, 1993, 81, 3052–3062 CAS
- M. Reich, T. Liefeld, J. Gould, J. Lerner, P. Tamayo and J. P. Mesirov, GenePattern 2.0, Nat. Genet., 2006, 38, 500–501 CrossRef CAS
- D. Bhojwani, H. Kang and N. P. Moskowitz,
et al., Biologic pathways associated with relapse in childhood acute lymphoblastic leukemia: a Children's Oncology Group study, Blood, 2006, 108, 711–717 CrossRef CAS
- F. J. Staal, D. de Ridder, T. Szczepanski, T. Schonewille, E. C. van der Linden and E. R. van Wering,
et al., Genome-wide expression analysis of paired diagnosis-relapse samples in ALL indicates involvement of pathways related to DNA replication cell cycle and DNA repair, independent of immune phenotype, Leukemia, 2010, 24, 491–499 CrossRef CAS
- H. Kang, I. M. Chen, C. S. Wilson and E. J. Bedrick,
et al., Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia, Blood, 2010, 115, 1394–1405 CrossRef CAS
- F. M. Uckun, W. E. Evans, C. J. Forsyth, K. G. Waddick, L. T. Ahlgren and L. M. Chelstrom,
et al., Biotherapy of B-cell precursor leukemia by targeting genistein to CD19-associated tyrosine kinases, Science, 1995, 267, 886–891 CrossRef CAS
- F. M. Uckun, K. J. Gajl-Peczalska, J. H. Kersey, L. L. Houston and D. A. Vallera, Use of a novel colony assay to evaluate the cytotoxicity of an immunotoxin containing pokeweed antiviral protein against blast progenitor cells freshly obtained from patients with common B-lineage acute lymphoblastic leukemia, J. Exp. Med., 1986, 163, 347–368 CrossRef CAS
- F. M. Uckun, P. Goodman, H. Ma, I. Dibirdik and S. Qazi, CD22 Exon 12 Deletion as a Novel Pathogenic Mechanism of Human B-Precursor Leukemia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16852–16857 CrossRef CAS
- F. M. Uckun, Z. Ozer, S. Qazi, L. Tuel-Ahlgren and C. Mao, Polo-like kinase 1 (PLK1) as a molecular target to overcome Syk-mediated resistance of B-lineage acute lymphoblastic leukemia cells to oxidative stress, Br. J. Haematol., 2010, 148, 714–725 CrossRef CAS
- A. Vassilev, Z. Ozer, C. Navara, S. Mahajan and F. M. Uckun, Bruton's tyrosine kinase as an inhibitor of the Fas/CD95 death-inducing signaling complex, J. Biol. Chem., 1999, 274, 1646–1656 CrossRef CAS
- F. M. Uckun, W. Jaszcz, J. L. Ambrus, A. S. Fauci, K. Gajl-Peczalska, C. W. Song, M. R. Wick, D. E. Myers, K. Waddick and J. A. Ledbetter, Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins, Blood, 1988, 71, 13–29 CAS
- F. M. Uckun, L. Tuel-Ahlgren, V. Obuz, R. Smith, I. Dibirdik and M. Hanson,
et al., Interleukin 7 receptor engagement stimulates tyrosine phosphorylation, inositol phospholipid turnover, proliferation, and selective differentiation to the CD4 lineage by human fetal thymocytes, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6323–6327 CrossRef CAS
- N. Dias and C. A. Stein, Antisense oligonucleotides: Basic concepts and mechanisms, Mol. Cancer Ther., 2002, 1, 347–355 CAS
- F. M. Uckun, Regulation of human B-cell ontogeny (Review), Blood, 1990, 76, 1723–1733 CAS
- F. M. Uckun, A. L. Burkhardt, L. Jarvis, X. Jun, B. Stealey, I. Dibirdik, D. E. Myers, L. Tuel-Ahlgren and J. B. Bolen, Signal transduction through the CD19 receptor during discrete developmental stages of human B-cell ontogeny, J. Biol. Chem., 1993, 268, 21172–21184 CAS
- O. W. Press, A. G. Farr, K. I. Borroz, S. K. Anderson and P. J. Martin, Endocytosis and Degradation of Monoclonal Antibodies Targeting Human B-Cell Malignancies, Cancer Res., 1989, 49, 4906–4912 CAS
- G. S. Ingle, P. Chan, J. M. Elliott, W. S. Chang, H. Koeppen, J.-P. Stephan and S. J. Scales, High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19 drug conjugate, Br. J. Haematol., 2007, 140, 46–58
- S. Pulczynski, A. M. Boesen and O. M. Jensen, Antibody-induced modulation and intracellular transport of CD10 and CD19 antigens in human B-cell lines: An immunofluorescence and immunoelectron microscopy study, Blood, 1993, 81, 1549–1557 CAS
- X. Ming, Cellular delivery of siRNA and antisense oligonucleotides via recepto-mediated endocytosis, Expert Opin. Drug Delivery, 2011, 8, 435–449 Search PubMed
- R. L. Juliano, X. Ming and O. Nakagawa, Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides, Bioconjugate Chem., 2012, 23, 147–157 CrossRef CAS
- D. Haussecker, The Business of RNA Therapeutics in 2012, Mol. Therapy-Nucleic Acids, 2012, 2, e8, DOI:10.1038/mtna.2011.9
- K. A. Whitehead, R. Langer and D. G. Anderson, Knocking down barriers: advances in siRNA delivery, Nat. Rev. Drug Discovery, 2009, 8, 129–133 CrossRef CAS
- B. Yu, X. Zhao, L. J. Lee and R. J. Lee, Targeted delivery systems for oligonucleotide therapeutics, AAPS J., 2009, 1, 195–203 Search PubMed
- J. Wang, Z. Lu, M. Wientjes and J. Au, Delivery of siRNA therapeutics: barriers and carriers, AAPS J., 2010, 12, 492–503 Search PubMed
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. L. Mui and C. K. Cho,
et al., Rational design of cationic lipids for siRNA delivery, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS
- J. Winkler, Nanomedicines based on recombinant fusion proteins for targeting siRNA oligonucleotides, Ther. Delivery, 2011, 2, 891–905 Search PubMed
- E. Song, P. Zhu and S. K. Lee,
et al., Antibody mediated in vivo delivery of small interfering RNAs via cell surface receptors, Nat. Biotechnol., 2005, 23, 709–717 CrossRef CAS
- A. Eguchi, B. R. Meade and Y.-C. Chang,
et al., Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein, Nat. Biotechnol., 2009, 27, 567–571 CrossRef CAS
- S. W. Kim, N. Y. Kim, Y. B. Choi, S. H. Park, J. M. Yang and S. Shin, RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system, J. Controlled Release, 2010, 143, 335–343 CrossRef CAS
- N. Manjunath and D. M. Dykxhoorn, Advances in Synthetic siRNA Delivery, Discovery Med., 2010, 9, 418–430 Search PubMed
| This journal is © The Royal Society of Chemistry 2013 |