Synthesis of antimalarial trioxanes via continuous photo-oxidation with 1O2 in supercritical CO2†
Jessica F. B.
Hall
,
Richard A.
Bourne
,
Xue
Han
,
James H.
Earley
,
Martyn
Poliakoff
* and
Michael W.
George
*
School of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: martyn.poliakoff@nottingham.ac.uk; michael.george@nottingham.ac.uk; Tel: +44 (0)115 951 3386
First published on 15th November 2012
Abstract
The oxidation of an allylic alcohol to its hydroperoxides represents a key step in the synthesis of a series of spirobicyclic, antimalarial trioxanes. Herein, we investigate the continuous photo-oxidation of an allylic alcohol with 1O2 in scCO2, as a ‘green’ alternative to conventional methods, and examine the remaining two steps in the synthesis of antimalarial trioxanes from readily available starting materials.
Malaria is one of the most devastating infectious diseases in the world, killing an estimated 655
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people in 2010 alone.1 Of crucial concern is the fact that the virus Plasmodium falciparum, responsible for the most lethal form of malaria, malaria tropica, is becoming increasingly resistant to former front-line quinine based antimalarials2 and there is a need for new antimalarial compounds to be produced.
000 people in 2010 alone.1 Of crucial concern is the fact that the virus Plasmodium falciparum, responsible for the most lethal form of malaria, malaria tropica, is becoming increasingly resistant to former front-line quinine based antimalarials2 and there is a need for new antimalarial compounds to be produced.
One example is artemisinin,3,4 a naturally occurring lactone with a 1,2,4-trioxane core structure.5,6 Synthetic derivatives of artemisinin also possess excellent antimalarial activity and, despite concerns that P. falciparum is also showing signs of resistance to these compounds, artemisinin and its analogues are still the primary treatments for falciparum malaria.3,7 Artemisinin is still commonly extracted from the plant Artemisia annua because its synthesis is labour-intensive3 and unsuited to large-scale production.
A feature common to artemisinin and several other potential antimalarials is the trioxane moiety, shown in Scheme 1. Photochemically generated 1O2 offers a convenient way of introducing such a motif. However, the excitation of ground state molecular oxygen, 3O2 to 1O2, requires the use of photocatalysts. The highly reactive nature of 1O2 can mean that there are problems with the scale-up of such processes as industrially acceptable solvents must be non-flammable and inefficient 1O2 quenchers.
![Synthetic route to spirobicyclic trioxanes, 4a and 4b, highlighting the photo-oxidation step. 1: mesityl oxide; 2: 4-methylpent-3-en-2-ol; 3: hydroperoxides. Here, n = 1, cyclopentanone, or 2, cyclohexanone to give 8-isopropenyl-9-methyl-6,7,10-trioxa-spiro[4.5]decane (4a) or -isopropenyl-4-methyl-1,2,5-trioxa-spiro[5.5]undecane (4b) respectively.](/image/article/2013/GC/c2gc36711d/c2gc36711d-s1.gif) | ||
| Scheme 1 Synthetic route to spirobicyclic trioxanes, 4a and 4b, highlighting the photo-oxidation step. 1: mesityl oxide; 2: 4-methylpent-3-en-2-ol; 3: hydroperoxides. Here, n = 1, cyclopentanone, or 2, cyclohexanone to give 8-isopropenyl-9-methyl-6,7,10-trioxa-spiro[4.5]decane (4a) or -isopropenyl-4-methyl-1,2,5-trioxa-spiro[5.5]undecane (4b) respectively. | ||
Recent work has resulted in the partial scale-up of the photo-oxidation of the natural product artemisinic acid to artemisinin using CH2Cl2 as the solvent.8 Apart from the nature of the solvent, the problem of obtaining the artemisinic acid still remains; like artemisinin, it is usually extracted from A. annua, although it can now be produced by engineered yeast.9
A number of new antimalarial trioxanes, including 4a and 4b, Scheme 1, have also been synthesised using 1O2 as a key step. In this work, the photo-oxidation procedure was conducted in a batch reactor using either non-polar solvents or a solvent-free approach with porphyrin loaded polystyrene beads.10,11 The addition of spiroadamantane motifs was found to increase antimalarial activity, however the products were hardly water soluble, indicating that a diverse approach to a variety of such compounds is necessary and that more synthetic work is required to evaluate the best antimalarial compounds.12
We have previously shown that photo-oxidation using 1O2 can be conducted continuously and safely in supercritical carbon dioxide, scCO2, which provides a non-toxic and non-flammable reaction medium and which can be used close to ambient temperature.13–15 The photo-oxidation proceeds somewhat faster in scCO2 than in many conventional solvents, probably because gaseous O2 and scCO2 are fully miscible, allowing for single phase reaction conditions. In addition, 1O2 has a relatively long lifetime in CO2 (5.1 ms at 14.7 MPa, 314 K, compared to 59 ms for CCl4).16,17 By using a flow system, the reactor volume is reduced compared to a batch reactor of similar productivity, giving a higher degree of safety in these potentially dangerous systems. Recently we also demonstrated that the photocatalyst could be recycled in a closed loop whilst maintaining a homogeneous reaction system18 by incorporating a fluorous biphase into our existing process.
