Palladium supported on an acidic metal–organic framework as an efficient catalyst in selective aerobic oxidation of alcohols
Gongzhou
Chen
,
Shijian
Wu
,
Hongli
Liu
,
Huanfeng
Jiang
and
Yingwei
Li
*
Key Laboratory of Fuel Cell Technology of Guangdong Province and The Key Lab of Enhanced Heat Transfer and Energy Conservation, Ministry of Education, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: liyw@scut.edu.cn
First published on 22nd November 2012
Abstract
Highly dispersed palladium nanoparticles were deposited on an acidic MOF (MIL-101) by using a simple colloidal method. The resulting Pd/MIL-101 catalyst was highly active in the liquid-phase aerobic oxidation of a wide range of alcohols including benzyl, allylic, aliphatic and heterocyclic alcohols as well as diols, affording the desired oxidation products in high yields under base-free and mild conditions. The catalyst was shown to be able to efficiently catalyze aerobic oxidation even at ambient temperature using air instead of pure O2. The solvent-free oxidation of benzyl alcohol gave a remarkably high turnover frequency (TOF) of approximately 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 h−1. However, the catalytic activity was significantly suppressed when ethylenediamine was grafted on the uncoordinated Cr sites of the MIL-101 support, which suggests that the open Cr sites might play an important role in promoting the oxidation of alcohols in the present catalytic system.
900 h−1. However, the catalytic activity was significantly suppressed when ethylenediamine was grafted on the uncoordinated Cr sites of the MIL-101 support, which suggests that the open Cr sites might play an important role in promoting the oxidation of alcohols in the present catalytic system.
Introduction
Selective oxidation of alcohols is an important transformation in the chemical industry.1,2 The traditional oxidizing reagents, which have usually required stoichiometric amounts of metal oxidants such as chromate or permanganate, are expensive and have serious toxicity issues associated with them.3,4 With ever-increasing environmental concerns, there is substantial interest in the development of heterogeneous catalytic systems that use molecular oxygen as the oxidant.5 Accordingly, a remarkable number of transition-metal nanoparticles such as palladium,6–8 gold,9–17 platinum,18 and rhodium,19 have been used as catalysts for the aerobic alcohol oxidation processes. Although some of the metal catalysts have been shown to be effective for the aerobic oxidation reactions, in most cases they have been applied at temperatures above 100 °C or in the presence of a large excess of base additives.20–26 Moreover, most of the catalysts can be applied only to a very limited range of substrates under base-free conditions.20–22 Therefore, the development of excellent reusable catalysts for the liquid-phase aerobic alcohol oxidation that exhibit a wide range of substrate tolerance under mild and base-free conditions is still challenging in both green chemistry and organic synthesis.Metal–organic frameworks (MOFs) are a new class of porous materials assembled with metal ions and organic linkers.27 Owing to their high surface area, porosity, and chemical tunability, the applications of MOFs in heterogeneous catalysis have recently attracted tremendous attention, especially, those using MOFs as a support for metal (e.g., Pd, Au, Ru, and Pt) nanoparticles.28–41 A few MOF-supported noble-metal (e.g., Au, Pt) nanoparticles have also been investigated as active heterogeneous catalysts in the aerobic oxidation of alcohols,42,43 even under base-free conditions.35
