DOI:
10.1039/C3GC41839A
(Paper)
Green Chem., 2014,
16, 357-364
N-Chlorosuccinimide-promoted synthesis of thiophosphates from thiols and phosphonates under mild conditions†
Received
4th September 2013
, Accepted 26th October 2013
First published on 28th October 2013
Abstract
A very simple N-chlorosuccinimide-promoted synthesis of thiophosphates through the coupling of thiols and phosphonates is reported. Notably, the reactions were carried out in the absence of a base. Functional groups including fluoro, bromo and trifluoromethyl are all tolerated by the reaction conditions employed. Both aryl and alkyl thiols are coupled smoothly with a broad spectrum of phosphonates to afford the corresponding thiophosphates in good to excellent yields.
Introduction
Thiophosphates are important skeletons in biology1 and organic synthesis,2 as a result, the synthesis of thiophosphates has gained much attention. Timperley and coworkers reported the formation of thiophosphates through the coupling of the corresponding phosphorochloridates or phosphorobromidates with thiols in the presence of a base; however, the preparation of phosphorochloridates and phosphorobromidates is required [Scheme 1(a)]. A typical procedure for preparing phosphorochloridates and phosphorobromidates relied on reacting H-phosphonates with chlorine and bromine in an organic solvent, respectively. Chlorine and bromine are both toxic and difficult to control.3 Kaboudin described a one-pot procedure by the treatment of diethyl phosphonate with ammonium acetate/sulfur/Al2O3 followed by addition of alkyl halides to give thiophosphates; unfortunately, the substrate is limited to diethyl phosphonate, and only alkyl substituents are introduced to the sulfur atom in this system [Scheme 1(b)], moreover, microwave-heating is required in this approach.4 Jensen et al. reported that thiophosphates can be synthesized through the coupling of sodium diethyl phosphonate with organic disulfides [Scheme 1(c)],5 but again there are several limitations to using this approach. First, a strong base (i.e., NaH) is necessary to generate the sodium diethyl phosphonate. Notably, a strong base will generally reduce the functional group compatibility. Second, one equiv. of disulfide is required to produce the thiophosphate as the target molecule; however, one equiv. of sodium thiolate6,7 will be generated as a waste. Recently, a copper-catalyzed coupling of dialkyl phosphonates with organic disulfides was reported by Zhao and coworkers [Scheme 1(d)];8 again several synthetic limitations have been observed by using this approach. First, the reaction is limited to dialkyl phosphonates. Second, no alkyl disulfides are involved in this system. Third, an additional base is required. In addition, thiophosphates have shown potential applications in chemical biology; and transition metals are generally avoided for use in the final step for the preparation of biological molecules in order to exclude the presence of transition metal-contaminant.
 |
| | Scheme 1 Preparation of thiophosphates. | |
From the substrate generality and conditions employed, a general direct coupling of thiols with H-phosphonates in transition metal-free and mild conditions is highly desirable for preparing thiophosphates. Building on our previous work,9 we now report a general method to prepare thiophosphates through N-chlorosuccinimide-promoted10–12 P–S bond formation between phosphonates and thiols. Importantly, this approach is simple and the reactions are carried out in one pot without an additive base [Scheme 1(e)].
Results and discussion
Thiophenol 1a and diphenyl phosphonate 2a were used as model substrates to determine the optimized reaction conditions. The results are summarized in Table 1. A trace amount of desired product was detected using 1.1 equiv. of N-iodosuccinimide (Table 1, entry 1), because the intermediate sulfenyliodide is too reactive to react with diphenyl phosphonate; this results in the formation of diphenyl disulfide as a side product. Interestingly, a 23% yield of the target, 3a was afforded when N-bromosuccinimide was used instead of N-iodosuccinimide (Table 1, entry 2). To our delight, a 78% yield of 3a was achieved when the reaction was carried out by using 1.1 equiv. of N-chlorosuccinimide (NCS, Table 1, entry 3). No improvement was observed when Et3N was added as a base (Table 1, entry 4).9 The control experiment showed that no product was formed in the absence of N-chlorosuccinimide (Table 1, entry 5). We then studied the influence of solvent (Table 1, entries 6–11) and found that MeCN was superior to THF, ether, dioxane, DMF and DMSO.
Table 1 Optimize the reaction conditionsa
|

|
| Entry |
X |
Solvent |
Yieldb (%) |
|
Reaction conditions: thiophenol (1.0 mmol), N-halosuccinimide (1.1 mmol), under a nitrogen atmosphere in solvent (1.5 mL) at rt for 20 min for the first step; diphenyl phosphonate (1.0 mmol) was added, and reacted at rt for 10 min for the second step.
Isolated yield.
Et3N (1.0 mmol) was added in the second step.
|
| 1 |
I |
Toluene |
Trace |
| 2 |
Br |
Toluene |
23 |
| 3 |
Cl |
Toluene |
78 |
| 4c |
Cl |
Toluene |
76 |
| 5 |
— |
Toluene |
— |
| 6 |
Cl |
THF |
81 |
| 7 |
Cl |
Ether |
75 |
| 8 |
Cl |
Dioxane |
79 |
| 9 |
Cl |
DMF |
76 |
| 10 |
Cl |
DMSO |
Trace |
| 11 |
Cl |
MeCN |
85 |
We examined the scope of the substrates based on the optimized reaction conditions above as demonstrated in Table 2. Aryl thiols bearing electron-donating and electron-withdrawing groups reacted with a variety of phosphonates to give the corresponding thiophosphates in good to excellent yields. Notably, this system shows good functional group compatibility. Functional groups including fluoro (Table 2, entries 2 and 19), bromo (Table 2, entries 3 and 7), trifluoromethyl (Table 2, entries 9, 10, 16 and 17) are all tolerated by the reaction conditions. Sterically demanding substituted aryl thiols did not decrease the reactivity of the reactions, and afforded the products with satisfying yields (Table 2, entries 7, 8, 14 and 18).
Table 2 NCS-promoted synthesis of thiophosphates from aryl thiols and phosphonatesa
Based on the promising results for aryl thiols in Table 2, we then turned our attention to the coupling reaction of alkyl thiols with phosphonates. The results are summarized in Table 3. Alkyl thiols such as cyclohexanethiol (4a), 2-methylbutane-1-thiol (4b), hexanethiol (4c) and benzylthiol (4d) were coupled smoothly with phosphonates to give the corresponding thiophosphates in good to excellent yields.