In this paper, we report the synthesis of the spirobicyclic trioxanes (4a and 4b), with particular emphasis on the continuous photo-oxidation of the allylic alcohol (2), to its hydroperoxides (3), conducted in scCO2, see Scheme 1.11 The continuous conversion of mesityl oxide (1) to the allylic alcohol (2) via heterogeneously catalysed, high temperature transfer hydrogenation at ca. 20 atm pressure has also been investigated. In the final step, Lewis acid catalysed condensation of 3 with either cyclopentanone or cyclohexanone yields 4a and 4b respectively. Before scale-up of the photo-oxidation of 2, we carried out the reaction in the small scale batch reactor which had been used in our previous work.13 As before, we used the photosensitiser 5,10,15,20-tetrakis-(pentafluorophenyl)porphyrin (TPFPP) to generate 1O2. TPFPP is insoluble in 2 but, conveniently, we found that the cyclic ketones, used for the final transformation to 4, could also act as suitable co-solvents.
TPFPP, 2 and the cyclic ketone were injected into the cell prior to pressurisation to 18 MPa, to ensure single phase reaction conditions, at 40 °C with 6% O2 in CO2. A twofold excess of the cyclic ketone, with respect to 2, was used. The reaction was found to follow zero-order kinetics with respect to 2, as determined by FTIR monitoring, Fig. 1. In our scCO2 system, the reaction ran with quantitative conversion of 2 to 3, in the presence of either cyclopentanone or cyclohexanone; however 20% of 2 remained unreacted under these conditions. The oxidation of 2, Scheme 2, leads to both the syn and anti isomers of 3, the ratio of which is solvent dependent.19 With either cyclopentanone or cyclohexanone, the syn selectivity in scCO2 (d.r. 85:15) was found to be higher than in MeOH (d.r. 73:27) but lower than in CCl4 (d.r. 93:7), in agreement with the observation that non-polar solvents favour formation of the syn isomer and that the hydroxyl group of the allylic alcohol enhances selectivity.19,20
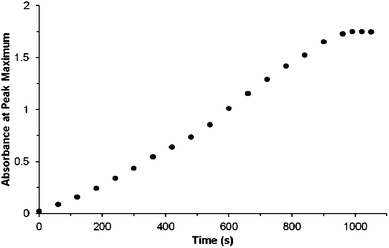 | ||
| Fig. 1 FTIR monitoring of the overall formation of the isomers of 3via the ν(C–H) band at 3083 cm−1 during the photo-oxidation of 2 in an unstirred batch reaction. The cell was irradiated with a single white 1000 lumen LED12 for 60 s and an IR spectrum was recorded before further irradiation until the peak height of the band at 3083 cm−1 no longer changed. 50 μL of 2 + cyclopentanone (1:2, mol:mol) was used with a 2 mm path length, 0.78 mL volume, cell. NMR confirms a ca. 80% yield of 3 for this experiment. Time indicates the overall irradiation time; the reaction reaches a maximum of 80% yield of 3; ca. 20% of 2 remained unconverted. | ||
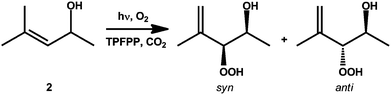 | ||
| Scheme 2 The photo-oxidation of 2 to its syn- and anti-hydroperoxides. | ||
Following these successful batch experiments, the photo-oxidation of 2 could be scaled up to a continuous flow process. Fig. 2 shows a schematic of our continuous 1O2 photo-reactor with twin irradiation vessels. As the reaction was found to proceed more slowly than in the examples we have published previously,13,14 the total system flow rate had to be lowered accordingly. In this work, the flow rate of CO2 was maintained at 0.3 mL min−1 and 2 was pumped with TPFPP and the cyclic ketone, again in a twofold excess, at a flow rate of 0.03 mL min−1. In this way, the residence time was increased to ca. 40 min with the aim to maximise conversion of 2. Under these continuous flow conditions, at 18 MPa and with 6% O2 in CO2, we obtained an 86% maximum yield of 3, slightly higher than in batch but with the same selectivity for the syn-isomer of 3 (d.r. 85:15), as determined by 1H-NMR.