Here we report a new and highly active heterogeneous Pd catalyst, which was deposited on a zeolite-type MOF (i.e. MIL-101) using a simple colloid method. The supported Pd nanoparticles have been shown to be highly efficient in the liquid-phase aerobic oxidation of a variety of alcohols in the absence of base under mild conditions. We have chosen MIL-101, Cr3F(H2O)2O[(O2C)–C6H4–(CO2)]3,44 because it has a large surface area and pore size as well as a good water stability, which jointly make it an attractive support for depositing small palladium nanoparticles. On the other hand, MIL-101 possesses numerous potentially unsaturated chromium sites (up to 3.0 mmol g−1) upon removal of the terminal water molecules.30,45,46 The significant Lewis acidity of the MIL-101 support has been demonstrated to play significant roles in promoting the reactivity of aromatic substrates.40,46 The effect of the open chromium sites of the support on the reactivity of alcohol oxidation was also investigated in this work.
Results and discussion
The powder XRD pattern of the as-synthesized MIL-101 matches well with the already published XRD patterns (Fig. 1). No apparent loss of crystallinity in the XRD patterns was found upon ethylenediamine grafting on MIL-101. After the incorporation of Pd nanoparticles, the structure of MIL-101 is mostly preserved regardless of the metal content in the materials. Moreover, no diffractions for Pd nanoparticles were detected, which imply that the Pd content is too low or Pd particles are too small in all samples. As indicated in the TEM images, Pd nanoparticles were well dispersed on MIL-101 with a uniform size of 2.5 ± 0.5 nm (Fig. 2).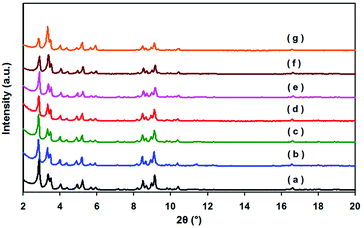 | ||
| Fig. 1 Powder XRD patterns of MIL-101 samples: (a) MIL-101; (b) 0.15% Pd/MIL-101; (c) 0.35% Pd/MIL-101; (d) 0.59% Pd/MIL-101; (e) 0.94% Pd/MIL-101; (f) 0.35% Pd/MIL-101 after five runs; (g) 0.35% Pd/En-MIL-101. | ||
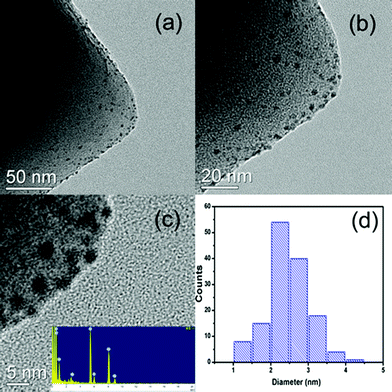 | ||
| Fig. 2 TEM images (a–c) of 0.35% Pd/MIL-101 and corresponding size distribution of Pd nanoparticles (d). The inset in (c) is the EDX pattern. | ||
The specific surface areas of the samples were measured by N2 adsorption at 77 K and the results are presented in Fig. 3 and Table 1. The appreciable decreases in nitrogen adsorption amount and surface area indicate that the cavities of MIL-101 are occupied by highly dispersed Pd nanoparticles or/and blocked by Pd nanoparticles that are deposited at the pore surface. In addition, the possible residual PVA (added as a protecting agent in the course of preparation) in the samples could also have some impact on the gas adsorption capacities of the catalysts.
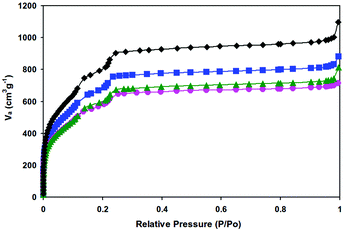 | ||
| Fig. 3 Nitrogen adsorption isotherms at 77 K of the as-synthesized MIL-101 (◆), 0.35% Pd/MIL-101 (■), 0.59% Pd/MIL-101 (●), and 0.35% Pd/En-MIL-101 (▲). | ||
| Sample | S BET (m2 g−1) | S Langmuir (m2 g−1) | V pore (cm3 g−1) |
|---|---|---|---|
| MIL-101 | 2869 | 4874 | 1.59 |
| 0.15% Pd/MIL-101 | 2506 | 3947 | 1.36 |
| 0.35% Pd/MIL-101 | 2412 | 3875 | 1.25 |
| 0.59% Pd/MIL-101 | 2084 | 2648 | 1.11 |
| 0.94% Pd/MIL-101 | 2029 | 2581 | 1.07 |
| 0.35% Pd/En-MIL-101 | 2173 | 2836 | 1.18 |
The XPS spectra of the 0.35% Pd/MIL-101 and 0.35% Pd/En-MIL-101 samples are shown in Fig. 4. The two samples both exhibit Pd 3d5/2 bands at ca. 335–336 eV (Fig. 4a), typical of Pd metal,47 indicating that the Pd(II) cations were transformed to Pd(0) after reduction by sodium borohydride. No appreciable differences were observed in the binding energies and intensities of the Pd(3d) spectra for the two materials. The results suggest that the ethylenediamine molecules were not grafted on the Pd nanoparticles nor had a strong interaction with Pd.
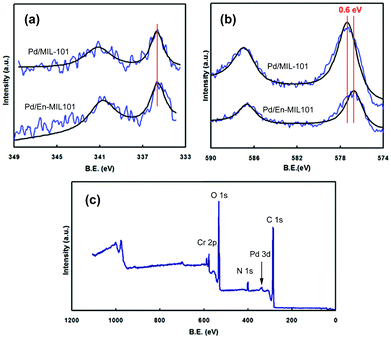 | ||