Table 3 NCS-mediated coupling of alkyl thiols with phosphonatesa
Conclusions
In conclusion, we report a convenient protocol for the synthesis of thiophosphates through the N-chlorosuccinimide-promoted coupling of thiols with phosphates. Both aryl and alkyl thiols are coupled smoothly with a variety of phosphates to provide the corresponding thiophosphates in good to excellent yields in the absence of a base. Importantly, it is not necessary to functionalize the starting materials, and this one-pot procedure can be completed within 30 min. Functional groups including fluoro, bromo and trifluoromethyl are all tolerated by the reaction conditions employed. Applications of N-chlorosuccinimide-promoted coupling reaction for carbon–carbon and carbon–heteroatom bond formations are underway in our laboratory.
Experimental
General information
All chemicals were purchased from commercial suppliers and used without further purification. TLC analyses were performed on Merck DC-Alufolien with Kieselgel 60F-254, and were visualized with UV light. Purifications were performed by flash chromatography on silica gel 60 (Merck, 230–400 mesh ASTM). NMR spectra were recorded on a Varian Unity Inova-600 or a Varian Mercury-400 instrument using CDCl3 as the solvent. Chemical shifts are reported in parts per million (ppm) and referenced to the residual solvent resonance. Coupling constants (J) are reported in hertz (Hz). Standard abbreviations indicating multiplicity were used as follows: s = singlet, d = doublet, t = triplet, dd = double doublet, q = quartet, m = multiplet. IR spectra were measured using a Bruker Equinox 55 or Bruker Tensor 27 spectrophotometer. High resolution mass spectra (HRMS) were recorded on an electron ionization time-of-flight (EI-TOF) mass spectrometer at the National Chung Hsing University.
General procedure for Table 1
A Schlenk tube equipped with a magnetic stir bar was charged with N-chlorosuccinimide (0.15 g, 1.1 mmol), thiophenol 1a (1.0 mmol) and solvent (1.5 mL). After the mixture was stirred for 20 min, diphenylphosphonate 2a (1.0 mmol) was added by a syringe under a nitrogen-filled balloon. After being stirred for another 10 min at room temperature, the resulting solution was directly filtered through a pad of silica gel and then washed with ethyl acetate (20 mL) and concentrated to give the crude material which was then purified by column chromatography (SiO2, hexane) to afford 3a.
Representative example of Table 1
O,O,S-Triphenylphosphorothioate (entry 11, 3a).
Following the general procedure for Table 1, using thiophenol (0.103 mL, 1.0 mmol, 1.0 equiv.), N-chlorosuccinimide (0.15 g, 1.1 mmol, 1.1 equiv.), MeCN (1.5 mL) and diphenylphosphonate (0.192 mL, 1.0 mmol) and then purification by column chromatography (SiO2, hexane) provides 3a as a colorless oil (290 mg, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.16–7.30 (m, 6 H), 7.31–7.37 (m, 7 H), 7.48–7.51 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 115.3, 120.3, (d, J = 2.7 Hz), 125.5, 129.2, 129.4, 129.5, 129.7, 135.1 (d, J = 5.4 Hz), 150.2 (d, J = 9.1 Hz); 31P NMR (162 MHz, CDCl3) δ 15.52; IR (CHCl3): ν 3064, 2959, 2929, 2871, 1949, 1868, 1775, 1718, 1593, 1489, 1457, 1442, 1367, 1270, 1157, 1071, 1025, 1007, 920, 827, 746 cm−1; HRMS-EI calcd for C18H15O3PS: 342.0480, found: 342.0484.
General procedure for Table 2
A Schlenk tube equipped with a magnetic stir bar was charged with N-chlorosuccinimide (0.15 g, 1.1 mmol), aryl thiol 1 (1.0 mmol) and MeCN (1.5 mL). After the mixture was stirred for 20 min, phosphonate 2 (1.0 mmol) was added by a syringe under a nitrogen-filled balloon. After being stirred for another 10 min at room temperature and diluted with ethyl acetate (20 mL), the resulting solution was directly filtered through a pad of silica gel and then washed with ethyl acetate (20 mL) and concentrated to give the crude material which was then purified by column chromatography (SiO2, hexane) to yield 3.
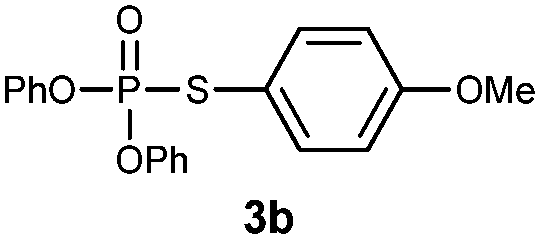
S-(4-Methoxyphenyl) O,O-diphenylphosphorothioate (3b).
Following the general procedure for Table 2, using 4-methoxythiophenol (0.123 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3b as a colorless oil (301 mg, 81% yield). 1H NMR (400 MHz, CDCl3) δ 3.77 (s, 3 H), 6.83–6.85 (m, 2 H), 7.17–7.22 (m, 6 H), 7.31–7.39 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 55.2, 114.7 (d, J = 8.2 Hz), 115.0 (d, J = 2.7 Hz), 120.3 (d, J = 5.4 Hz), 125.4, 129.6, 136.8 (d, J = 5.5 Hz), 150.2 (d, J = 8.2 Hz), 160.8 (d, J = 2.7 Hz); 31P NMR (162 MHz, CDCl3) δ 16.17; IR (CHCl3): ν 3069, 3013, 2919, 2839, 1592, 1491, 1459, 1408, 1271, 1251, 1206, 1184, 1161, 1107, 1071, 1026, 1007, 938, 829, 769, 689 cm−1; HRMS-EI calcd for C19H17O4PS: 372.0585, found: 372.0595.
S-(4-Fluorophenyl) O,O-diphenylphosphorothioate (3c).