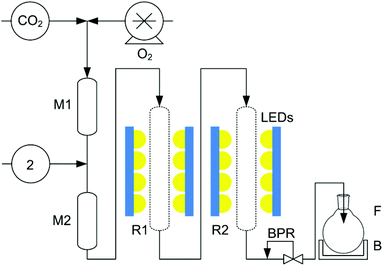 | ||
| Fig. 2 The twin photochemical flow reactor,‡ used to maximise the yield of 3. CO2 is delivered by a Jasco PU-1580-CO2 pump at 0.3 mL min−1, O2 is added using a Rheodyne dosage unit. 2 is pumped with the cyclic ketone (2:1 ketone:2, mol:mol) and TPFPP (2 mg mL−12) at 0.03 mL min−1 using a Jasco PU-980 HPLC pump. B: ice bath; BPR: back-pressure regulator (Jasco BP-1580-81); F: round bottomed flask with or without 100 mL CH2Cl2 + 0.2 mL BF3·Et2O; LEDs: light source (4 × 5 × Citizen Electronics Co. Ltd CL-L233-C13 N on an Al heat sink); M1 & M2: mixers; R1 & R2: sapphire tube reactors. The total pressure was 18 MPa. | ||
With a modification of the equipment, we found that our perfluorinated photosensitiser18 could be pumped in a fluorous solvent, HFE-7500, separately from 2 and could be recycled. The yield and selectivity for 3 was unaffected by this recycling at the same flow rates and pressure used for the single pass experiment. This biphasic technique should allow a wider range of trioxanes to be synthesised because one no longer requires a ketone which dissolves TPFPP.
The obvious next step was to investigate the possibility of carrying out each stage of the synthesis continuously. Therefore, we briefly investigated the continuous reduction of 1 to 2 and the Lewis acid catalysed condensation of 3 to yield 4, the aim being to identify any problems in the development of a ‘greener’ route to 4.
The reduction of 1 to 2 is traditionally conducted21 using LiAlH4, see ESI† for details. However, LiAlH4 is inconvenient to handle in continuous processes. Hydrogenation with H2 in scCO2 is highly efficient22,23 but is unlikely to have sufficiently high selectivity towards hydrogenation of the carbonyl group in this system. Therefore, we have focussed on the H-transfer reduction of 1, which has previously been investigated over a variety of metal oxide catalysts.24,25 It has been reported that 1 can be selectively reduced to 2 over basic MgO with a high surface area, using iso-propanol (IPA) as the H-transfer agent, with yields of 2 of 28%.24 It was suggested that the weak acid–strong base Mg2+–O2− pair sites of MgO promote a Meerwein–Ponndorf–Verley mechanism between 1 and IPA.25
We have investigated the continuous reduction of 1 with IPA with a view to improving the yield of 2 as an inexpensive alternative to LiAlH4 and we synthesised MgO with a BET surface area of ca. 84 m2 g−1. Continuous experiments† were conducted using a high pressure flow rig incorporating a reactor filled with 1 g MgO. The effect of changing the reactor parameters was investigated and it was found that, at our optimum conditions, 2 could be obtained in >50% yield over a relatively long period, as determined by online GLC, see Fig. 3. As the by-product of this reaction, acetone, cannot safely be used with peroxides in the planned next step, 2 must be separated from the IPA and acetone prior to photo-oxidation.