| Fig. 4 XPS spectra of 0.35% Pd/MIL-101 and 0.35% Pd/En-MIL-101 catalysts: (a) Pd3d; (b) Cr2p; (c) survey spectra of 0.35% Pd/En-MIL-101. | ||
Fig. 4b presents the Cr(2p) spectra of Pd/MIL-101 and Pd/En-MIL-101. It can be seen that the Cr(2p) spectrum of the amine-grafted material is less intense compared to Pd/MIL-101. In addition, the Cr(2p) peaks of Pd/En-MIL-101 are shifted by ca. 0.6 eV toward lower binding energies, compared to those of the pristine Pd/MIL-101. Such shifts reflect an increase in the electron density of Cr that may be attributed to the metal–ethylenediamine interactions on the Pd/En-MIL-101 sample. The incorporation of ethylenediamine in the sample was also confirmed by the presence of a well-defined band corresponding to N(1s) in the survey spectrum (Fig. 4c).
The infrared spectra of the 0.35% Pd/MIL-101 and Pd/En-MIL-101 samples are shown in Fig. 5. The γ(N–H) and γ(C–H) stretching regions observed for Pd/En-MIL-101 indicate the presence of ethylenediamine by comparing with the spectrum of ethylenediamine in the liquid phase (not shown here). The aliphatic C–H stretching vibrations (at 2800–3000 cm−1) are shifted to higher wavenumbers (from 2848 and 2924 cm−1 to 2860 and 2931 cm−1, respectively), as observed when the molecule is coordinated to a Lewis acid center,48 further demonstrating the successful grafting of ethylenediamine onto the coordinately unsaturated Cr sites of the MIL-101.
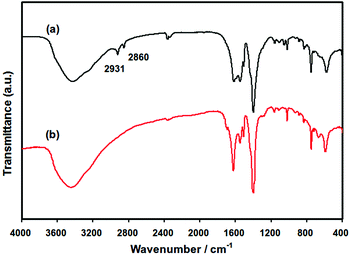 | ||
| Fig. 5 Infrared spectra of (a) Pd/En-MIL-101 and (b) Pd/MIL-101. | ||
The aerobic oxidations of alcohols were carried out under atmospheric O2 pressure and the prepared Pd/MIL-101 samples with different Pd loadings were used as catalysts. The results of the oxidation of cinnamyl alcohol are summarized in Table 2. The use of the parent MIL-101 gave essentially no conversion in this reaction system (Table 2, entry 1), confirming as expected the need of a metal to perform the oxidation of alcohol. The incorporation of Pd nanoparticles significantly promoted the reaction. Results of the aerobic oxidation pointed to an optimized performance of 0.35% Pd/MIL-101 (Table 2, entry 3), which provided a complete conversion of cinnamyl alcohol to cinnamyl aldehyde within 0.5 h at 80 °C. With the 0.35% Pd/MIL-101 catalyst, various solvents were subsequently screened, which indicated that toluene was the best solvent for this transformation under the conditions (Table 2, entries 6–10). In principle, the conversion increased with a decrease in the polarity of solvent. The relatively low activity of the reaction in isooctane (with a low polarity) may be attributed to the poor solubility of cinnamyl alcohol in this solvent. The activity could be enhanced remarkably when toluene was added in this system to improve the solubility of cinnamyl alcohol in the mixed solvents (Table 2, entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Conv./sel. (%) |
| a Reaction conditions: cinnamyl alcohol (1 mmol), catalyst (Pd 1 mol%), solvent (10 mL), 80 °C, 1 atm, O2 bubbling rate (20 mL min−1). b 1 eq. NaOH was added. | ||||
| 1 | MIL-101 | Toluene | 3 | <3/98 |
| 2 | 0.15% Pd/MIL-101 | Toluene | 0.5 | 75/97 |
| 3 | 0.35% Pd/MIL-101 | Toluene | 0.5 | 99/99 |
| 4 | 0.59% Pd/MIL-101 | Toluene | 0.75 | 93/99 |
| 5 | 0.94% Pd/MIL-101 | Toluene | 0.75 | 90/99 |
| 6 | 0.35% Pd/MIL-101 | DMF | 1 | 74/99 |
| 7 | 0.35% Pd/MIL-101 | o-Xylene | 0.5 | 97/99 |
| 8 | 0.35% Pd/MIL-101 | DMSO | 1 | 6/95 |
| 9 | 0.35% Pd/MIL-101 | Isooctane | 0.5 | 61/98 |
| 10 | 0.35% Pd/MIL-101 | Isooctane + toluene (1:1) |
0.5 | 78/99 |
| 11 | 0.35% Pd/En-MIL-101 | Toluene | 3 | 45/99 |
| 12b | 0.35% Pd/En-MIL-101 | Toluene | 3 | 95/99 |
| 13 | 0.35% Pd/AC | Toluene | 2 | 23/99 |