Following the general procedure for Table 2, using 4-fluorothiophenol (0.107 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3c as a colorless oil (190 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 7.00–7.05 (m, 2 H), 7.19–7.26 (m, 6 H), 7.33–7.37 (m, 4 H), 7.43–7.47 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 115.3, 116.7 (dd, J = 2.4, 21.8 Hz), 120.4 (d, J = 4.8 Hz), 125.7, 129.8, 137.4 (q, J = 4.7 Hz), 150.2 (d, J = 8.6 Hz), 163.7 (dd, J = 3.8, 250.1 Hz); 31P NMR (162 MHz, CDCl3) δ 15.19; 19F NMR (376 MHz, CDCl3) δ −111.8 (s); IR (CHCl3): ν 3069, 1590, 1488, 1457, 1399, 1272, 1182, 1158, 1093, 1071, 1025, 1008, 939, 833, 766, 688 cm−1; HRMS-EI calcd for C18H14FO3PS: 360.0385, found: 360.0376.
S-(4-Bromophenyl) O,O-diphenylphosphorothioate (3d).
Following the general procedure for Table 2, using 4-bromothiophenol (0.189 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3d as a colorless oil (212 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ 7.18–7.23 (m, 6 H), 7.31–7.35 (m, 6 H), 7.42–7.44 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 115.3, 120.3 (d, J = 4.6 Hz), 125.7, 129.2, 129.8, 132.5 (d, J = 2.8 Hz), 136.6 (d, J = 5.4 Hz), 150.1; 31P NMR (162 MHz, CDCl3) δ 14.49; IR (CHCl3): ν 3068, 2919, 2851, 1942, 1899, 1778, 1590, 1488, 1387, 1273, 1181, 1085, 1070, 1025, 1009, 939, 817, 765, 689, 619, 590, 545, 512 cm−1; HRMS-EI calcd for C18H14BrO3PS: 419.9585, found: 419.9592.
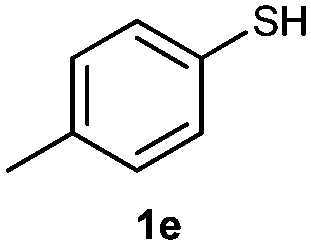
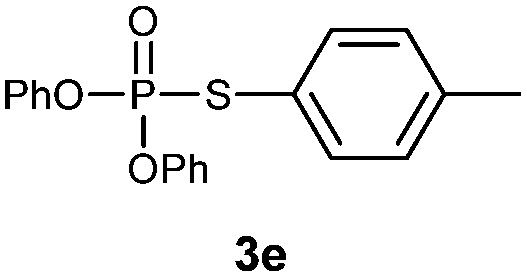
O,O-Diphenyl S-p-tolylphosphorothioate (3e).
Following the general procedure for Table 2, using 4-methylbenzenethiol (0.124 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3e as a yellow oil (160 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ 2.33 (s, 3 H), 7.11–7.21 (m, 8 H), 7.30–7.38 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 21.1, 115.4, 120.4 (d, J = 5.5 Hz), 125.5, 129.2, 129.7, 130.2 (d, J = 2.8 Hz), 135.2 (d, J = 5.5 Hz), 140.0, 150.3 (d, J = 9.1 Hz); 31P NMR (162 MHz, CDCl3) δ 15.93; IR (CHCl3): ν 3097, 3072, 3043, 2962, 2922, 2869, 1942, 1906, 1865, 1639, 1594, 1488, 1456, 1400, 1378, 1263, 1158, 1071, 1025, 919, 809, 757, 689 cm−1; HRMS-EI calcd for C19H17O3PS: 356.0636, found: 356.0630.
O,O-Dibutyl S-phenyl phosphorothioate (3f)8.
Following the general procedure for Table 2, using thiophenol (0.103 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3f as a yellow oil (257 mg, 85% yield). 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 7.4 Hz, 6 H), 1.30–1.40 (m, 4 H), 1.59–1.66 (m, 4 H), 4.06–4.17 (m, 4 H), 7.33–7.36 (m, 3 H), 7.55–7.58 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 13.4, 18.5, 31.9 (d, J = 7.3 Hz), 67.6 (d, J = 6.3 Hz), 126.5, 128.8 (d, J = 2.8 Hz), 129.1 (d, J = 1.9 Hz), 134.3 (d, J = 4.5 Hz); 31P NMR (162 MHz, CDCl3) δ 23.60; IR (CHCl3): ν 3063, 2961, 2934, 2874, 1641, 1583, 1471, 1442, 1383, 1258, 1194, 1120, 1060, 1021, 819, 783, 747, 692 cm−1.
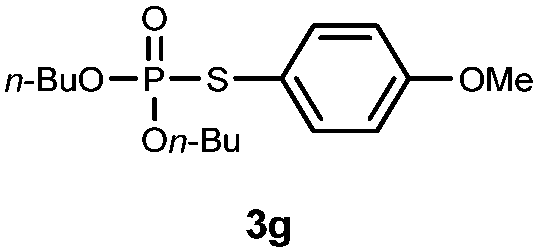
O,O-Dibutyl S-(4-methoxyphenyl)phosphorothioate (3g).
Following the general procedure for Table 2, using 4-methoxythiophenol (0.123 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3g as a yellow oil (310 mg, 94% yield). 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.4 Hz, 6 H), 1.33–1.41 (m, 4 H), 1.60–1.66 (m, 4 H), 3.80 (s, 3 H), 4.06–4.15 (m, 4 H), 6.86–6.88 (m, 2 H), 7.46–7.48 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 13.4, 18.5, 32.0 (d, J = 9.4 Hz), 55.2, 67.6 (d, J = 7.3 Hz), 114.8 (d, J = 1.8 Hz), 116.4 (d, J = 8.2 Hz), 136.2 (d, J = 2.7 Hz), 160.3; 31P NMR (162 MHz, CDCl3) δ 24.32; IR (CHCl3): ν 3070, 2960, 2937, 1634, 1593, 1574, 1494, 1463, 1383, 1290, 1245, 1176, 1043, 1016, 1008, 993, 906, 830, 798, 731 cm−1; HRMS-EI calcd for C15H25O4PS: 332.1211, found: 332.1204.
S-(2-Bromophenyl) O,O-dibutylphosphorothioate (3h).