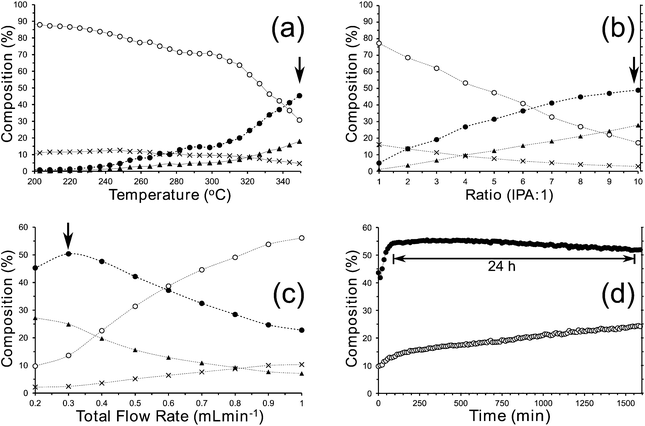 | ||
| Fig. 3 Summary of results obtained for the high pressure (2 MPa) continuous conversion of 1 to 2 using IPA over an MgO catalyst in the absence of added solvent with a ratio of IPA:1 of 10:1 unless otherwise stated. (a) Increasing the temperature where a maximum yield of 2 was observed at the highest temperature; (b) increasing the ratio IPA:1 where the highest ratio gave the highest yield; (c) increasing the flow rate of the IPA + 1 mixture where the yield reaches a maximum at 0.3 mL min−1 and (d) an extended run at 350 °C demonstrating near stable performance of the catalyst for >24 h. In (a)–(d), the traces are labelled as follows; ●: 2; ○: 1; ▲: 4-methylpent-4-en-2-ol; X: iso-mesityl oxide. | ||
The final step, conversion of 3 to 4, requires a Lewis acid catalyst. We approached this conversion in two ways. Initially, the product stream from the 1O2 flow experiment, containing 3 plus the cyclic ketone, was collected directly into a round bottomed flask containing CH2Cl2 (100 mL) and BF3·Et2O (0.2 mL) so as to quench the peroxide products immediately. The flask was immersed in an ice bath, the temperature was gradually increased to room temperature and the mixture was stirred for a further 12 h. The product, 4b, was purified by thick layer column chromatography, see ESI† for details. This gave an overall yield of 4b of ca. 25%, similar to that reported previously for this step.11
Since CH2Cl2 is not a particularly ‘green’ solvent, we tried a solvent-free approach to converting 3 to 4a. A stainless steel tubular reactor (1/4′′ o.d., 2.5 mL volume) was packed with silica/BF3 (Aldrich) and the solution of 3 in cyclopentanone was pumped through at a rate of 0.05 mL min−1. Similar conversion was achieved as with CH2Cl2, 30% yield of 4a, but BF3 leached into the solution and the conversion dropped off after 20 min.
Nevertheless, this demonstrates that an additional solvent is not required and raises the prospect of using a more robustly supported Lewis acid.
Conclusions
We have synthesised two spirobicyclic trioxanes, 4a and 4b, which have previously been shown to exhibit antimalarial activity11 and have demonstrated that the photo-oxidation of an allylic alcohol, a key step in this synthesis, can be scaled up to continuous flow in scCO2. We have also investigated the continuous H-transfer reduction of 1 to 2 and the Lewis acid catalysed final step of the transformation, with the aim of developing a fully continuous, sustainable process for the synthesis of antimalarial trioxanes.Additionally, we have indicated how the photo-oxidation can be developed further by applying our recently developed fluorous biphase technique18 for photo-oxidations, which allows the photosensitiser to be pumped into the system despite being insoluble in the starting material.