For comparison, the amine grafted sample 0.35% Pd/En-MIL-101 was also tested in the aerobic oxidation of cinnamyl alcohol under the optimized reaction conditions. The use of En-grafted MIL-101 as support led to inferior activity in the oxidation (Table 2, entry 11), giving only 45% yield of cinnamyl aldehyde even though the reaction time was prolonged to 3 h. Nevertheless, when a small amount of NaOH was added to the reaction system, the activity was restored significantly, furnishing 95% conversion of alcohol within 3 h (Table 2, entry 12). As is well known, the addition of base additives may promote the oxidation of alcohols by facilitating the dehydrogenation of alcohols.43 Therefore, this result implies that most of the Pd active sites were not covered by the ethylenediamine molecules, in agreement with the XPS data (Fig. 4a). The remarkably reduced activity of Pd/MIL-101 upon amine grafting on the unsaturated chromium sites of MIL-101 suggests that the uncoordinated Lewis acidic Cr sites have played an important role in the alcohol oxidation, which correlates well with the low activity observed for Pd/AC under the same reaction conditions (Table 2, entry 13). These findings also support previous findings of the group related to a promoting effect observed in MIL-101-supported metal nanoparticles for the conversions of aromatics.40,46
The scope of the presented catalyst system was then subsequently extended to the aerobic oxidation of a large variety of alcohols under base-free conditions using the optimum 0.35% Pd/MIL-101 catalyst. The results summarized in Table 3 show that Pd/MIL-101 was highly active and extremely selective for the oxidation of all substrates, indicating a high versatility of the MOF-supported Pd catalyst. Substituted benzyl alcohols containing electron-donating groups such as CH3, OCH3, or OH, (Table 3, entries 1–4) are more easily oxidized than those containing electron-withdrawing groups (Table 3, entry 5). Secondary benzylic alcohols were also converted to the corresponding ketones in quantitative yields (Table 3, entry 6). A complete conversion of allylic alcohols, for example, cinnamyl alcohol and trans-2-hexen-1-ol, to the desired aldehydes was achieved within 1 h (Table 3, entries 7 and 8). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds remained intact without an intramolecular hydrogen transfer over Pd/MIL-101 under the investigated conditions.
C double bonds remained intact without an intramolecular hydrogen transfer over Pd/MIL-101 under the investigated conditions.
| Entry | Substrate | Time (h) | Conv./sel. (%) |
|---|---|---|---|
| a Reaction conditions: alcohol (1 mmol), 0.35% Pd/MIL-101 (Pd 1 mol%), toluene (10 mL), 80 °C, 1 atm, O2 bubbling rate (20 mL min−1). b Pd 1.5 mol %, 100 °C. c Lactones were the predominant oxidation products. d Pd 3.0 mol %, 25 °C, air was used as the oxidant instead of pure O2. e Alcohol (10 mmol), toluene (30 mL), O2 bubbling rate (30 mL min−1). | |||
| 1 |