Following the general procedure for Table 2, using 2-bromothiophenol (0.124 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3h as a yellow oil (366 mg, 96% yield). 1H NMR (400 MHz, CDCl3) δ 0.88–0.93 (m, 6 H), 1.32–1.41 (m, 4 H), 1.61–1.67 (m, 4 H), 4.11–4.20 (m, 4 H), 7.17–7.21 (m, 1 H), 7.28–7.32 (m, 1 H), 7.32–7.63 (m, 1 H), 7.79–7.81 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3, 18.4, 31.9 (d, J = 7.3 Hz), 67.9 (d, J = 6.4 Hz), 127.8 (d, J = 1.8 Hz), 128.1 (d, J = 7.3 Hz), 128.5 (d, J = 5.5 Hz), 129.9, 133.3, 136.1 (d, J = 4.5 Hz); 31P NMR (162 MHz, CDCl3) δ 22.03; IR (CHCl3): ν 3062, 2962, 2934, 2874, 1633, 1562, 1452, 1428, 1383, 1260, 1149, 1109, 1060, 1018, 900, 836, 753 cm−1; HRMS-EI calcd for C17H21BrO3PS: 380.0211, found: 380.0207.
O,O-Dibutyl S-naphthalen-1-yl phosphorothioate (3i).
Following the general procedure for Table 2, using 1-naphthalenethiol (0.140 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3i as a yellow oil (290 mg, 82% yield). 1H NMR (400 MHz, CDCl3) δ 0.79–0.83 (m, 6 H), 1.18–1.27 (m, 4 H), 1.45–1.52 (m, 4 H), 3.97–4.10 (m, 4 H), 7.42 (t, J = 7.8 Hz, 1 H), 7.50 (t, J = 7.2 Hz, 1 H), 7.56 (t, J = 7.6 Hz, 1 H), 7.82–7.91 (m, 3 H), 8.52 (d, J = 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3, 18.3, 32.0 (d, J = 6.4 Hz), 67.7 (d, J = 7.3 Hz), 123.5 (d, J = 8.2 Hz), 125.4 (d, J = 3.6 Hz), 125.8, 126.2, 126.8, 128.3, 130.0 (d, J = 3.6 Hz), 134.0 (d, J = 1.8 Hz), 134.5 (d, J = 3.6 Hz), 135.0 (d, J = 5.4 Hz); 31P NMR (162 MHz, CDCl3) δ 23.32; IR (CHCl3): ν 3056, 2961, 2933, 2873, 1267, 1590, 1564, 1432, 1381, 1256, 1146, 1120, 1060, 985, 903, 799, 772, 730 cm−1; HRMS-EI calcd for C18H25O3PS: 352.1262, found: 352.1256.
O,O-Dibutyl S-(4-(trifluoromethyl)phenyl)phosphorothioate (3j).
Following the general procedure for Table 2, using 4-trifluoromethylthiophenol (0.137 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3j as a colorless oil (226 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 0.88–0.94 (m, 6 H), 1.26–1.40 (m, 4 H), 1.61–1.68 (m, 4 H), 4.07–4.21 (m, 4 H), 7.60 (d, J = 8.4 Hz, 1 H), 7.72 (d, J = 8.0 Hz, 1 H), 7.78–7.84 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 13.3, 18.4, 31.9 (d, J = 6.8 Hz), 67.9 (d, J = 6.6 Hz), 123.6 (q, J = 270.9 Hz), 125.9, 130.7 (q, J = 32.8 Hz), 131.9 (d, J = 6.6 Hz), 134.1 (d, J = 5.4 Hz); 31P NMR (162 MHz, CDCl3) δ 22.06; 19F NMR (376 MHz, CDCl3) δ −64.5 (s); IR (CHCl3): ν 2963, 2936, 2876, 1640, 1608, 1465, 1401, 1326, 1264, 1168, 1130, 1106, 1091, 1063, 1015, 889, 836, 772, 702 cm−1; HRMS-EI calcd for C15H22F3O3PS: 370.0979, found: 370.0970.
O,O-Dibutyl S-(3-(trifluoromethyl)phenyl)phosphorothioate (3k).
Following the general procedure for Table 2, using 3-trifluoromethylthiophenol (0.140 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3k as a yellow oil (316 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ 0.88–0.96 (m, 6 H), 1.30–1.40 (m, 4 H), 1.60–1.67 (m, 4 H), 4.08–4.20 (m, 4 H), 7.49 (t, J = 7.8 Hz, 1 H), 7.61–7.63 (m, 1 H), 7.78–7.84 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 13.1, 18.4, 31.8 (d, J = 6.8 Hz), 67.8 (d, J = 6.8 Hz), 123.3 (q, J = 271.1 Hz), 125.4, 128.2 (d, J = 6.8 Hz), 129.5, 130.8, 131.4 (q, J = 32.0 Hz), 137.5 (d, J = 4.8 Hz); 31P NMR (162 MHz, CDCl3) δ 22.27; 19F NMR (376 MHz, CDCl3) δ −64.5 (s); IR (CHCl3): ν 3076, 2963, 2876, 1465, 1423, 1384, 1324, 1263, 1169, 1130, 1069, 986, 899, 799, 697 cm−1; HRMS-EI calcd for C15H22F3O3PS: 370.0979, found: 370.0970.
O,O-Dibutyl S-(4-fluorophenyl)phosphorothioate (3l).
Following the general procedure for Table 2, using 4-fluorothiophenol (0.107 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3l as a colorless oil (190 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.4 Hz, 6 H), 1.31–1.41 (m, 4 H), 1.60–1.67 (m, 4 H), 4.04–4.18 (m, 4 H), 7.03–7.07 (m, 2 H), 7.53–7.57 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 13.3, 18.4, 31.9 (d, J = 7.4 Hz), 67.7 (d, J = 6.6 Hz), 116.3 (dd, J = 2.6, 22.5 Hz), 121.6 (q, J = 9.2 Hz), 136.5 (q, J = 4.5 Hz), 163.1 (dd, J = 3.0, 248.1 Hz); 31P NMR (162 MHz, CDCl3) δ 23.37; 19F NMR (376 MHz, CDCl3) δ −113.3 (s); IR (CHCl3): ν 2962, 2935, 2875, 1640, 1590, 1491, 1465, 1383, 1258, 1232, 1158, 1060, 1017, 988, 891, 834, 783, 727 cm−1; HRMS-EI calcd for C14H22FO3PS: 320.1011, found: 320.1020.
O,O-Dibutyl S-p-tolylphosphorothioate (3m).