Furthermore, the ketone for the final step could be added after the photo-oxidation, opening up the possibility of using ketones with functionalities which would be destroyed in the presence of 1O2. Also, adding the ketone downstream of the reactor could be used to generate a whole series of spirobicyclic compounds by sequentially pulsing different ketones into the flowing stream of 3. Ultimately, we hope that our approach can lead to libraries of antimalarial trioxanes and an exploration of such compounds synthesised from readily available starting materials.
We thank the EPSRC DICE project for support and 3M for a sample of HFE-7500. M.W.G. acknowledges a Royal Society Wolfson Merit Award. We thank M. Dellar, M. Guyler, D. Litchfield, R. Wilson and P. Fields for technical support, S. Aslam for NMR assistance, E. Masika for BET analysis and Z. Amara for helpful discussions.
Notes and references
- M. Aregawi, R. Cibulskis, M. Lynch and R. Williams, World Malaria Report 2011.
- S. Foote and A. Cowman, Acta Trop., 1994, 56, 157 CrossRef CAS.
- S. Mok, M. Imwong, M. J. Mackinnon, J. Sim, R. Ramadoss, P. Yi, M. Mayxay, K. Chotivanich, K. Liong, B. Russell, D. Socheat, P. N. Newtone, N. P. J. Day, N. J. White, P. R. Preiser, F. Nosten, A. M. Dondorp and Z. Bozdech, BMC Genomics, 2011, 12, 391 CrossRef CAS.
- D. Klayman, Science, 1985, 228, 1049 CAS.
- Y. Wu, Acc. Chem. Res., 2002, 35, 255 CrossRef CAS.
- R. Haynes and S. Vonwiller, Acc. Chem. Res., 1997, 30, 73 CrossRef CAS.
- N. J. White, Science, 2008, 320, 330 CrossRef CAS.
- F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706 CrossRef.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940 CrossRef CAS.
- A. G. Griesbeck and A. Bartoschek, Chem. Commun., 2002, 1594 RSC.
- A. G. Griesbeck, T. T. El-Idreesy, M. Fiege and R. Brun, Org. Lett., 2002, 4, 4193 CrossRef CAS.
- A. G. Griesbeck, T. T. El-Idreesy, L. Höinck, J. Lex and R. Brun, Bioorg. Med. Chem. Lett., 2005, 15, 595 CrossRef CAS.
- R. A. Bourne, X. Han, A. O. Chapman, N. J. Arrowsmith, H. Kawanami, M. Poliakoff and M. W. George, Chem. Commun., 2008, 4457 RSC.
- R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322 CrossRef CAS.
- X. Han, R. A. Bourne, M. Poliakoff and M. W. George, Green Chem., 2009, 11, 1787 RSC.
- D. R. Worrall, A. A. Abdel-Shafi and F. Wilkinson, J. Phys. Chem. A, 2001, 105, 1270 CrossRef CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663 CrossRef CAS.
- J. F. B. Hall, X. Han, M. Poliakoff, R. A. Bourne and M. W. George, Chem. Commun., 2012, 48, 3073 RSC.
- A. G. Griesbeck, W. Adam, A. Bartoschek and T. T. El-Idreesy, Photochem. Photobiol. Sci., 2003, 2, 877 CAS.
- W. Adam and B. Nestler, J. Am. Chem. Soc., 1992, 114, 6549 CrossRef CAS.
- M. E. Cain, J. Chem. Soc., 1964, 3532 RSC.
- M. G. Hitzler, F. R. Smail, S. K. Ross and M. Poliakoff, Org. Process Res. Dev., 1998, 2, 137 CrossRef CAS.
- X. Han and M. Poliakoff, Chem. Soc. Rev., 2012, 41, 1428 RSC.
- J. Kaspar, A. Trovarelli, M. Lenarda and M. Graziani, Tetrahedron Lett., 1989, 30, 2705 CrossRef CAS.
- J. I. Di Cosimo, A. Acosta and C. R. Apesteguía, J. Mol. Catal. A: Chem., 2004, 222, 87 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36711d |
| ‡ Safety warning: These reactions involve high pressures and require an appropriately rated apparatus and with due regard to the potentially explosive reaction between O2 and organic compounds. |
| This journal is © The Royal Society of Chemistry 2013 |