|
1.5 | 99/99 |
| 2 |

|
0.75 | 99/99 |
| 3 |

|
1 | 99/99 |
| 4 |

|
1.5 | 99/99 |
| 5 |

|
4.5 | 97/99 |
| 6 |

|
1.5 | 99/99 |
| 7 |

|
0.5 | 99/99 |
| 8 |

|
1 | 99/99 |
| 9b |

|
10 | 95/96 |
| 10b |

|
5 | 99/98 |
| 11b |

|
6 | 96/99 |
| 12b |

|
4 | 99/98 |
| 13b |

|
4 | 99/98 |
| 14c |

|
4 | 99/90 |
| 15c |

|
6 | 99/88 |
| 16d |

|
24 | 99/99 |
| 17e |

|
3 | 99/98 |
Aliphatic alcohols have been reported to be difficult compounds to undergo oxidation.9,35 Our Pd/MIL-101 catalyst was also applicable to the oxidation of various aliphatic alcohols including primary and secondary linear, and cyclic aliphatic alcohols, to afford the corresponding aldehydes or ketones in excellent yields within short reaction times under mild and base-free conditions (Table 3, entries 9–11). Heterocyclic alcohols, such as 2-pyridinemethanol and furfuryl alcohol, were also smoothly oxidized into the corresponding aldehydes in excellent yields (Table 3, entries 12 and 13). The Pd/MIL-101 catalyst was also active in the oxidation of diols, furnishing the corresponding lactones in high yields (Table 3, entries 14 and 15).
Encouraged by these promising results, we further examined the catalytic activity of Pd/MIL-101 for the aerobic oxidation of cinnamyl alcohol using air instead of pure O2 at ambient temperature. Cinnamyl alcohol was selectively oxidized into cinnamyl aldehyde in the absence of water or base, furnishing a complete conversion with 99% selectivity after 24 h of reaction (Table 3, entry 16). We also investigated the oxidation of cinnamyl alcohol on the 10 mmol scale, which gave a complete conversion with 98% selectivity for cinnamyl aldehyde (isolated yield: 94%) within 3 h (Table 3, entry 17).
The recyclability of the Pd/MIL-101 catalyst was examined in the aerobic oxidation of cinnamyl alcohol under the optimized conditions. After the catalytic reaction, the catalyst was isolated from the liquid phase by centrifugation, thoroughly washed with ethanol, and then reutilized as catalyst in subsequent runs under identical reaction conditions. The results included in Table 4 indicate that no efficiency loss was observed in the aerobic oxidation of alcohol for up to five runs. These findings were in accordance with AAS experiments for which no Pd traces (below the detection limit) were detected in the reaction solution as well as with the PXRD experiments of the catalyst after the fifth run which showed no appreciable changes in the crystallinity of the MIL-101 material (Fig. 1).