Following the general procedure for Table 2, using 4-methylbenzenethiol (0.124 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3m as a colorless oil (262 mg, 83% yield). 1H NMR (400 MHz, CDCl3) δ 0.88–0.92 (m, 6 H), 1.31–1.40 (m, 4 H), 1.59–1.66 (m, 4 H), 2.33 (s, 3 H), 4.05–4.17 (m, 4 H), 7.12–7.15 (m, 2 H), 7.42–7.45 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 13.3, 18.4, 20.9, 31.9 (d, J = 7.3 Hz), 67.5 (d, J = 6.4 Hz), 122.6 (d, J = 6.3 Hz), 129.9, 134.4 (d, J = 5.4 Hz), 139.0; 31P NMR (162 MHz, CDCl3) δ 24.00; IR (CHCl3): ν 3025, 2961, 2934, 2874, 1493, 1464, 1382, 1257, 1119, 1060, 1018, 893, 810, 728 cm−1; HRMS-EI calcd for C15H25O3PS: 316.1262, found: 316.1260.
O,O-Dimethyl S-phenyl phosphorothioate (3n).
Following the general procedure for Table 2, using thiophenol (0.103 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3n as a yellow oil (157 mg, 72% yield). 1H NMR (400 MHz, CDCl3) δ 3.80–3.84 (m, 6 H), 7.34–7.38 (m, 3 H), 7.55–7.58 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 54.1 (d, J = 5.4 Hz), 125.8 (d, J = 7.3 Hz), 129.0 (d, J = 2.8 Hz), 129.3 (d, J = 2.7 Hz), 134.4 (d, J = 5.5 Hz); 31P NMR (162 MHz, CDCl3) δ 26.81; IR (CHCl3): ν 3060, 3004, 2954, 2919, 2851, 1962, 1887, 1810, 1721, 1638, 1580, 1474, 1443, 1257, 1182, 1020, 918, 831, 796, 768, 694 cm−1; HRMS-EI calcd for C8H11O3PS: 218.0167, found: 218.0160.
O,O-Dimethyl S-naphthalen-1-yl phosphorothioate (3o).
Following the general procedure for Table 2, using 1-naphthalenethiol (0.140 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3o as a yellow oil (191 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ 3.71–3.76 (m, 6 H), 7.42 (t, J = 7.8 Hz, 1 H), 7.50 (t, J = 7.4 Hz, 1 H), 7.59 (t, J = 7.4 Hz, 1 H), 7.81–7.88 (m, 3 H), 8.50 (d, J = 8.4 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 54.3 (d, J = 6.4 Hz), 122.8 (d, J = 8.2 Hz), 125.5 (d, J = 5.5 Hz), 126.4, 127.1, 128.5, 130.3 (d, J = 3.7 Hz), 134.1 (d, J = 1.9 Hz), 134.5 (d, J = 3.7 Hz), 135.1 (d, J = 5.4 Hz); 31P NMR (162 MHz, CDCl3) δ 26.31; IR (CHCl3): ν 3056, 3006, 2954, 2851, 1722, 1637, 1590, 1564, 1503, 1456, 1380, 1336, 1256, 1183, 1020, 914, 831, 801, 771, 601, 569 cm−1; HRMS-EI calcd for C12H13O3PS: 268.0323, found: 268.0320.
S-(4-Methoxyphenyl) O,O-dimethyl phosphorothioate (3p).
Following the general procedure for Table 2, using 4-methoxythiophenol (0.123 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3p as a yellow oil (154 mg, 62% yield). 1H NMR (400 MHz, CDCl3) δ 3.79–3.84 (m, 9 H), 6.87–6.90 (m, 2 H), 7.45–7.48 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 54.0 (d, J = 6.3 Hz), 55.2, 115.0 (d, J = 1.8 Hz), 115.8 (d, J = 6.4 Hz), 136.2 (d, J = 4.5 Hz), 160.5; 31P NMR (162 MHz, CDCl3) δ 27.46; IR (CHCl3): ν 3070, 3007, 2955, 2849, 1639, 1592, 1494, 1462, 1408, 1291, 1250, 1177, 1024, 832, 791, 768 cm−1; HRMS-EI calcd for C9H13O4PS: 248.0272, found: 248.0269.
O,O-Dimethyl S-(3-(trifluoromethyl)phenyl)phosphorothioate (3q).
Following the general procedure for Table 2, using 3-trifluoromethylthiophenol (0.140 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3q as a colorless oil (216 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 3.82–3.87 (m, 6 H), 7.50 (t, J = 7.8 Hz, 1 H), 7.63–7.65 (m, 1 H), 7.77–7.82 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 54.3 (d, J = 6.0 Hz), 123.3 (q, J = 271.1 Hz), 125.8, 127.5 (d, J = 2.7 Hz), 129.8 (d, J = 1.8 Hz), 131.0 (t, J = 4.3 Hz), 131.6 (q, J = 32.8 Hz), 137.7 (d, J = 4.8 Hz); 31P NMR (162 MHz, CDCl3) δ 25.47; 19F NMR (376 MHz, CDCl3) δ −64.4 (s); IR (CHCl3): ν 2958, 2855, 1640, 1449, 1423, 1325, 1263, 1169, 1127, 1022, 898, 833, 799, 766, 697 cm−1; HRMS-EI calcd for C9H10F3O3PS: 286.0040, found: 286.0033.
O,O-Dimethyl S-(4-(trifluoromethyl)phenyl)phosphorothioate (3r).
Following the general procedure for Table 2, using 4-trifluoromethylthiophenol (0.137 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3r as a colorless oil (265 mg, 93% yield). 1H NMR (400 MHz, CDCl3) δ 3.84–3.89 (m, 6 H), 7.20–7.25 (m, 1 H), 7.30–7.34 (m, 1 H), 7.64–7.66 (m, 1 H), 7.74–7.77 (m, 1 H); 13C NMR (150 MHz, CDCl3) δ 54.3 (d, J = 6.0 Hz), 128.0 (d, J = 2.4 Hz), 128.0 (dd, J = 7.8, 106.2 Hz), 130.3 (d, J = 3.0 Hz), 133.4 (d, J = 2.4 Hz), 136.4 (d, J = 4.4 Hz); 31P NMR (162 MHz, CDCl3) δ 25.14; 19F NMR (376 MHz, CDCl3) δ −64.4 (s); IR (CHCl3): ν 3062, 3003, 2954, 2851, 1636, 1562, 1450, 1427, 1257, 1182, 1109, 1030, 833, 757, 720, 646 cm−1; HRMS-EI calcd for C9H10F3O3PS: 286.0040, found: 286.0044.
S-(2-Bromophenyl) O,O-dimethyl phosphorothioate (3s).
Following the general procedure for Table 2, using 2-bromothiophenol (0.124 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3s as a yellow oil (208 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 3.83–3.87 (m, 6 H), 7.60–7.62 (m, 2 H), 7.70–7.72 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 54.2 (d, J = 5.5 Hz), 122.2, 124.9, 126.0, 130.7, 131.1, 134.2 (d, J = 5.4 Hz); 31P NMR (162 MHz, CDCl3) δ 25.30; IR (CHCl3): ν 3097, 3046, 3005, 2958, 2855, 1688, 1576, 1496, 1459, 1401, 1328, 1264, 1173, 1091, 1011, 837, 759, 767 cm−1; HRMS-EI calcd for C8H10BrO3PS: 295.9272, found: 295.9265.
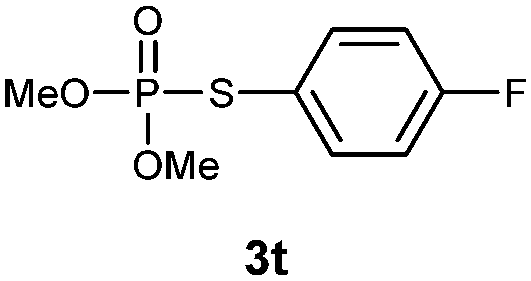
S-(4-Fluorophenyl) O,O-dimethyl phosphorothioate (3t).
Following the general procedure for Table 2, using 4-fluorothiophenol (0.107 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 3t as a colorless oil (156 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ 3.80–3.85 (m, 6 H), 7.04–7.09 (m, 2 H), 7.53–7.57 (m, 2 H); 13C NMR (150 MHz, CDCl3) δ 54.1 (d, J = 6.6 Hz), 116.5 (d, J = 21.5 Hz), 120.9 (q, J = 3.5 Hz), 136.6 (q, J = 12.8 Hz), 163.2 (dd, J = 3.2, 248.9 Hz); 31P NMR (162 MHz, CDCl3) δ 26.51; 19F NMR (376 MHz, CDCl3) δ −112.8 (s); IR (CHCl3): ν 2957, 2853, 1642, 1590, 1491, 1460, 1399, 1258, 1232, 1183, 1160, 1022, 835, 792, 767 cm−1; HRMS-EI calcd for C8H10FO3PS: 236.0072, found: 236.0081.
General procedure for Table 3
A Schlenk tube equipped with a magnetic stir bar was charged with N-chlorosuccinimide (1.1 mmol), alkyl thiol 4 (1.0 mmol) and MeCN (1.5 mL). After the mixture was stirred for 20 min, phosphonate 2 (1.0 mmol) was added by a syringe under a nitrogen-filled balloon. After being stirred for another 10 min at room temperature and diluted with ethyl acetate (20 mL) the resulting solution was directly filtered through a pad of silica gel and then washed with ethyl acetate (20 mL) and concentrated to give the crude material which was then purified by column chromatography (SiO2, hexane) to yield 5.
S-Cyclohexyl O,O-diphenylphosphorothioate (5a).
Following the general procedure for Table 3, using cyclohexanethiol (0.126 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5a as a colorless oil (306 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 1.19–1.37 (m, 3 H), 1.41–1.53 (m, 3 H), 1.64–1.69 (m, 2 H), 1.96–2.00 (m, 2 H), 3.44–3.47 (m, 2 H), 7.17–7.21 (m, 2 H), 7.25–7.36 (m, 8 H); 13C NMR (100 MHz, CDCl3) δ 24.9, 25.6, 34.9 (d, J = 4.4 Hz), 46.9 (d, J = 3.6 Hz), 120.5 (d, J = 4.5 Hz), 125.3, 129.5, 150.0 (d, J = 8.2 Hz); 31P NMR (162 MHz, CDCl3) δ 21.64; IR (CHCl3): ν 3069, 3043, 2934, 2855, 2793, 1592, 1496, 1480, 1451, 1340, 1275, 1241, 1193, 1180, 1166, 1157, 1100, 1025, 1006, 996, 950, 940, 903, 814, 772, 689 cm−1; HRMS-EI calcd for C18H21O3PS: 348.0949, found: 348.0956.
S-(2-Methylbutyl) O,O-diphenylphosphorothioate (5b).
Following the general procedure for Table 3, using 2-methyl-1-butanethiol (0.136 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5b as a colorless oil (291 mg, 87% yield). 1H NMR (400 MHz, CDCl3) δ 0.81 (t, J = 7.4 Hz, 3 H), 0.88–0.90 (m, 3 H), 1.11–1.18 (m, 1 H), 1.35–1.41 (m, 1 H), 1.55–1.60 (m, 1 H), 2.76–2.84 (m, 1 H), 2.91–2.99 (m, 1 H), 7.17–7.21 (m, 2 H), 7.28–7.36 (m, 8 H); 13C NMR (100 MHz, CDCl3) δ 10.9, 18.2, 28.0, 35.4 (d, J = 5.4 Hz), 38.1 (d, J = 3.6 Hz), 120.4 (d, J = 4.6 Hz), 125.4, 129.6, 150.0 (d, J = 8.2 Hz); 31P NMR (162 MHz, CDCl3) δ 22.58; IR (CHCl3): ν 3070, 3044, 2963, 2930, 2876, 1591, 1489, 1458, 1430, 1380, 1333, 1272, 1185, 1161, 1071, 1025, 1007, 929, 770, 689 cm−1; HRMS-EI calcd for C17H21O3PS: 336.0949, found: 336.0952.
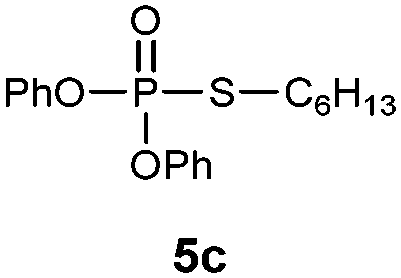
S-Hexyl O,O-diphenylphosphorothioate (5c).
Following the general procedure for Table 3, using 1-hexanethiol (0.145 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5c as a colorless oil (261 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 0.83–0.87 (m, 3 H), 1.17–1.33 (m, 6 H), 1.55–1.62 (m, 2H), 2.89–2.96 (m, 2 H), 7.18–7.25 (m, 2 H), 7.28–7.38 (m, 8 H); 13C NMR (100 MHz, CDCl3) δ 13.8, 22.2, 27.8, 30.4 (d, J = 5.4 Hz), 30.9, 31.6 (d, J = 3.7 Hz), 120.5 (d, J = 4.6 Hz), 125.4, 129.6, 150.0 (d, J = 8.2 Hz); 31P NMR (162 MHz, CDCl3) δ 22.23; IR (CHCl3): ν 3069, 3044, 2957, 2930, 2857, 1591, 1489, 1457, 1378, 1272, 1185, 1161, 1071, 1025, 930, 770, 689 cm−1; HRMS-EI calcd for C18H23O3PS: 350.1106, found: 350.1101.
S-Benzyl O,O-diphenylphosphorothioate (5d).
Following the general procedure for Table 3, using α-toluenethiol (0.120 mL, 1.0 mmol) and diphenylphosphonate (0.192 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5d as a yellow oil (263 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ 4.11–4.15 (m, 2 H), 7.18–7.28 (m, 11 H), 7.30–7.35 (m, 4 H); 13C NMR (100 MHz, CDCl3) δ 35.7 (d, J = 3.6 Hz), 120.6 (d, J = 4.6 Hz), 125.6, 127.8, 128.7, 128.9, 129.7, 136.4 (d, J = 6.4 Hz), 150.1 (d, J = 8.2 Hz); 31P NMR (162 MHz, CDCl3) δ 20.69; IR (CHCl3): ν 3065, 3032, 2942, 2851, 1950, 1867, 1637, 1592, 1488, 1455, 1272, 1241, 1160, 1071, 1025, 1007, 922, 703, 691 cm−1; HRMS-EI calcd for C19H17O3PS: 356.0636, found: 356.0644.
O,O-Dibutyl S-cyclohexylphosphorothioate (5e).
Following the general procedure for Table 3, using cyclohexanethiol (0.126 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5e as a colorless oil (226 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ 0.86–0.96 (m, 6 H), 1.24–1.60 (m, 10 H), 1.65–1.77 (m, 6 H), 2.05–2.09 (m, 2 H), 3.26–3.29 (m, 1 H), 4.01–4.012 (m, 4 H); 13C NMR (100 MHz, CDCl3) δ 13.4, 18.6, 25.1, 25.7, 32.0 (d, J = 7.3 Hz), 35.1 (d, J = 5.5 Hz), 45.3 (d, J = 3.6 Hz), 66.9 (d, J = 6.4 Hz); 31P NMR (162 MHz, CDCl3) δ 28.01; IR (CHCl3): ν 2960, 2933, 2873, 2856, 1252, 1148, 1121, 1061, 1020, 979, 888, 784, 727 cm−1; HRMS-EI calcd for C14H29O3PS: 308.1575, found: 308.1570.
O,O-Dibutyl S-(2-methylbutyl)phosphorothioate (5f).
Following the general procedure for Table 3, using 2-methyl-1-butanethiol (0.136 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5f as a colorless oil (271 mg, 92% yield). 1H NMR (400 MHz, CDCl3) δ 0.89–1.00 (m, 12 H), 1.21–1.28 (m, 1 H), 1.38–1.52 (m, 5 H), 1.65–1.72 (m, 5 H), 2.66–2.74 (m, 1 H), 2.82–2.88 (m, 1 H), 4.02–4.16 (m, 4 H); 13C NMR (100 MHz, CDCl3) δ 10.8, 13.2, 18.2, 18.4, 28.0, 31.8 (d, J = 7.3 Hz), 35.3 (d, J = 6.4 Hz), 37.1 (d, J = 4.5 Hz), 66.7 (d, J = 6.3 Hz); 31P NMR (162 MHz, CDCl3) δ 29.45; IR (CHCl3): ν 2962, 2934, 2875, 1258, 1064, 1020, 980, 784, 729 cm−1; HRMS-EI calcd for C13H29O3PS: 296.1575, found: 296.1576.
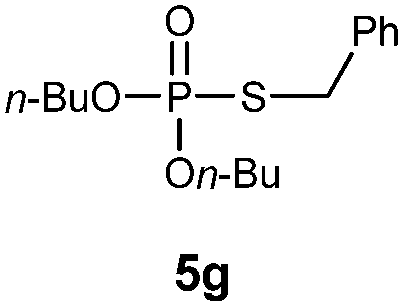
S-Benzyl O,O-dibutylphosphorothioate (5g).
Following the general procedure for Table 3, α-toluenethiol (0.120 mL, 1.0 mmol) and dibutylphosphonate (0.203 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5g as a yellow oil (224 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.4 Hz, 6 H), 1.32–1.41 (m, 4 H), 1.57–1.64 (m, 4 H), 3.92–4.08 (m, 6 H), 7.23–7.37 (m, 5 H); 13C NMR (100 MHz, CDCl3) δ 13.4, 18.5, 31.9 (d, J = 7.3 Hz), 34.7 (d, J = 3.6 Hz), 67.0 (d, J = 5.4 Hz), 127.4, 128.4, 128.7, 137.4 (d, J = 5.4 Hz); 31P NMR (162 MHz, CDCl3) δ 27.49; IR (CHCl3) : ν 3089, 3064, 3031, 2961, 2935, 2874, 1639, 1496, 1457, 1432, 1383, 1260, 1149, 1120, 1061, 980, 894, 780, 730, 701 cm−1; HRMS-EI calcd for C15H25O3PS: 316.1262, found: 316.12627.
S-Cyclohexyl O,O-dimethyl phosphorothioate (5h).
Following the general procedure for Table 3, using cyclohexanethiol (0.126 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5h as a yellow oil (153 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 1.22–1.44 (m, 3 H), 1.48–1.61 (m, 3 H), 1.73–1.79 (m, 2 H), 2.06–2.10 (m, 2 H), 3.23–3.31 (m, 1 H), 3.75–3.78 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 24.9, 25.6, 34.9 (d, J = 5.4 Hz), 45.3 (d, J = 2.8 Hz), 53.4 (d, J = 5.5 Hz); 31P NMR (162 MHz, CDCl3) δ 31.73; IR (CHCl3): ν 2934, 2854, 1641, 1449, 1342, 1250, 1182, 1019, 888, 827, 773, 728 cm−1; HRMS-EI calcd for C8H17O3PS: 224.0636, found: 224.0633.
O,O-Dimethyl S-(2-methylbutyl)phosphorothioate (5i).
Following the general procedure for Table 3, using 2-methyl-1-butanethiol (0.136 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5i as a yellow oil (189 mg, 89% yield). 1H NMR (400 MHz, CDCl3) δ 0.92 (t, J = 6.8 Hz, 3 H), 1.00 (d, J = 6.4 Hz, 3 H), 1.21–1.28 (m, 1 H), 1.46–1.53 (m, 1 H), 1.66–1.71 (m, 1 H), 2.67–2.75 (m, 1 H), 2.82–2.89 (m, 1 H), 3.78–3.83 (m, 6 H); 13C NMR (100 MHz, CDCl3) δ 10.9, 18.3, 28.0, 35.4 (d, J = 5.5 Hz), 37.2 (d, J = 3.6 Hz), 53.4 (d, J = 5.5 Hz); 31P NMR (162 MHz, CDCl3) δ 32.80; IR (CHCl3): ν 2961, 2931, 2877, 2853, 1638, 1461, 1380, 1334, 1258, 1182, 1022, 925, 829, 794, 774, cm−1; HRMS-EI calcd for C7H17O3PS: 212.0636, found: 212.0629.
S-Benzyl O,O-dimethyl phosphorothioate (5j).
Following the general procedure for Table 3, using α-toluenethiol (0.120 mL, 1.0 mmol) and dimethylphosphonate (0.094 mL, 1.0 mmol), and then purification by column chromatography (SiO2, hexane) provides 5j as a yellow oil (156 mg, 67% yield). 1H NMR (400 MHz, CDCl3) δ 3.66–3.70 (m, 6 H), 4.02 (d, J = 14.4 Hz, 2 H), 7.27–7.37 (m, 5 H); 13C NMR (100 MHz, CDCl3) δ 34.7 (d, J = 3.7 Hz), 53.5 (d, J = 5.5 Hz), 127.5, 128.5, 128.7, 137.2 (d, J = 5.4 Hz); 31P NMR (162 MHz, CDCl3) δ 30.94; IR (CHCl3): ν 3063, 3030, 2953, 2851, 1638, 1496, 1455, 1261, 1183, 1015, 920, 829, 771, 702 cm−1; HRMS-EI calcd for C9H13O3PS: 232.0323, found: 232.0318.
Acknowledgements
The National Science Council, Taiwan (NSC 101-2113-M-005-008-MY3), the National Chung Hsing University and the Center of Nanoscience and Nanotechnology (NCHU) are gratefully acknowledged for financial support. We also thank Prof. Fung-E Hong (NCHU) for sharing his GC-MS instruments. C.F.L. is a Golden-Jade Fellow of Kenda Foundation, Taiwan.
References
-
(a) T. S. Kumar, T. Yang, S. Mishra, C. Cronin, S. Chakraborty, J.-B. Shen, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2013, 56, 902–914 CrossRef CAS PubMed;
(b) R. Xie, Q. Zhao, T. Zhang, J. Fang, X. Mei, J. Ning and Y. Tang, Bioorg. Med. Chem., 2013, 21, 278–282 CAS.
-
(a) M. Piekutowska and Z. Pakulski, Carbohydr. Res., 2008, 343, 785–792 CrossRef CAS PubMed;
(b) M. Piekutowska and Z. Pakulski, Tetrahedron Lett., 2007, 48, 8482–8486 CrossRef CAS PubMed , and more references cited therein.
- T. M. Timperley, S. A. Saunders, J. Szpalek and M. J. Waters, J. Fluorine Chem., 2003, 119, 161–171 CrossRef.
- B. Kaboudin, Tetrahedron Lett., 2002, 43, 8713–8714 CrossRef CAS.
- R. Harveyh, E. Jacobson and E. Jensen, J. Am. Chem. Soc., 1963, 85, 1623–1626 CrossRef.
- A. J. Parker and N. Kharasch, Chem. Rev., 1959, 59, 583–628 CrossRef CAS.
-
O. Foss, Organic Sulfur Compounds, Pergamon Press, London, 1959, vol. I Search PubMed.
- Y.-X. Gao, G. Tang, Y. Cao and Y.-F. Zhao, Synthesis, 2009, 1081–1086 Search PubMed.
- J.-H. Cheng, C. Ramesh, H.-L. Kao, Y.-J. Wang, C.-C. Chan and C.-F. Lee, J. Org. Chem., 2012, 77, 10369–10374 CrossRef CAS PubMed.
-
(a) L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474–15476 CrossRef CAS PubMed;
(b) Y. Wei, S. Lin and F. Liang, Org. Lett., 2012, 14, 4202–4205 CrossRef CAS PubMed;
(c) L. Zhou, C. K. Tan, J. Zhou and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 10245–10247 CrossRef CAS PubMed;
(d) D. Kalyani, A. R. Dick, W. Q. Anani, S. Melanie and M. S. Sanford, Org. Lett., 2006, 8, 2523–2526 CrossRef CAS PubMed;
(e) D. W. Kim, H. Y. Choi, K. J. Lee and D. Y. Chi, Org. Lett., 2001, 3, 445–447 CrossRef CAS.
-
(a) X. Liu, Y. Zhang, L. Wang, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2008, 73, 6207–6212 CAS;
(b) T. J. Barker and E. R. Jarvo, Angew. Chem., Int. Ed., 2011, 50, 8325–8328 CrossRef CAS PubMed;
(c) M. Jithunsa, M. Ueda and O. Miyata, Org. Lett., 2011, 13, 518–521 CAS;
(d) M. Sai and S. Matsubara, Org. Lett., 2011, 13, 4676–4679 CrossRef CAS PubMed;
(e) J. A. Tunge and S. R. Mellegaard, Org. Lett., 2004, 6, 1205–1207 CrossRef CAS PubMed.
-
(a) K. M. Schlosser, A. P. Krasutsky, H. W. Hamilton, J. S. Reed and K. Sexton, Org. Lett., 2004, 6, 819–821 CrossRef CAS PubMed;
(b) F. Kroll, R. Morphy, D. Rees and D. Gani, Tetrahedron Lett., 1997, 38, 8573–8576 CrossRef CAS;
(c) J. S. Yadav, B. V. S. Reddy, R. Jain and G. Baishya, Tetrahedron Lett., 2008, 49, 3015–3018 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data for new products. See DOI: 10.1039/c3gc41839a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.