The high reactivity of the present catalyst system was further highlighted by a 50 mmol scale oxidation of benzyl alcohol employing 1 × 10−3 mol% Pd under base- and solvent-free conditions. The Pd/MIL-101 catalyst provided an extremely high turnover frequency (TOF) of 16900 h−1, with greater than 99% selectivity for the desired product. It is noteworthy that the TOF value achieved by Pd/MIL-101 is comparable to those reported for most other active catalysts, e.g., Pd/7Mn3Ce-C that afforded a TOF of 15235 h−1 for the conversion of benzyl alcohol under similar conditions as used in this study (Scheme 1).49
 | ||
| Scheme 1 TOF for the aerobic oxidation of benzyl alcohol for the initial 1 h of reaction (ratio of moles of benzaldehyde per mole of Pd per hour). Reaction conditions: 0.35% Pd/MIL-101 (1 × 10−3 mol%), benzyl alcohol (50 mmol). | ||
Conclusions
In summary, we have developed a highly efficient heterogeneous catalyst system for liquid-phase aerobic oxidation of a variety of alcohols using MIL-101-supported Pd nanoparticles as the catalyst. The catalyst can even efficiently catalyze the aerobic oxidation under ambient conditions in the absence of any basic additives. Moreover, the catalyst is recyclable. The combination of high activity and selectivity, wide substrate tolerance, and good stability and recyclability makes Pd/MIL-101 a potential material for practical applications in the aerobic oxidation of alcohols. Work is underway to investigate the catalytic mechanism and understand the acidic effect of MIL-101 on oxidations in our laboratories.Experimental
Catalyst preparation
Synthesis of Pd/MIL-101: a series of supported palladium catalysts were synthesized using a sol–gel method reported in our previous work for the preparation of Au nanoparticles on MIL-101.35 Using the synthesis of 0.35 wt% Pd/MIL-101 as an example, an aqueous solution of PdCl2 (200 mg L−1, 15.4 mL) was first prepared, and PVA was added as a protecting agent (PVA monomer/metal = 10:1 molar ratio, 7.6 mg). The mixed solution was placed in an ice bath and vigorously stirred for 1 h. Then, a freshly prepared aqueous solution of NaBH4 (NaBH4/metal = 5:1 (molar ratio), 0.1 M, 0.87 mL) was added under vigorous stirring to obtain a dark colloidal dispersion. The pH value of the solution was adjusted to 7–8 with diluted hydrochloric acid. After the Pd-PVA colloids were formed, the activated MIL-101 (0.5 g, dispersed in 5 mL of water) was immediately added. The suspension was stirred at 0 °C for 4 h, followed by washing thoroughly with deionized water. The sample was finally dried at 100 °C under vacuum for 2 h. The Pd loading on the sample was 0.35 wt% based on atomic absorption spectroscopy (AAS) analysis. An identical protocol was applied for the preparation of other Pd/MIL-101 catalysts except using different amounts of Pd reagents (calculated loadings: 0.16, 0.63, and 0.97 wt%). The actual Pd contents measured by AAS in these Pd/MIL-101 catalysts were 0.15, 0.59, and 0.94 wt%, respectively.
Synthesis of Pd/En-MIL-101: amine-functionalized MIL-101 was prepared according to the reported protocol.45 Identical procedures were followed to deposit Pd nanoparticles onto the En-MIL-101 as for the synthesis of Pd/MIL-101. The Pd content in the sample was ca. 0.35 wt% based on AAS analysis.
Synthesis of Pd/AC: prior to use, the activated carbon (DARCO®, SIGMA-ALDRICH, BET surface area: 981 m2 g−1) was washed with acetone and dehydrated at 150 °C for 12 h. Then, identical procedures were followed to deposit Pd nanoparticles onto the AC as for the synthesis of Pd/MIL-101. The Pd content in the sample was ca. 0.36 wt% based on AAS analysis.
Characterization
Surface area measurements were performed with N2 adsorption/desorption isotherms at 77 K on a Micromeritics ASAP 2020 instrument. Before the analysis, the samples were evacuated at 150 °C for 12 h. Powder X-ray diffraction patterns of the samples were obtained by a Rigaku diffractometer (D/MAX-IIIA, 3 kW) using Cu Kα radiation (40 kV, 40 mA, λ = 0.1542 nm). The palladium contents of the samples were determined quantitatively by atomic absorption spectroscopy (AAS) analysis on a HITACHI Z-2300 instrument. X-Ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos Axis Ultra DLD system with a base pressure of 10−9 Torr. Infrared spectra of the samples were performed in a Thermo Scientific Nicolet iS10 spectrometer using a Smart OMNI-Transmission accessory.Catalytic reactions
Typical procedures for aerobic oxidation of alcohol: Alcohol (1 mmol) and supported palladium catalyst (1 mol%) were added to 10 mL of toluene. The reaction mixture was stirred at 80 °C under 1 atm of O2 (O2 bubbling rate, 20 mL min−1). Upon reaction completion, the catalyst particles were removed from the solution by filtration and washed with toluene. The liquid phase was subsequently analyzed by GC/MS (Shimadzu GCMS-QP5050A equipped with a 0.25 mm × 30 m DB-WAX capillary column).For the recyclability tests, the reactions were performed under the same reaction conditions as described above, except using the recovered catalyst. Each time, the catalyst was isolated from the reaction solution at the end of the reaction, washed with ethanol, and then heated at 150 °C under vacuum.
Acknowledgements
This work was supported by the NSF of China (20803024, 20936001, and 21073065), the NSF of Guangdong (S2011020002397, and 10351064101000000), the program for New Century Excellent Talents in Universities (NCET-08-0203), and the Fundamental Research Funds for the Central Universities (2011ZG0009).References
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- M. Hudlicky, Oxidations in Organic Chemistry, ACS, Washington, DC, 1990 Search PubMed.
- G. Cainelli and G. Cardillo, Chromium Oxidants in Organic Chemistry, Springer, Berlin, 1984 Search PubMed.
- D. G. Lee and U. A. Spitzer, J. Org. Chem., 1970, 35, 3589 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS.
- A. Villa, D. Wang, N. Dimitratos, D. S. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8 CrossRef CAS.
- E. V. Johnston, O. Verho, M. D. Kärkäs, M. Shakeri, C. K. Tai, P. Palmgren, K. Eriksson, S. Oscarsson and J. E. Bäckvall, Chem.–Eur. J., 2012, 18, 12202 CrossRef CAS.
- F. Z. Su, Y. M. Liu, L. C. Wang, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2008, 47, 334 CrossRef CAS.
- A. Villa, C. E. Chan-Thaw, G. M. Veith, K. L. More, D. Ferri and L. Prati, ChemCatChem, 2011, 3, 1612 CrossRef CAS.
- M. Boronat, A. Corma, F. Illas, J. Radilla, T. Ródenas and M. J. Sabater, J. Catal., 2011, 278, 50 CrossRef CAS.
- D. Saio, A. Amaya and T. Hirao, Adv. Synth. Catal., 2010, 352, 2177 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- M. Haruta, Nature, 2005, 437, 1098 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- R. W. J. Scott, C. Sivadinarayana, O. M. Wilson, Z. Yan, D. W. Goodman and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 1380 CrossRef CAS.
- Y. H. Ng, S. Ikeda, T. Harada, Y. Morita and M. Matsumura, Chem. Commun., 2008, 3181 RSC.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538 CrossRef CAS.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768 RSC.
- V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2007, 9, 267 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- N. F. Zheng and G. D. Stucky, Chem. Commun., 2007, 3862 RSC.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS.
- H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151 CrossRef CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS.
- B. Z. Yuan, Y. Y. Pan, Y. W. Li, B. L. Yin and H. F. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054 CrossRef CAS.
- Y. Y. Pan, B. Z. Yuan, Y. W. Li and D. H. He, Chem. Commun., 2010, 46, 2280 RSC.
- Y. Zhao, J. Zhang, J. Song, J. Li, J. Liu, T. Wu, P. Zhang and B. Han, Green Chem., 2011, 13, 2078 RSC.
- M. S. El-Shall, V. Abdelsayed, A. S. Khder, H. A. Hassan, H. M. El-Kaderi and T. E. Reich, J. Mater. Chem., 2009, 19, 7625 RSC.
- H. Li, Z. Zhu, F. Zhang, S. Xie, H. Li, P. Li and X. Zhou, ACS Catal., 2011, 1, 1604 CrossRef CAS.
- Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 2899 CrossRef CAS.
- H. L. Liu, Y. L. Liu, Y. W. Li, Z. Y. Tang and H. F. Jiang, J. Phys. Chem. C, 2010, 114, 13362 CAS.
- H. L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304 CrossRef CAS.
- H. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302 CrossRef CAS.
- F. Schröder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, G. Van Tendeloo, B. Walaszek, G. Buntkowsky, H. H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119 CrossRef.
- Y. K. Park, S. B. Choi, H. J. Nam, D. Y. Jung, H. C. Ahn, K. Choi, H. Furukawad and J. Kim, Chem. Commun., 2010, 46, 3086 RSC.
- H. Liu, Y. W. Li, H. Jiang, C. Vargas and R. Luque, Chem. Commun., 2012, 48, 8431 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2011, 1, 856 CAS.
- S. Proch, J. Herrmannsdorfer, R. Kempe, C. Kern, A. Jess, L. Seyfarth and J. Senker, Chem.–Eur. J., 2008, 14, 8204 CrossRef CAS.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS.
- H. Liu, Y. W. Li, R. Luque and H. Jiang, Adv. Synth. Catal., 2011, 353, 3107 CrossRef CAS.
- V. I. Nefedov, Y. V. Salyn, I. I. Moiseev, A. P. Sadovskii, A. S. Berenbljum, A. G. Nizhnik and S. L. Mund, Inorg. Chim. Acta, 1979, 35, L343 CrossRef CAS.
- D. A. Young, T. B. Freedman, E. D. Lipp and L. A. Nafie, J. Am. Chem. Soc., 1986, 108, 7255 CrossRef CAS.
- Y. Chen, H. J. Zheng, Z. Guo, C. M. Zhou, C. Wang, A. Borgna and Y. H. Yang, J. Catal., 2011, 283, 34 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |